
Abstract
In small molecule drug discovery projects, the receptor structure is not always available. In such cases it is enormously useful to be able to align known ligands in the way they bind in the receptor. Here we shall present an algorithm for the alignment of multiple small molecule ligands. This algorithm takes pre-generated conformers as input, and proposes aligned assemblies of the ligands. The algorithm consists of two stages: the first stage is to perform alignments for each pair of ligands, the second stage makes use of the results from the first stage to build up multiple ligand alignment assemblies using a novel iterative procedure. The scoring functions are improved versions of the one mentioned in our previous work. We have compared our results with some recent publications. While an exact comparison is impossible, it is clear that our algorithm is fast and produces very competitive results.
Similar content being viewed by others
Introduction
When designing ligands to target a binding site, the receptor structure is not always available. In order to perform analysis like 3D QSAR, CoMFA [1], pharmacophore analysis, virtual screening, it is necessary or desirable to superimpose known ligands the way they bind in the active site. Over the years, a lot of effort has been spent tackling this problem of aligning small molecule ligands. Nevertheless, there are not many fully automated algorithms for aligning multiple ligands. Many publications are about aligning pairs of ligands. Others require the user to specify a rigid template ligand. A comprehensive review of the early methods has been written by Lemmen and Langauer [2]. More recent reviews have been provided by Wolber et al. [3] and by Leach et al. [4]. These two publications were portrayed as reviews on pharmacophore methods. But the problems of pharmacophore elucidation and of aligning ligands are very closely related. When aligning ligands, the use of pharmacophore representation can help by reducing the complexity of the problem compared to atom-based approaches, and thus speed up calculation. On the other hand, pharmacophore models can be readily derived once there is a reliable alignment of the ligands. However, the two problems are not exactly equivalent. Once an alignment of the ligands is obtained, any ligand based design technique can be employed. On the other hand, using the pharmacophore representation is not a must for tackling the alignment problem.
Readers are referred to the three reviews mentioned above [2,3,4] for descriptions of small molecule alignment algorithms published before 2009, including the widely mentioned commercial algorithms of GASP [5], CATALYST [6], FLEXS [7], MOE [8], ROCS [9], and PHASE [10]. Here we give a brief characterization of some algorithms published since 2009. MOGA, by Gardiner et al. [11], GAPE, by Jones [12], and the more recent publication by Taylor et al. [13], can all be considered extensions or improvements of GASP [5]. They were built upon the conventional pharmacophore representation of hydrogen bond acceptors/donors, hydrophobes, etc., and use genetic algorithm to obtain alignments of flexible ligands. PharmACOphore, by Korb et al. [14], and PENG, by Moser et al. [15], are also based on the conventional pharmacophore approach. Instead of using genetic algorithm for optimization, like references [5, 11,12,13], pharmACOphore uses ant colony optimization, while PENG uses a neural gas approach. For PENG, it is not guaranteed that all ligands will be aligned. Instead of using the conventional pharmacophore approach, the FAP algorithm by Shin et al. [16], and Open3DAlign, by Tosco et al. [17], use formal or partial atomic charges from empirical force fields to score the alignment of ligand pairs. FAP aligns a query ligand onto a rigid template ligand. Poses are generated systematically, followed by a relaxation of the query ligand with respect to the energy and the alignment score. Open3DAlign aligns rigid conformers onto a rigid template ligand. Alignment poses are generated either using pharmacophore elements or from a matching of atoms based on the scoring function. Avalanche, by Diller et al. [18], provides a method for aligning pairs of rigid ligands. The score is based on the pharmacophoric similarity between each atom pair. Alignment poses are generated using pairs of similar atoms on the two ligands. FKCOMBU, by Kawabata and Nakamura [19], aligns ligands onto a rigid template. A mapping of atoms is derived from the common 2D substructures (can be more than one connected piece) between the query ligand and the template. A score is derived from this mapping, and is optimized by varying the torsion angles and the position of the query ligand. It is not clear how the method performs for ligands with little chemical similarity. Instead of focusing on the ligands themselves through pharmacophore elements or the atoms, like the previously mentioned algorithms, SHAEP, by Vainio et al. [20], and FLAPpharm, by Cross et al. [21], focus on the molecular field around the ligands. SHAEP uses the electric potential around the ligands derived from empirical force field charges to perform pairwise alignments. In FLAPpharm, the propensities of hydrogen bonding, hydrophobic, and other interactions are measured on a grid surrounding the ligands. Leherte and Vercauteren [22] compared the performance of a few different molecular fields towards the pairwise alignment of rigid ligands. PHASE-shape, by Sastry et al. [23], generates alignment poses between pairs of rigid conformers using triplets of similar atom pairs, and scores them based on the volume overlap. Both DIFGAPE [24], by Jones et al., and MARS [25], by Klabunde et al., use pairwise alignment results from ROCS [9] to generate multiple ligand alignments. DIFGAPE uses genetic algorithm and its own scoring function. MARS uses helper ligands to build up multiple ligand alignments, which are evaluated using ROCS score.
Methods
The current algorithm takes pre-generated conformers as input. It does not alter their geometries. This is similar to many existing algorithms, including references [6, 9, 10, 15, 17, 18, 20,21,22,23, 25], and one of the two algorithms mentioned by Jones in ref. [12]. While PHASE [10] and FLAPpharm [21] do provide their own conformer generators, the alignment algorithms are really uncoupled from the generation of conformers.
Our algorithm consists of two stages. The first stage is to perform an alignment for each pair of ligands, using the available conformers. The second stage is to build up multiple ligand assemblies based on the pairwise alignment poses from the first stage, using a novel iterative process. Here we shall first describe the scoring schemes. We shall then introduce some definitions and concepts. After this, we shall describe the pairwise alignment procedure, followed by the procedure to obtain multiple ligand alignment assemblies. Finally, we shall describe the mechanism for removing duplicated solutions.
The scoring functions
Different scoring schemes are used for different stages of the algorithm. Simple pairwise scoring is used in the pairwise alignment stage. Fast scoring is used during the generation of the multiple ligand alignment assemblies. Precision scoring is used for the final ranking of the assemblies. All scoring functions are based on our previous work [26], which we shall refer to as the old scoring scheme. Changes have been made to improve the treatment of hydrogen bond projected features (the expected positions of the hydrogen bonding partners on the binding pocket, based on ligand geometry). In order to improve efficiency and to make the situation more tractable, projected features are not taken into account until the very last step, when the multiple ligand alignment assemblies are scored for ranking using precision scoring.
In the old scoring scheme, the score for a pair of ligands consists of a summation of terms, Tk, each of which has the form
where k denotes a particular pair of atom/feature types (e.g., hydrogen bond acceptors), w k and α k are parameters associated with that atom/feature type pair, A k and B k correspond to the lists of atoms/features in ligand 1 and ligand 2 belonging to that atom/feature type pair, r ij is the distance between the atom/feature pair (i, j). For more detail, refer to Table 1 of ref. [26].
In short, the old scoring scheme rewards atomic overlap in general. Overlaps of atoms of the same type (hydrogen bond donor/acceptor, or hydrophobic atom) are particularly encouraged. Hydrogen bond projected features are taken into account in a similar way. They are treated like the hydrogen bond donor and acceptor atoms, except that they have a more diffuse character (a smaller value of α in Eq. (1)), i.e., the score is more forgiving towards deviations from the ideal overlap.
In simple pairwise scoring, which is used during the pairwise alignment stage, we take out the terms involving projected features (T8 and T9 of Table 1 in ref. [26]) to simplify the calculation. Otherwise we simply employ the old scoring scheme.
When multiple ligand alignment assemblies are being generated in stage two, fast scoring is used. The score of an assembly is the sum of the simple pairwise scores for all ligand pairs.
The rest of this section describes the precision scoring scheme.
In precision scoring, the score of an assembly is still the sum of all pairwise scores, and based on the old scoring scheme. The old scheme is modified to allow for a better treatment of the hydrogen bond projected features. It was prompted by visual inspections of multiple ligand alignment results during the early stages of development of this project.
For a pair of ligands to form the same hydrogen bond with the binding pocket, their corresponding hydrogen bonding atoms must have at least one pair of projected features in proximity, and this neighborhood must be accessible (not blocked by any of the ligands in the alignment assembly). In order to quantify this, we use the following mathematical construct.
Let q be a point in space. Let k be a heavy (non-hydrogen) atom, with van der Waals radius r k , in the alignment assembly. Let d qk be the distance between q and the centre of atom k. The inaccessibility of q due to atom k is given by a bell shaped function of the form \(\frac{1}{{1+{x^4}}}\). Here x is a normalized distance between q and k. The accessibility of q due to atom k, \({S_{acc,k}}\), is one minus the inaccessibility, i.e.,
where
where r k is the van der Waals radius of atom k, r p is the average of the van der Waals radii of a nitrogen and an oxygen atom (nitrogen and oxygen are typical hydrogen bonding atoms, which may be present on the receptor to fulfill the projected feature), which is 1.535 Å. The overall accessibility \({S_{acc}}\) of point q is the minimum of all accessibilities, \({S_{acc,k}}\), calculated using each heavy atom k in the alignment assembly except for the hydrogen bonding atoms associated with the project features. The \(\frac{1}{{1+{x^4}}}\) form is used instead of the Gaussian form of \(\exp ( - \propto {x^2})\) because it gives a wider plateau which we believe better reflects the requirement for this function.
Let A and B be a pair of compatible hydrogen bonding atoms from the two ligands. Let PAi be one projected feature from atom A, and PBj be one from atom B. Let d ij be the distance between PAi and PBj. For this pair of projected features, the proximity score \({S_{prox}}\) is the same as the term for projected features in the old scoring scheme (T8 and T9 of Table 1 in ref. [26]), namely,
where α is 0.125 Å−2. The accessibility score, \({S_{acc}}\), at the mid-point of PAi and PBj is calculated. The overall score \({S_{ij}}\) is the product of \({S_{prox}}\) and \({S_{acc}}\). As for the atom pair A and B, the hydrogen bonding score, S HB , is the best score \({S_{ij}}\) amongst all of their projected feature pairs, (PAi, PBj)’s. For a summary, see Fig. 1.

A summary to aid the explanation of precision scoring. See text for details. HB hydrogen bonding or hydrogen bond, proj feat projected feature
The score of a multiple ligand alignment assembly is the sum of all scores between all pairs of ligands. The current scoring scheme for each pair of ligands is obtained from the old scoring scheme (see Table 1 in ref. [26], and Eq. (1)) after applying the changes described in this paragraph. First of all, terms T0 (heavy atoms) and T3 (hydrophobic atoms) remain unchanged, while the projected feature terms, T8 and T9, are discarded. In order to take into account effects of the projected features, the weights (w k in Eq. (1)) of the terms involving hydrogen bond acceptors and donors have been modified. For the donor–donor and acceptor–acceptor terms (T1 and T2), instead of using a simple constant weight of 4 for w k , w k is now set to 12SHB (SHB has a value of between 0 and 1). For the cross terms between hydrogen bonding and hydrophobic atoms (T4 and T5), instead of using a constant value of −1 for w k , w k is now set to the negative value of the highest accessibility \({S_{acc}}\) amongst all projected features of the hydrogen bonding atom (see summary in Fig. 1). In other words, if at least one projected feature of the hydrogen bonding atom is fully accessible, w k will be −1 for terms T4 and T5, which would be the same as in the old scoring scheme. Generally, if all projected features of a hydrogen bonding atom are inaccessible, it will not contribute to any term other than T0, the generic volumetric overlap term.
Comparing the current precision scoring scheme with the old scheme, projected features are now generated for each hydrogen bonding atom and checked for accessibility. In the old scheme, all pairs of projected features that are in proximity contribute to the score, regardless of the accessibility of the feature; and all pairs of acceptor (and donor) atoms contribute an equal amount to the score, regardless of the accessibility and proximity of their projected features.
To summarize, in precision scoring, the score is the sum of all pairwise scores. Compared to the old scheme, these pairwise scores now include better consideration of the hydrogen bond projected features and can depend on other ligands in the assembly.
Representative points
Representative points are used for generating pairwise alignments, and for removing duplicates amongst the final solutions. Given a ligand, representative points are generated as follows. First of all, hydrogen atoms are discarded. Then each hydrogen bond donor or acceptor atom is chosen as a representative, covering the atom itself as well as all atoms bonded to it. The ring centre for each ring of size 7 or less is a representative, covering all atoms in the ring. Of the atoms that are not yet covered, branching atoms (atoms bonded to three or more heavy atoms) are chosen as representatives, covering the atoms themselves as well as all atoms directly bonded to them. If there are still any atoms left uncovered, they would reside within chains of bonds. These chains are split into groups of two or three atoms, and the (unweighted) centroid of each group becomes a representative, covering all atoms in the group. During each of the above stages, all representatives are chosen in parallel (i.e. not affected by other representatives chosen in the same stage). The chain splitting mechanism is symmetric with respect to the two ends of the chain. The generation of representative points is thus canonical.
A pair of representative points from different ligands is defined to be compatible if they are both hydrogen bond donors, or both hydrogen bond acceptors, or neither is a hydrogen bonding atom (which includes non-atom representative points like ring centres). A set of n representative points from ligand A is considered compatible to a set of n representative points from ligand B if, (1), each of the n pairs of corresponding points (one from each set) is compatible; and, (2), the distance between each pair of representative points in one set is within a tolerance value of the corresponding distance in the other set. In simpler terms, a set of representative points from one ligand is compatible with a set from another ligand if they resemble each other in characteristic and in geometry.
In a sense, these representative points are similar to the pharmacophore elements in many publications which are used directly to generate and score alignments. Using pharmacophore/representative points instead of atoms speeds up calculation by substantially reducing the size of the search space. Moreover, the pharmacophore model can make a simple, tractable representation of the essential characteristics important for binding, and can also be used for virtual screening. However, our experience showed that scoring schemes based on them do not give enough resolution to distinguish between good and bad alignments. One problem is that hydrophobic or steric pharmacophore features are difficult to be defined unambiguously. Table 3 in the review by Wolber et al. [3] gives a good illustration of how different programs place hydrophobic features on ligands. Indeed, even aromatic ring centres do not always superimpose exactly on top of each other in alignments of crystallographic structures, especially for multiple ring systems. Therefore although we use them to sample the solution space, we do not use them for score evaluation. Nevertheless, for the sampling of the solution space, efficiency is greatly improved compared with an atom-based approach.
Generating pairwise alignments
There are two major methods for generating pairwise alignments, and a random method that works as a last resort. The systematic method is mostly deterministic, but is relatively slow. It is used when there are fewer than 1000 conformers in total. Otherwise the simultaneous multiple pose generation method is used. All these pose generators make use of the pair pose registers, which we shall describe now.
The pair pose registers
For each pair of ligands, a pair pose register is set up to record the \({n_{slot}}\) best scoring pairwise alignment poses. Instead of using a constant number of slots for all registers, there are more slots for bigger ligands and for ligands with more conformers. It is also desirable that for a pair of small ligands with few conformers, there is still a minimum number of slots. Let \({c_1}\) and \({c_2}\) be the numbers of conformers, and \({r_1}\) and \({r_2}\) be the numbers of representative points, for the two ligands. The number of slots, \({n_{slot}}\), for the register for this ligand pair is given by
so that the above requirements can be satisfied without taking up too much memory for big ligands with many conformers. Experiments showed that the quality of the results is not very sensitive to variations in the factor of “5” in the formula.
Whatever the method for generating pairwise poses is, the method proposes poses, one pose at a time, of the ligand pair to the pair pose register of that ligand pair. Upon receiving a pairwise pose, the pose register calculates its pairwise alignment score. If all slots in the register have been filled and the score for the current proposed pose is inferior to the worst scoring pose in the register, the current pose is ignored. Otherwise, if the current proposed pose is similar (within 2 Å rmsd for the heavy atoms) to a pose in the register, only the better scoring pose is kept. Otherwise the current pose displaces the pose with the worst score in the register.
Note that there is one pair pose register for each pair of ligands. Some conformers may not appear in a register.
Systematic pose generation
In systematic pose generation, for each conformer, the distances between all pairs of representative points are calculated. Then the algorithm processes each pair of conformers independently. The next paragraph describes the processing of one pair of conformers.
Based on the distance matrices with respect to the representative points, maximal cliques corresponding to compatible sets of three or more representative points are obtained using the Bron and Kerbosch algorithm [27]. The distance tolerance value (see “Representative points” section above) is set to 1.0 Å. Each maximal clique gives a mapping between a pair of compatible sets of the representative points from the two conformers. The Kabsch algorithm [28] is employed to obtain a rotation-translation to minimize the rmsd between the two compatible sets of representative points. The resulting pairwise pose is transferred to the pair pose register of the corresponding ligand pair. In order to avoid generating a large number of pairwise alignments due to local similarities, there is a geometric size limit for the compatible sets of representative points. Let’s define the diameter of a set of points to be the maximum distance between any pair of points within the set. The diameter of a compatible set is required to be at least half the length of the smaller diameter of the two representative point sets of the two conformers. Otherwise the compatible set is not considered.
After considering all maximal cliques of all conformers of a ligand pair, ten or more register slots would usually have been filled. If fewer than ten slots have been filled, the process is repeated using a tolerance value of 1.5 Å (instead of 1.0 Å). If still fewer than ten slots have been filled, more alignment poses will be obtained using compatible sets of two representative points. Each set of two representative points gives rise to eight alignment poses, separated by 45° of rotation about the axis through the two representative points. These eight poses are transferred to the pair pose register.
For cliques with more than three vertices, it is possible that the chiralities of the compatible sets of representative points are different. But we do not compare the chiralities. We just let the pairwise alignment score do its work. Unfavorable poses do not score well, and are automatically rejected by the pair pose register.
Simultaneous multiple pose generation
This pose generation method has a random element. It is carried out in cycles until there is convergence, as explained in the “Termination criterion” section in the “Appendix”. Each cycle is based on one query triplet of representative points. This query triplet is chosen randomly according to the protocol described in the “Choosing random query triplets” section in the “Appendix”. An absolute position for this triplet is set up, based on the source of the triplet. For each conformer of each ligand, all triplets of representative points compatible with the query triplet are identified. For each compatible triplet, a pose of the conformer is generated by performing a least square fit of the compatible triplet onto the query triplet using the Kabsch algorithm [28]. Hence, the number of poses of a conformer is the same as the number of compatible triplets it has. Some conformers may have no pose while others may have multiple poses. After processing all conformers, a grand assembly of conformer poses will have been generated. For each pair of poses in this assembly, unless the two poses are from conformers of the same ligand, the pair of poses is transferred to the pair pose register of the corresponding ligand pair.
After the process terminates, some pair pose registers may still have fewer than ten slots filled. Typically, these registers involve at least one small ligand. Larger ligands, with more representative points, are unlikely to be missed by the random sampling. Systematic pose generation will be run for the relevant ligand pairs to fill up these registers.
Random pose generation
When the above methods fail to generate ten or more poses for a pair of ligands, the random method is employed. A random triplet of heavy atoms on a random conformer of one ligand is superimposed onto a random triplet of heavy atoms on a random conformer of the other ligand, using the Kabsch algorithm [28], which minimizes the rmsd of these three atom pairs. The alignment pose is transferred to the pair pose register. This process is repeated until the register does not change for 250 consecutive attempts (i.e., no good scoring pose that differ substantially from the existing ones in the register has been found after 250 random triplets).
After the application of the random method, if required, there will not be any empty pair pose registers.
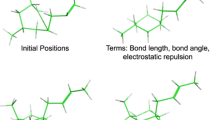
Figure 2 gives a simplified scheme illustrating the pairwise alignment procedure.

A simplified scheme illustrating the pairwise alignment stage. See main text for details. confs conformers, nconf total number of conformers, RPs representative points
Generating multiple ligand assemblies
To construct a multiple ligand alignment assembly, we start by picking one of the conformers as the base template. Generally, for each of the other ligands, the best scoring pose found from the pairwise alignment with the template is transferred to the assembly. This generates a starting point assembly. For more detail, see “Starting points for multiple ligand alignment” section in the “Appendix”. For ligand pairs that do not involve the template, their positions relative to each other may not be optimal. An iterative process is carried out to optimize the assembly. In each iterative cycle, we attempt to replace the ligands, one at a time, to improve the overall score of the assembly. Given an assembly, for each ligand, we analyze the pairwise alignment scores between this ligand and each of the other ligands. Let s ij be the pairwise alignment score between ligands i and j in the assembly. Let b ij be the best alignment score between (any conformer of) i and j found in the pairwise alignments. The score deficit, d ij , for this ligand pair is defined to be the difference between s ij and b ij . The ligand i with the largest total score deficit, \(\sum\nolimits_{j} {d_{{ij}} }\), is earmarked for replacement. New poses for the earmarked ligand are generated by using each of the other ligands in the assembly, one ligand at a time, as a helper. For each of the poses from the pairwise alignment results between the earmarked ligand (any conformer) and the helper conformer, a new pose for the earmarked ligand is generated based on the coordinates of the helper in the assembly and in the pairwise alignment results. Out of all these assemblies with a new pose for the earmarked ligand, the best scoring assembly is chosen. This cycle is repeated until no more score improvement is possible, as explained in the next few sentences. If no score improvement can be found in an iterative step, the earmarked ligand will be marked as unavailable. The ligand with the next largest total score deficit will be earmarked for replacement. When all ligands become unavailable, the iterative process will stop. The final assembly becomes one solution. Note that once a replacement is successful (i.e., a new assembly with an improved score is found), all ligands will be marked available again. This whole iterative process is illustrated in Fig. 3.

Generating multiple ligand assemblies from pairwise alignment results
Fast scoring is used during the iterative process. Since the fast score between a pair of ligands does not depend on the other ligands, when the earmarked ligand is re-posed, only the pairwise scores involving the earmarked ligand need to be recalculated. After the iteration process finishes, the assemblies are scored and ranked using precision scoring.
Duplication removal
Very similar assemblies can be generated from different starting points. After all (up to 250) solution assemblies have been generated, there is a duplication removal step. For each ligand, the positions of all representative points are noted. The distances between all pairs of representative points in the assembly are calculated. To compare two assemblies, corresponding distances are compared across the two. If all corresponding distances differ by <2 Å, the two assemblies are deemed equivalent and the one with the less favorable score is eliminated. In principle, it is possible that two assemblies that are almost mirror images of one another can be considered equivalent by this method. But this is unlikely to happen unless all ligands are achiral, in which case the mirror images of all solutions are automatically equally viable solutions.
Geometric optimization
During our research we also experimented with a rigid-body geometric optimization step. After the pairwise alignment stage, each registered pose pair was subjected to a rigid-body optimization to improve the score of the pair. Separately, we also tried adding a rigid-body optimization step to the final multiple ligand alignment assemblies. While these do improve the scores, the improvements do not justify the time spent. Geometric optimization is relatively slow compared to other components of our algorithm. There are more efficient ways of spending time for score improvement, e.g., using more starting points for the multiple alignment stage. Therefore we do not include any geometric optimization in our algorithm.
Results and discussion
The main test sets and conformer generation
While there have been many publications on small molecule alignments, there has previously not been a suite of systematically derived validation sets for calibrating alignment algorithms, until the publication of ref. [29] in 2012 and [30] in 2013. This is perhaps due to the limited amount of available high quality experimental data before entries of the Protein Data Bank (PDB) [31] took off exponentially in the last decade. The validation sets constructed by both publications were derived from the PDB. Not surprisingly, there is a lot of overlap between these two suites of sets. Both suites are available to the public. We found that the sets from ref. [29], in its current version, contain a lot of errors (e.g. bonding data, formal charge). Recently, Giangreco et al. [32] applied the molecular alignment algorithm of Taylor et al. [13] to the validation sets of ref. [30]. Here we shall compare our results on these validation sets with theirs. There are 121 sets. The maximum number of ligands in one set is 39, but only ten sets have more than 24 ligands. The whole suite is available to the public through the web site of the Cambridge Crystallographic Data Centre, as the AstraZeneca Overlays Validation Test Set, at http://www.ccdc.cam.ac.uk/support-and-resources/downloads/. The data comes in the SD format.
Before conformers were generated, ligand structures were converted to 2D Smiles strings using the RDKit [33]. Conformers were generated using Balloon [34] and Confect [35].
Preliminary work suggested that using the default settings, Balloon [34] has a tendency to generate comparatively curled up conformers due to electrostatic interactions. Since apparently there is no simple way to turn off electrostatic interactions, the dielectric constant was set to a large value of 10,000 to reduce the electrostatic effect. The initial population size for the genetic algorithm was set to 100. Default values were used for all other settings. Balloon generated 87,890 conformers for the 1464 ligands. All ligands have conformers generated. For 95% of the 1464 ligands, at least one conformer with heavy atom rmsd of under 1.5 Å was generated. Only 5 ligands did not have a conformer within 2.0 Å.
For Confect [35], different quality levels can be set to control the granularity to which conformational space is sampled. At the default quality level of q = 1, Confect did not generate any conformers for 98 out of the 1464 ligands. Using a quality level of q = 21, only 21 out of the 1464 ligands did not have conformers. For our alignment runs, we primarily used conformers from the q = 1 runs. For ligands with no conformers, conformers from the q = 21 runs were used. For the 21 ligands that still did not have any conformers, Balloon conformers were used. Altogether there were 40,724 conformers for the 1464 ligands. For 91% of the 1464 ligands, at least one conformer with heavy atom rmsd of under 1.5 Å was generated. Only 37 ligands did not have a conformer within 2.0 Å.
Success criteria
As mentioned time and again in the literature, it is not unusual for calculated alignments to look more reasonable than the experimental alignment. It is impossible to derive a scoring function that always ranks the experimental alignment the highest. In a drug development project, it is not unreasonable to expect a chemist to inspect and explore a handful of alignment results. Hence we believe that as long as we can obtain a good alignment within the top couple of spots, the algorithm can be considered useful. To measure the quality of a single alignment solution, we used two criteria, as we shall describe now.
Geometric criterion
Naturally, many publications concerning small molecule alignment algorithms use the heavy atom rmsd from the experimental answer as one measure of success. In ref. [12] an alignment is considered successful if half or more of the ligands can be fitted onto the crystal alignment to within 2 Å heavy atom rmsd. Under this criterion, individual ligands, especially those considerably smaller than the average, can still have a heavy atom rmsd of over 2 Å. Ref. [32] tightens this criterion by requiring that only ligands with a heavy atom rmsd of no more than 2 Å are considered correct. A calculated alignment is considered successful if it can be fitted onto the experimental alignment with at least half the ligands correct. Here we follow this more stringent criterion as one measure of success.
We shall also report the sizes (number of ligands) of the largest correctly aligned groups. Each ligand in a correctly aligned group must be within 2 Å heavy atom rmsd compared to the experimental answer. Note that the content of the largest correctly aligned group may not be unique. For example, there may be a way to fit ligands 1, 2, 3 onto the experimental alignment such that they are all correct, and a way to fit ligands 1, 2, 4, while it is impossible to fit all four ligands.
Topological criterion
Even short of an accurate geometric prediction, an alignment can be useful if it gives a good matching between atoms from different ligands. Based on a good atom match, synthetic chemists can make modifications to probe various parts of the binding pocket. Our second measure of success is based on the atom correspondence between aligned ligands. Basically, we would like atom neighbors in the experimental alignment to remain atom neighbors in the calculated alignment. The following paragraph describes a quantitative measure.
Let A and B be two ligands in a test set. A pair of atoms, one from each ligand, is considered neighbors in the experimental alignment if they are <1.5 Å apart. Suppose there are n AB such atom neighbor pairs. In the calculated alignment, we go through each of these n AB pairs to see if they remain neighbors. To allow for small movements, the pair is considered neighbors in the calculated alignment if they are <2.5 Å apart. Out of the n AB pairs, the fraction that remains neighbors in the calculated alignment is noted. Let f AB be this fraction, which we shall also term the topological similarity between the calculated and the experimental alignments for this ligand pair. (In the special case where n AB is 0, i.e., no overlap between A and B, f AB is considered 1, since it is unreasonable to expect an alignment algorithm to be able to reproduce this.) As can be inferred, this fraction is asymmetric with respect to the experimental and the calculated alignments. Our construction is not unreasonable. Consider an experimental alignment where the tails of two ligands occupy different parts of the binding pocket. One cannot expect an alignment algorithm to reproduce this. In other words, we believe that a good alignment algorithm must recognize neighbors as neighbors, but may still make non-neighbors neighbors.
A group of ligands is considered correctly aligned if, (1), for all pairs of ligands, (A, B), in the group, f AB is at least 0.75; and, (2), the average of all f AB ’s is at least 0.80. A set is considered successfully aligned by the topological criterion if there exists a group of correctly aligned ligands comprising at least half of all ligands. Similar to the geometric criterion, the content of the largest (by the number of ligands) correctly aligned group may not be unique.
While the topological criterion is usually easier to achieve than the geometric criterion, it is not a loose criterion. As we shall see, it is not unusual for a group of ligands to be considered correctly aligned using the geometric criterion but not the topological criterion (see Table 1 or the bottom region of Table 2). An example will be given when we analyze our results (Fig. 8).
Results on the main test sets
Ref [32] classified the 121 test set into four levels of difficulty. 22 sets were classified as easy, 73 as moderate, 18 as hard, and 8 as unfeasible.
In order to provide a baseline ideal scenario where the conformer generation procedure does not come into play, we first ran MolAlign using only the experimental conformers. This provides an upper limit of how well an algorithm can perform. Table 1 gives our results. Only the best scoring alignment was considered. Using the geometric criterion, in 7 of the 22 easy sets, all but one ligands were correctly aligned. For the remaining 15 easy sets, all ligands were correctly aligned. As for the 73 moderate sets, on average, 76% of the ligands were correctly aligned. In all but five sets, at least half the ligands were correctly aligned. For the 18 hard sets, the average percentage of correctly aligned ligands was 54%. We were not able to align half or more ligands for any unfeasible set. All in all, we correctly aligned at least half the ligands for 100 of the 121 sets, when only the top one scoring solution was considered.
Supplementary Tables S1 and S2 (Online Resource) give our results using conformers from Balloon [34] and Confect [35], respectively. Here we always considered the top five solutions. The assemblies with the largest (in terms of number of ligands) correctly aligned group are reported. Recall that there are two algorithms for generating the pairwise alignments. For test sets with a total of fewer than 1000 conformers, the Systematic Pose Generation method was used. This method is mostly deterministic. For other sets, the Simultaneous Multiple Pose Generation method was used. This method involves a random seed. Five runs were performed. Results for the first three runs are presented in the tables.
From Tables S1 and S2 (Online Resource), according to the topological criterion and considering the five top solutions, almost all runs managed to correctly align at least a quarter of the ligands (anything other than a dash in the table entry). The only exceptions are P59071 for the Balloon runs, and P00811 for the Confect runs. Both sets were classified as unfeasible. As for the 22 easy sets, out of the 44 runs, half resulted in all ligands being correctly aligned. Only four runs resulted in more than one incorrect ligand. For these four runs, three had two incorrect ligands, while the Balloon run for P56658 had three incorrect ligands. P56658 will be discussed in more details below.
Table 2 gives a summary of our results on a more general level. The result for a test set was considered either successful or not. As described above, an alignment was successful (according to either the geometric or the topological criterion) if at least half the ligands were correctly aligned in one group. A run was successful if there is a successful alignment within the top 5/10/15 (see Table for more details) solutions. As mentioned above, for test sets with a total of more than 999 conformers, five runs were performed. Success means success in at least three of the five runs. Results from ref. [32] are also shown in the table for comparison. Since their algorithm is stochastic, for each test set, ten runs were performed. A run was considered successful if the top one scoring alignment was successful, as defined using the geometric criterion. The numbers of successful runs are given in the table. Three scoring schemes were used, and the results are given in separate columns. One of their scoring schemes, the AlignScore, is retrospective. It measures the similarity between a solution and the experimental answer, i.e., when this score was used, a run was considered successful if a correct solution was amongst the output, which contained up to 20 solutions.
Table 3 summarizes the results on an even higher level. It reports the overall statistics of Table 2 for each of the four categories of difficulty. Columns AS10, BT10, SE10 correspond to results from ref. [32], using AlignScore, Borda Tally, and the strain energy, respectively. Their runs are stochastic. A set was considered successful if the top one solution in at least one out of ten runs was successful, i.e., any non-zero entry in the corresponding column in Table 2. Column BTSE gives the numbers of successes covered by either BT10 or SE10, i.e., any non-zero entry in either BT10 or SE10 of Table 2. Our results are also given in Table 3, when considering the top five (columns B5, C5), top ten (columns B10, C10), and top fifteen (columns B15, C15) solutions for each run. For stochastic runs, a success means at least three successes out of five runs.
According to the topological criterion, if we consider our top five solutions (letter “A” in Table 2, columns B5, C5, BC5 in Table 3), we have successfully aligned all easy sets. From Table 3 column BC5, if we consider the top five solutions from both the Balloon and Confect runs (i.e. ten solutions), 90% of the 73 moderate sets were successful, while the overall success rate for all 121 sets was 80%. From column BC15, if we consider the top 15 solutions from both runs (i.e. 30 solutions), all but 4 of the 73 moderate sets were successful, and 61% of the 18 hard sets were successful.
Table 4 is a summary by run time. For sets with multiple runs, the average run times are reported. For flexible alignments using the Confect conformers, 46% of the test sets finished within 1 min on a Pentium T4400 2.2 GHz CPU.
Case studies
Considering the top five solutions per run, according to the geometric criterion, out of the 22 easy sets, five were unsuccessful (Table 2 entry not “A”) in the Balloon runs and four in the Confect runs. Out of these nine cases, three were sets of dihydrofolate reductase (DHFR) ligands. The Balloon runs did not successfully align P00374 and P0A017. The Confect runs did not successfully align P16184. The situation with P16184 is given in Fig. 4, while the experimental alignments of P00374 and P0A017 are given in Supplementary Figure S1 (Online Resource). The ligands typically contain a nitrogen rich single or double aromatic ring system with two amines in ortho positions (e.g., pyrimidine-diamine or quinazoline-diamine or diaminopyridopyrimidine). This rigid ring system is attached to a bulky part of the ligand that probes a relatively flat region of the binding pocket. The relative orientations between these two flat parts are somewhat flexible in the ligands. Hence it is easy to get the two parts correctly aligned individually, but with a wrong relative orientation. The good results using the topological criterion (see Supplementary Tables S1 and S2, Online Resource) reflect this.

DHFR ligands in the set P16184. The left figures give the experimental alignment. The ligands generally consist of two flat pieces connected by a flexible linkage. The right figures give the third best scoring alignment using the Confect conformers. Three of the seven ligands can be superimposed onto the experimental alignment with a heavy atom rmsd of under 2 Å
Considering the top five solutions per run, according to the geometric criterion, out of the 22 easy sets, only one was unsuccessful (Table 2 entry not “A”) in both the Balloon and the Confect runs. This was P56658, consisting of adenosine deaminase ligands. This was also the only easy set with more than two incorrect ligands (three incorrect ligands for the Balloon run) according to the topological criterion. Figure 5 shows the situation with this set. To start with, ligand 2 (1krm) looks a bit out of place with the other ligands. Each of the remaining 8 ligands consists of two or more rigid parts joined by flexible regions. While it may not be difficult to match the atoms (in the Confect run, all but ligand 2 were correctly aligned topologically), it is not easy to get the correct conformations. Moreover, three ligands are very flexible. Ligands 5, 6 and 7 (1ndz, 1o5r and 1uml) all have 11 rotatable bonds. Confect generated 17, 19 and 19 conformers for these ligands. The best heavy atom rmsd’s for them were 2.48, 1.80 and 1.90 Å. In other words, the conformers were not of excellent quality. As for Balloon, 118, 111 and 100 conformers were generated for these ligands. The best heavy atom rmsd’s for these 3 ligands were 1.59, 1.20 and 0.80 Å. It seems that there were some good conformers after all. In order to gain some insight into what happened, we picked the best conformers for these three ligands and performed an alignment of these three conformers. Figure 5e shows the result of this exercise. Even with these relatively good conformers, the flexible tail part did not align very well. When the tails were roughly aligned volumetrically, as in Fig. 5e, the corresponding hydrogen bonding features in the flexible tails did not align. As a result, the score would not have been able to provide a strong incentive to choose the correct conformers and align them. On the other hand, the incentive to precisely align the feature-rich head part of the ligands is strong. Hence the alignment of the tails was sacrificed for getting a high quality alignment of the head part.

The set P56658, consisting of adenosine deaminase ligands. a and b show the experimental alignment. Ligand 2 (1krm) is highlighted in (a). The best scoring alignments using the Confect and Balloon conformers are shown in c and d, respectively. e gives the best scoring solution from aligning the single best conformer from each of ligands 5, 6 and 7 (1ndz, 1o5r and 1uml), generated by Balloon. In c, all but ligand 2 were correctly aligned topologically. Because Confect uses a knowledge-based approach, corresponding torsion angles in congeneric ligands can have exactly the same values. This is why c may seem to contain fewer than nine ligands
There are 23 heat shock protein 90-alpha ligands in the moderate set P07900. Other than P56658 mentioned above, this is the only case where we were not successful (considering top five solutions, Table 2 entry not “A”) using either the Balloon or the Confect conformers, but ref. [32] consistently performed well even without using the retrospective AlignScore (considering top one solution, with more than two successes out of ten runs in either BT10 or SE10 of Table 2). Figure 6 gives the alignments of this set. The ligands can be classified into two groups, with “group A” containing an aminopyrimidine or an aminotriazine (middle row of Fig. 6), and “group B” containing a dihydroxyphenyl moiety (bottom row of Fig. 6). While these groups may individually be easy to align, aligning the two groups may not be so. As Fig. 6a shows, the alignment between the dihydroxyphenyl and the aminopyrimidine is not trivial. Figure 6d shows our second best scoring result from the Balloon conformers. The dichlorophenyl moieties of group A were matched to the dihydroxyphenyl moieties of group B, while the azole rings of group B were matched to the aminopyrimidine/aminotriazine rings of group A. If the two groups were considered separately, according to the geometric criterion, five of the ten ligands in group A (Fig. 6e) and ten of the thirteen ligands in group B (Fig. 6 f) were correctly aligned.

The set P07900, consisting of HSP 90-alpha ligands. The experimental alignment is shown on the left (a–c). The second best scoring alignment using the Balloon conformers is shown on the right (d–f). The top row (a, d) shows all 23 ligands in the set. The middle row (b, e) shows the 10 ligands containing an aminopyrimidine or an aminotriazine. The bottom row (c, f) shows the 13 ligands containing a dihydroxyphenyl moiety. 5 of the 10 ligands in (e) and 10 of the 13 ligands in (f) can be fitted onto the experimental alignment with a heavy atom rmsd of under 2 Å
Considering our top five solutions, for the hard set P68400 (casein kinase II) and the unfeasible set P14174 (macrophage migration inhibitory factor), both our Balloon and Confect runs were successful (Table 2 entry is “A”) using the geometric criterion, but neither of the runs was successful using the topological criterion. Figure 7 shows the alignments of P68400. The binding site is quite flat. Other than the shape, the main pharmacophore elements include a carboxylate group and an acceptor atom, as highlighted in Fig. 7b, e. Fig. 8 uses two ligands in this set to illustrate how two ligands can be included in a correctly aligned group using the geometric criterion but not the topological criterion. Similar to set P68400, the binding site of set P14174 has a narrow planar shape, although the plane is now slightly curved. Supplementary Figure S2 (Online Resource) shows the alignments.

The set P68400, consisting of casein kinase II ligands. The experimental alignment is shown on the left (a–c) while our best scoring alignment using the Balloon conformers is shown on the right (d–f). The top four figures (a, b, d, e) show all 14 ligands. The bottom row (c, f) shows only the eight correctly aligned ligands according to the geometric criterion (heavy atom rmsd under 2 Å). The main pharmacophore elements include a carboxylate group and an acceptor atom, as highlighted by the circles and arrows

Alignment of the casein kinase II ligands 2zjw and 3amy in the set P68400. The upper left figure is (part of) the experimental alignment. The upper right figure is (part of) the top scoring alignment using the Balloon conformers. The lower figures compare the experimental and the calculated positions of these two ligands. Oxygen atoms are coloured red. The carbon atoms of each ligand are coloured uniquely. The heavy atom rmsd for 2zjw is 1.96 Å while that for 3amy is 1.84 Å. The topological similarity, f AB , between the calculated and experimental alignments of this ligand pair is 0.344 (see “Topological criterion” section). Thus these two ligands can be included in a correctly aligned group according to the geometric criterion but not the topological criterion
Comparison with other publications
In addition to the detailed comparisons above, we have also compared our algorithm with all references published since 2009 which have quantitative alignment results for multiple molecules that do not require the user to supply a template conformer. Here, we used either the experimental conformers (rigid alignments), or conformers generated using Confect [35] (flexible alignments). In all cases, there were fewer than 1000 conformers in total, so our runs were generally deterministic.
Table 5 compares our results with Table 2 of ref. [24], for alignment using only the experimental conformers. Only the best scoring solution was considered. DIFGAPE [24] output solution assemblies that typically contain fewer ligands than the input. Our results are decisively better than those from DIFGAPE. Moreover, MolAlign ran extremely rapidly. As for flexible alignments, ref. [24] classified a set as a “pass” if at least half the ligands can be fit into an assembly with an all-atoms rmsd of under 2 Å. They obtained a pass in four sets: CDK2_focused, the two FXa sets, and trypsin. We obtained a pass in these four sets plus another four sets, as can be seen from Table 6.
Ref. [12] used the same test sets as ref. [24]. It does not have any results for rigid alignments. Table 6 compares our results on flexible alignment to Table 2 of ref. [12]. Following ref. [12], the number of correctly aligned ligands is the maximum number of ligands that can be superimposed onto the experimental alignment such that the all-atoms rmsd remains below 2 Å. This differs from the standard geometric criterion mentioned above, which requires each correctly aligned ligand to be under 2 Å. From Table 6, it can be seen that results from MolAlign are better than those from GAPE [12]. Averaging over the ten sets, GAPE correctly aligned 58.5% of the ligands, while MolAlign correctly aligned 67.0%. The GAPE runs used a 1 GHz CPU, while the MolAlign runs used a 2.2 GHz CPU. For GAPE, 100 runs were performed and the best scoring solution was considered (see next paragraph for reducing the number of runs). For MolAlign, one deterministic run was performed and the best scoring solution was considered. Hence MolAlign is a lot faster than GAPE.
In ref. [12], Table 2 was obtained using the best scoring solution over 100 runs. Table 5 of ref. [12] gives the quality of the results when 25 instead of 100 runs were used. Here, the average number of successfully aligned sets (success means at least half the ligands within 2 Å rmsd) drops from 8 to 7.
In ref. [12], Table 4 presents the results using the multiconformer version of GAPE, Table 6 presents results from GASP [5], Table 7 presents results from GALAHAD [36]. All these results were significantly inferior to the default version of GAPE (Table 2 of ref. [12]), which we have compared against in our Table 6.
Table 7 compares our results with MARS [25]. Only the top scoring solution was considered. Ref. [25] required each correctly aligned ligand to have an rmsd of under 2 Å compared to the experimental result. This is the same as our standard geometric criterion, and different from the criterion used in ref. [12] and our Table 6. Our results on rigid alignments are decisively better than MARS [25]. In four of the six sets, we correctly aligned all ligands. In the fifth set (ESR1), only 1 out of the 13 ligands was not correctly aligned. This ligand, with 20 heavy atoms, registered a heavy atom rmsd of only 2.6 Å. In the sixth set (cdk2), we had 8 incorrectly aligned ligands, while MARS had between 16 and 23 incorrectly aligned ligands. For flexible alignments, our results are comparable to MARS [25]. But if we consider the top five solutions, our results would be much better in two of the six test sets. We do not know how MARS performed if the top handful solutions were considered. But one would expect a mention in ref. [25] if they had significantly better solutions in the wings.
To summarize, our rigid alignment results are decisively better than refs. [12, 24, 25]. Our flexible alignment results are better than refs. [5, 12, 24, 36] and comparable to ref. [25]. Apparently we lose some advantage when going from rigid to flexible alignments. It is tempting to think that this is due to the quality of the input conformers. While this may be the case, the real reasons could be more complex. It could be due to a difference in the coverage of sampling the solution space, or the performance of the scoring schemes. To understand the reasons, one could mix and match the input conformers and/or the scoring schemes for the final results between the methods. Unfortunately we do not have access to ROCS to perform these experiments. But this should be an interesting study.
Conclusions
We have presented an algorithm for aligning multiple small molecule ligands. The input is the conformers of the ligands. The scoring functions are based on the overlap of atoms of similar types (hydrogen bond donor/acceptor, hydrophobic atoms) as well as general volumetric overlap. These scoring functions were built on the foundation of our previous work [24]. The algorithm consists of two stages. The first stage is the alignment of all pairs of ligands using a simplified scoring function. The best scoring pairwise poses are recorded, along with their scores. From this pool of pairwise results, the second stage of the algorithm constructs multiple ligand alignment assemblies and refines the assemblies iteratively by replacing the ligands, one at a time. Due to the modular nature of the algorithm, it is possible to use other scoring functions, or to employ other pairwise alignment algorithms in the first stage.
Results have been compared with several recent publications, including a suite of 121 publicly available, systematically derived data sets [30]. Our algorithm is very fast. For rigid alignments using the experimental conformers, our results are decisively better than available comparisons. For flexible alignments, our advantage has decreased. While an exact comparison is impossible, it is clear that our results are very competitive. It would be interesting to figure out the reasons behind the relative decline in performance, and improve the algorithm.
The author is willing to perform free alignment runs for anyone as much as his computational resources allow.
References
Cramer RD, Patterson DE, Bunce JD (1988) Comparative molecular field analysis (CoMFA). 1. effect of shape on binding of steroids to carrier proteins. J Am Chem Soc 110:5959–5967
Lemmen C, Langauer T (2000) Computational methods for the structural alignment of molecules. J Comput Aided Mol Des 14:215–232
Wolber G, Seidel T, Bendix F, Langer T (2008) Molecule-pharmacophore superpositioning and pattern matching in computational drug design. Drug Discov Today 13:23–39
Leach AR, Gillet VJ, Lewis RA, Taylor R (2010) Three-dimensional pharmacophore methods in drug discovery. J Med Chem 53:539–558
Jones G, Willett P, Glen RC (1995) A genetic algorithm for flexible molecular overlay and pharmacophore elucidation. J Comput Aided Mol Des 9:532–549
Barnum D, Greene J, Smellie A, Sprague P (1996) Identification of common functional configurations among molecules. J Chem Inf Comput Sci 36:563–571
Lemmen C, Lengauer T, Klebe G (1998) FLEXS: a method for fast flexible ligand superposition. J Med Chem 41:4502–4520
Labute P, Williams C, Feher M, Sourial E, Schmidt JM (2001) Flexible alignment of small molecules. J Med Chem 44:1483–1490
ROCS, from OpenEye Scientific Software, http://www.eyesopen.com
Dixon SL, Smondyrev AM, Knoll EH, Rao SN, Shaw DE, Friesner RA (2006) PHASE: a new engine for pharmacophore perception, 3D QSAR model development, and 3D database screening: 1. Methodology and preliminary results. J Comput Aided Mol Des 20:647–671
Gardiner EJ, Cosgrove DA, Taylor R, Gillet VJ (2009) Multiobjective optimization of pharmacophore hypotheses: bias toward low-energy conformations. J Chem Inf Model 49:2761–2773
Jones G (2010) GAPE: an improved genetic algorithm for pharmacophore elucidation. J Chem Inf Model 50:2001–2018
Taylor R, Cole JC, Cosgrove DA, Gardiner EJ, Gillet VJ, Korb O (2012) Development and validation of an improved algorithm for overlaying flexible molecules. J Comput Aided Mol Des 26:451–472
Korb O, Monecke P, Hessler G, Stützle T, Exner TE (2010) pharmACOphore: multiple flexible ligand alignment based on ant colony optimization. J Chem Inf Model 50:1669–1681
Moser D, Wittmann SK, Kramer J, Blöcher R, Achenbach J, Pogoryelov D, Proschak E (2015) PENG: a neural gas-based approach for pharmacophore elucidation. Method design, validation, and virtual screening for novel ligands of LTA4H. J Chem Inf Model 55:284–293
Shin W, Hyun SA, Chae CH, Chon JK (2009) Flexible alignment of small molecules using the penalty method. J Chem Inf Model 49:1879–1888
Tosco P, Balle T, Shiri F (2011) Open3DALIGN: an open-source software aimed at unsupervised ligand alignment. J Comput Aided Mol Des 25:777–783
Diller DJ, Connell ND, Welsh WJ (2015) Avalanche for shape and feature-based virtual screening with 3D alignment. J Comput Aided Mol Des 29:1015–1024
Kawabata T, Nakamura H (2014) 3D flexible alignment using 2D maximum common substructure: dependence of prediction accuracy on target-reference chemical similarity. J Chem Inf Model 54:1850–1863
Vainio MJ, Puranen JS, Johnson MS (2009) ShaEP: molecular overlay based on shape and electrostatic potential. J Chem Inf Model 49:492–502
Cross S, Baroni M, Goracci L, Cruciani G (2012) GRID-based three-dimensional pharmacophores I: FLAPpharm, a novel approach for pharmacophore elucidation. J Chem Inf Model 52:2587–2598
Leherte L, Vercauteren DP (2012) Smoothed Gaussian molecular fields: an evaluation of molecular alignment problems. Theor Chem Acc 131:1259–1275
Sastry GM, Dixon SL, Sherman W (2011) Rapid shape-based ligand alignment and virtual screening method based on atom/feature-pair similarities and volume overlap scoring. J Chem Inf Model 51:2455–2466
Jones G, Gao Y, Sage CR (2009) Elucidating molecular overlays from pairwise alignments using a genetic algorithm. J Chem Inf Model 49:1847–1855
Klabunde T, Giegerich C, Evers A (2012) MARS: computing three-dimensional alignments for multiple ligands using pairwise similarities. J Chem Inf Model 52:2022–2030
Chan SL, Labute P (2010) Training a scoring function for the alignment of small molecules. J Chem Inf Model 50:1724–1735
Bron C, Kerbosch J (1973) Algorithm 457—finding all cliques of an undirected graph. Commun ACM 16:575–577
Kabsch W (1978) A discussion of the solution for the best rotation to relate two sets of vectors. Acta Crystallogr A 34:827–828
Cross S, Ortuso F, Baroni M, Costa G, Distinto S, Moraca F, Alcaro S, Cruciani G (2012) GRID-based three-dimensional pharmacophores II: pharmbench, a benchmark data set for evaluating pharmacophore elucidation methods. J Chem Inf Model 52:2599–2608
Giangreco I, Cosgrove DA, Packer MJ (2013) An extensive and diverse set of molecular overlays for the validation of pharmacophore programs. J Chem Inf Model 53:852–866
The Protein Data Bank. http://www.rcsb.org
Giangreco I, Olsson TSG, Cole JC, Packer MJ (2014) Assessment of a cambridge structural database-driven overlay program. J Chem Inf Model 54:3091–3098
The RDKit. http://www.rdkit.org, by Greg Landrum et al.
Vainio MJ, Johnson MS (2007) Generating conformer ensembles using a multiobjective genetic algorithm. J Chem Inf Model 47:2462–2474
Schärfer C, Schulz-Gasch T, Hert J, Heinzerling L, Schulz B, Inhester T, Stahl M, Rarey M (2013) CONFECT: conformations from an expert collection of torsion patterns. ChemMedChem 8:1690–1700
Richmond NJ, Abrams CA, Wolohan PR, Abrahamian E, Willett P, Clark RD (2006) GALAHAD: 1. pharmacophore identification by hypermolecular alignment of ligands in 3D. J Comput Aided Mol Des 20:567–587
Acknowledgements
The author would like to thank Dr. Christian Lemmen and the authors of Confect [35], and Dr. Mikko Vainio and other authors of Balloon [34]. This work would have been impossible without free access to these two conformer generators. Other indispensible freeware include the RDKit [33], the Yasara viewer (http://www.yasara.org), and the JMol Viewer (http://jmol.sourceforge.net). The author also thanks Drs. Alex Clark and Bill Myrtle for help with proof-reading.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Appendix
Appendix
Termination criterion for simultaneous multiple pose generation
After each cycle (each query triplet), the pair pose registers may be updated. The raw score improvement is defined to be the average (over all registers) of the improvement of the score at the fifth slot (i.e., the fifth best score in the register) over this cycle. The first slot is not used because it does not get updated very often (for the first slot to be updated, a new pose with a score better than any of the existing ones must be found). Position 5 is used because each register is guaranteed to have more than five slots, as per Eq. (2). In principle, we would terminate the iteration when the score improvement, SI, becomes too small. We noticed that some cycles produced very small SIs although there was still significant room for improvement. In order to smooth out the fluctuation, we average out the SI of the current cycle with the SIs of a few past cycles to create an effective SI, and use this to determine termination. Let \({n_{curr}}\) be the number of compatible triplets (which equals the number of conformer poses in the grand assembly) encountered in the current cycle. Let \({n_{avg}}\) be the hitherto average number of compatible triplets per cycle (i.e., \({n_{avg}}\) is the sum of all \({n_{curr}}\)′s encountered so far, divided by the number of cycles). The effective SI of the current cycle, \({s_{curr}}\), is given by
where \({s_{prev}}\) is the effective SI of the previous cycle, and \({s_0}\) is the raw score improvement, as described above. In other words, \({s_{curr}}\) is a weighted average of \({s_{prev}}\) and \({s_0}\). \({s_0}\) is weighted less if there are few compatible triplets in the current cycle. This should make the effective SI, \({s_{curr}}\), less prone to statistical fluctuations than the raw score improvement, \(~{s_0}\).
Iteration terminates when the effective SI drops below a cut-off value, \({s_{cut}}\). \({s_{cut}}\) is set to a value of 0.01. To put this in perspective, the pairwise score (according to any scoring scheme) of one hydrophobic atom overlapping on another is 2.0.
For the first cycle, the value of \({s_{prev}}\) in Eq. (3) is set to \({2^5} \times {s_{cut}}\). 5 is the index of the slot used for determining the raw score improvement, \({s_0}\), as mentioned above. This way, if \({s_0}\) is 0 and \({n_{curr}}\) remains unchanged over the cycles (implying that \({n_{avg}}\) always equals \({n_{curr}}\)), the iteration would go on for 5 cycles before terminating. Put in another way, if only one slot of each pair pose register is filled per iteration cycle, there is time for the register to be filled to the fifth slot, where \({s_0}\) is determined.
Choosing random query triplets for simultaneous multiple pose generation
Early in our research, we simply chose a random triplet from a random conformer of a random ligand. We found this inefficient. Since we would like each pair pose register to be at least partially filled, we can bias the choice of the ligands to encourage ligands with empty registers to be chosen. First, ligands having fewer than three representative points are not involved, because they do not have representative triplets. An array is constructed out of the indices of all ligands having at least three representative points. Each index appears once. We then go through all ligand pairs. For each ligand pair whose pair pose register is still empty, three copies of the index of each ligand in the pair are inserted into the array. (Experiments showed that using five instead of three has little effect on the results.) Finally, a random element from this array is selected to give a random ligand. Then a random conformer of this ligand is chosen, and a random triplet of representative points of this conformer is chosen. Similar to the systematic pose generation method, triplets that are geometrically too small are never chosen. The diameter (see “Systematic pose generation” section) of a chosen triplet has to be at least half that of the smallest diameter amongst the representative point sets of all conformers of all ligands.
Starting points for multiple ligand alignment
As described in the section “Generating multiple ligand assemblies”, one base template conformer gives rise to one starting point assembly, and subsequently one solution assembly. We discovered that using a maximum of 250 starting points strikes a balance between good results and speed. Before the iteration begins, each conformer of each ligand is used as a base template to generate a starting assembly. Given one base template conformer, for each of the other ligands, the best scoring pairwise alignment pose between the ligand and the base template is used to build the starting assembly. It is possible that no pairwise alignment pose exists in the pair pose register between the base template conformer and the ligand. If this is the case, the missing ligand count of this base template will be increased by one. After all the non-missing ligands have been positioned in the assembly, each missing ligand will be positioned using the non-missing ligands as helpers, one at a time. For each missing ligand, amongst all poses generated using the helpers, the one resulting in the best score for the assembly (all non-missing ligands plus the current ligand) is used. Any ligands still missing after this step will be positioned randomly. The multiple ligand alignment score of the starting point assembly will then be calculated, using fast scoring. The full list of starting point assemblies will then be sorted, first by the missing ligand count, then by the multiple ligand alignment score. If there are more than 250 assemblies, the 250 with the smallest missing ligand counts and the best scores are used as starting points. In a real run, there is of course no need to patch the missing ligands if there are already enough starting points without any missing ligands.
Rights and permissions
About this article

Cite this article
Chan, S.L. MolAlign: an algorithm for aligning multiple small molecules. J Comput Aided Mol Des 31, 523–546 (2017). https://doi.org/10.1007/s10822-017-0023-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10822-017-0023-8