
Abstract
Accurately predicting binding affinities between ligands and macromolecules has been a much sought-after goal. A tremendous amount of resources can be saved in the pharmaceutical industry through accurate binding-affinity prediction and hence correct decision-making for the drug discovery processes. Owing to the structural complexity of macromolecules, one of the issues in binding affinity prediction using molecular dynamics is the adequate sampling of the conformational space. Recently, the funnel metadynamics method (Limongelli et al. in Proc Natl Acad Sci USA 110:6358, 2013) was developed to enhance the sampling of the ligand at the binding site as well as in the solvated state, and offer the possibility to predict the absolute binding free energy. We apply funnel metadynamics to predict host–guest binding affinities for the cucurbit[7]uril host as part of the SAMPL4 blind challenge. Using total simulation times of 300–400 ns per ligand, we show that the errors due to inadequate sampling are below 1 kcal/mol. However, despite the large investment in terms of computational time, the results compared to experiment are not better than a random guess. As we obtain differences of up to 11 kcal/mol when switching between two commonly used force fields (with automatically generated parameters), we strongly believe that in the pursuit of accurate binding free energies a more careful force-field parametrization is needed to address this type of system.











Similar content being viewed by others
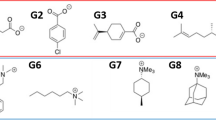

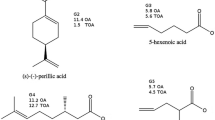
References
Limongelli V, Bonomi M, Parrinello M (2013) Funnel metadynamics as accurate binding free-energy method. Proc Natl Acad Sci USA 110:6358
Zwanzig RW (1954) J Chem Phys 22:1420
Christ CD, Fox T (2014) Accuracy assessment and automation of free energy calculations for drug design. J Chem Inf Model 54:108–120
Hou T, Wang J, Li Y, Wang W (2011) Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J Chem Inf Model 51(1):69–82
Warren GL, Andrews CW, Capelli AM, Clarke B, LaLonde J, Lambert MH, Lindvall M, Nevins N, Semus SF, Senger S, Tedesco G, Wall ID, Woolven JM, Peishoff CE, Head MS (2006) A critical assessment of docking programs and scoring functions. J Med Chem 49(20):5912–5931
Laio A, Parrinello M (2002) Proc Natl Acad Sci USA 20:12562
Gervasio FL, Laio A, Parrinello M (2005) Flexible docking in solution using metadynamics. J Am Chem Soc 127(8):2600–2607
Provasi D, Bortolato A, Filizola M (2009) Exploring molecular mechanisms of ligand recognition by opioid receptors with metadynamics. Biochemistry 48(42):10020–10029
Masetti M, Cavalli A, Recanatini M, Gervasio FL (2009) Exploring complex protein-ligand recognition mechanisms with coarse metadynamics. J Phys Chem B 113(14):4807–4816
Limongelli V, Bonomi M, Marinelli L, Gervasio FL, Cavalli A, Novellino E, Parrinello M (2010) Molecular basis of cyclooxygenase enzymes (COXs) selective inhibition. Proc Natl Acad Sci USA 107(12):5411–5416
Limongelli V, Marinelli L, Cosconati S, La Motta C, Sartini S, Mugnaini L, Da Settimo F, Novellino E, Parrinello M (2012) Sampling protein motion and solvent effect during ligand binding. Proc Natl Acad Sci USA 109(5):1467–1472
Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML (1983) J Chem Phys 79:926
Humphrey W, Dalke A, Schulten K (1996) J Mol Graphics 14.1:33
Vanommeslaeghe K, Hatcher E, Acharya C, Kundu S, Zhong S, Shim J, Darian E, Guvench O, Lopes P, Vorobyov I, MacKerell Jr AD (2010) J Comput Chem 31:671
Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA (2004) Development and testing of a general AMBER force field. J Comput Chem 25(9):1157–1174
Bayly CI, Cieplak P, Cornell WD, Kollman PA (1993) J Phys Chem 97:10269
Becke AD (1993) J Chem Phys 98:5648
Lee C, Yang W, Parr RG (1988) Phys Rev B 37:785
Hehre WJ, Ditchfield R, Pople JA (1972) J Chem Phys 56:2257
Hariharan PC, Pople JA (1973) Theor Chem Acc 28:213
Francl M, Pietro W, Hehre W, Binkley JS, Defrees DJ, Pople JA, Gordon M (1982) J Chem Phys 77:3654
Tomasi J, Mennucci B, Cammi R (2005) Chem Rev 105:2999
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2009) Gaussian 09 Revision D.01. Gaussian Inc., Wallingford, CT
Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K (2005) J Comput Chem 26:1781
Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG (1995) J Chem Phys 103:8577
Bonomi M, Branduardi D, Bussi G, Camilloni C, Provasi D, Raiteri P, Donadio D, Marinelli F, Pietrucci F, Broglia RA, Parrinello M (2009) Comp Phys Comm 180:1961
ACD/Labs (2013) http://www.acdlabs.com/products/percepta/predictors/pka/, Predict accurate acid/base dissociation constants from structure
Barducci A, Bussi G, Parrinello M (2008) Well-tempered metadynamics: a smoothly converging and tunable free-energy method. Phys Rev Lett 100(2):20603
Deng Y, Roux B (2009) Computations of standard binding free energies with molecular dynamics simulations. J Phys Chem B 113(8):2234–2246
Cao L, Isaacs L (2013) Absolute and relative binding affinity of cucurbit[7]uril towards a series of cationic guests. Supramol Chem. doi:10.1080/10610278.2013.852674
Liu S, Ruspic C, Mukhopadhyay P, Chakrabarti S, Zavalij PY, Isaacs L (2005) The cucurbit[n]uril family: prime components for self-sorting systems. J Am Chem Soc 127(45):15959
Muddana HS, Gilson MK (2012) Prediction of SAMPL3 host-guest binding affinities: evaluating the accuracy of generalized force-fields. J Comput Aided Mol Des 26(5):517–525
Åqvist J, Medina C, Samuelsson JE (1994) Protein Eng 7:385
Åqvist J, Luzhkov VB, Brandsdal BO (2002) Ligand binding affinities from MD simulations. Acc Chem Res 35:358
Khalili KF, Henni A, East ALL (2009) pKa values of some piperazines at (298, 303, 313, and 323) K. J Chem Eng Data 54:2914–2917
Moghaddam S, Yang C, Rekharsky M, Ko YH, Kim K, Inoue Y, Gilson MK (2011) New ultrahigh affinity host-guest complexes of cucurbit[7]uril with bicyclo[2.2.2]octane and adamantane guests: thermodynamic analysis and evaluation of M2 affinity calculations. J Am Chem Soc 133(10):3570–3581
Acknowledgments
We thank Dr. Vittorio Limongelli for advice on the funnel metadynamics setup. This investigation has been supported by the Swedish research council (agreement C0020401).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Hsiao, YW., Söderhjelm, P. Prediction of SAMPL4 host–guest binding affinities using funnel metadynamics. J Comput Aided Mol Des 28, 443–454 (2014). https://doi.org/10.1007/s10822-014-9724-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10822-014-9724-4