

Reaction of 5-dimethylaminotetrazolo[1,5-а][1,3,5]triazin-7-one tetrabutylammonium salt with allyl bromide in acetonitrile proceeds with the formation of 3-allyl-5-dimethylaminotetrazolo[1,5-a][1,3,5]triazin-7-one (N-alkylation), 6-allyloxy-4-azido-N,N-dimethyl-1,3,5-triazin-2-amine (O-alkylation), and 2-(1-allyl-1H-tetrazol-5-yl)-1,1-dimethylguanidine as the product of hydrolysis of N-alkylated tetrazolo[1,5-a][1,3,5]triazin-7-one. The structure of the reaction products was confirmed by IR, 1H and 13C NMR spectroscopy as well as elemental and X-ray structural analysis data.
Similar content being viewed by others

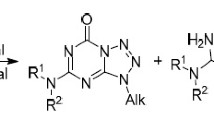
Nucleosides constructed from the aza analogs of natural heterocycles and cyclic or acyclic analogs of natural carbohydrates find uses as bioactive compounds with a broad spectrum of activity.1 One of the most common methods for the synthesis of anomalous nucleosides is the alkylation of nitrogen heterocycles with natural carbohydrates or their acyclic analogs.2 However, application of this method is complicated in cases where the anions of nitrogen heterocycles are of an ambident character. The presence in the molecule of the heterocycle of several nitrogen atoms and exocyclic oxygen atoms (usually in the form of carbonyl groups) leads to the formation of several isomers in the alkylation reaction.3
Until now only few examples of alkylation of the derivatives of tetrazolo[1,3,5]triazine heterocyclic system have been described in the literature.4
In the course of evaluating methods of synthesis of anomalous nucleosides that are based on tetrazolo[1,3,5]-triazine condensed system, the 5,8-diaza analog of purine, we investigated alkylation of 5-dimethylaminotetrazolo[1,5-a]-[1,3,5]triazin-7-one salt with allyl bromide.
The anion of 5-dimethylaminotetrazolo[1,5-a][1,3,5]triazin-7-one is ambident, whose distribution of electron density can be shown using five resonance structures (Fig. 1). This allows the possibility of alkylation taking place at five reactive sites: the exocyclic oxygen atom and the four nitrogen atoms of the ring system (atoms N-1 and N-3 of the tetrazole ring, and atoms N-4 and N-6 of the 1,3,5-triazine ring).

Major signals in IR spectra (in italics), 13С NMR spectra, and cross peaks in 2D HMBC spectra for compounds 2–4.
Tetrabutylammonium salt of 5-dimethylaminotetrazolo-[1,5-a][1,3,5]triazin-7-one (1) was used as the starting compound, the synthesis of which was described by us earlier.5 Alkylation with allyl bromide was carried out in acetonitrile at 20-22°C (control by TLC for the presence of the starting salt). As a result, three products were isolated: the product of alkylation at position 3 of the heterocyclic system 3-allyl-5-dimethylaminotetrazolo[1,5-a][1,3,5]triazin-7-one (2), product of alkylation at the exocyclic oxygen atom with opening of the tetrazole ring 6-allyloxy-4-azido-N,N-dimethyl-1,3,5-triazin-2-amine (3), and product of alkylation at position 3 of the heterocyclic system with opening of the 1,3,5-triazine ring 2-(1-allyl-1Н-tetrazol-5-yl)-1,1-dimethylguanidine (4) (Scheme 2).

Scheme 2
Regiochemistry of the alkylation products was determined based on the analysis of IR spectra, 1H and 13C NMR spectra, 2D HMBC and X-ray structural analysis data. In the IR spectrum of compound 2 an absorption band of the carbonyl group was present at 1724 cm−1. In the IR spectrum of compound 3 there was no absorption band of the carbonyl group, and an absorption band of the azide group was observed at 2121 cm−1. In the IR spectrum of compound 4, no absorption bands attributable to either carbonyl or azide groups were present, whereas an intense absorption bands characteristic of the primary amines (3467 and 3303 cm−1) appeared. In the 1H NMR spectrum of compound 4, a signal of an amino group was observed (7.55 ppm). Confirmation of the formation of an O-alkylation product was a shift of the signal of the carbon atom of CH2 group in the spectrum of compound 3 to 67.4 ppm (for comparison, in the spectra of compounds 2 and 4, the signals of these atoms appeared at 48.1 and 46.6 ppm, respectively). Cross peaks in 2D HMBC experiments (Fig. 1) allows conclusive assignment of all signals of the carbon atoms of the cyclic systems and guanidine.
According to X-ray structural analysis data for compound 2 (Fig. 2) its non-hydrogen atoms except the atoms of the allyl group lie in one plane with deviation of not more than 0.04 Å. Allyl group is situated near atom N(6) and tilts from the tetrazole plane by 99.1°. The lengths of bonds С(1)–N(1), N(4)–N(5), and C(2)–O(1) make it possible to reliably determine these bonds as being double. The lengths of bonds N(5)–N(6), C(2)–N(3), and N(6)–C(6) are close to that of a single bond. The rest of the C–N bonds in the molecule, including the exocyclic C(1)–N(7) bond have a character of a one and a half bond due to the conjugation effect.

Molecular structure of compound 2 with atoms represented by thermal displacement ellipsoids with 50% probability.
According to X-ray structural analysis data for compound 4 (Fig. 3) its non-hydrogen atoms except the atoms of the allyl group lie in one plane with deviation of not more than 0.05 Å. The allyl group is situated at the N(1) atom; it extends out of the tetrazole plane and demonstrates disordering of the С(2)–H group in two positions with a population of 0.9/0.1 (the minor component of the disordering of the allyl group is not shown in Fig. 3). The lengths of C–N bonds in the molecule are largely equal due to conjugation effects: the difference between double and single bonds in the guanidine moiety is not more than 0.03 Å. Both protons of the NH groups are localized on the unsubstituted nitrogen atom N(6). One of the protons is involved in the formation of intramolecular hydrogen bonding with nitrogen atom N(4) of the heterocycle fixing the guanidine moiety in the plane of the heterocycle, whereas the other hydrogen atom forms an intermolecular hydrogen bond with an adjacent heterocycle molecule. Spatial dimers are formed as a result, constituting the basic structural element of the molecular packing.

Molecular structure of compound 4 with atoms represented by thermal displacement ellipsoids with 50% probability.
The formation of tetrazolylguanidine 4 can be explained by the presence of trace amounts of water in the reaction mixture and by susceptibility of the alkylation product 2 to hydrolysis. To test this hypothesis, we subjected compound 2 to aqueous acetonitrile. As a result, tetrazolylguanidine 4 was obtained in quantitative yield. The hydrolysis proceeds via an addition of a water molecule to the double bond of the carbonyl group with the following opening of the 1,3,5-triazine ring and decarboxylation of the resulting carbamic acid (Scheme 3).

Scheme 3
The reason for the ease of cleaving the С(7)–N(8) bond in our opinion is the presence in the 1,3,5-triazine ring of the carbonyl group which is conjugated with the neighboring annulated tetrazole cycle. A similar hydrolytic cleavage of an azine ring followed by decarboxylation is observed for certain condensed azoloazines,6 whereas no such examples have been described for condensed tetrazoloazines. Similar cleavage of bond effected by nucleophiles is known for acyl derivatives of noncondensed tetrazoles.7
To conclude, the studies of alkylation of 5-dimethylaminotetrazolo[1,5-a][1,3,5]triazine tetrabutylammonium salt revealed the susceptibility of 3-allyl-5-dimethylaminotetrazolo[1,5-a][1,3,5]triazin-7-one to hydrolysis, which forms 2-(1-allyl-1Н-tetrazol-5-yl)-1,1-dimethylguanidine in the presence of water.
Experimental
IR spectra were registered on a Avatar 360ESP FT-IR spectrophotometer using the attenuated total reflectance (ATR) attachment. 1H and 13C NMR spectra were acquired on a JEOL JNM ECX-400 spectrometer (400 and 100 MHz, respectively) in DMSO-d6, with TMS as internal standard.
Elemental analysis was performed on a Eurovector EA 3000 apparatus. Melting points were determined on a Gallenkamp apparatus and are uncorrected. Monitoring of the reaction progress and assessment of the purity of synthesized compounds was done by TLC on Silufol UV-254 plates with EtOAc as eluent, visualization with 254-nm UV light. MN Kieselgel 60 sorbent was used for column chromatography.
Alkylation of 5-dimethylaminotetrazolo[1,5- a ][1,3,5]-triazin-7-one tetrabutylammonium salt (1). Allyl bromide (2.61 ml, 30 mmol) was added to a solution of 5-dimethylaminotetrazolo[1,5-а][1,3,5]triazin-7-one tetrabutylammonium salt (1) (4.22 g, 10 mmol) in MeCN (35 ml), and the resulting mixture stirred at room temperature for 5 days (the progress of the reaction was assessed by TLC by monitoring the remaining amount of starting salt). Acetonitrile was evaporated under reduced pressure, and the residue was treated with Н2О (30 ml). The water-insoluble precipitate was filtered off and dried in air. The precipitate was stirred magnetically with hexane (25 ml) for 10 min and the insoluble precipitate filtered to afford 1.17 g of 3-allyl-5-dimethylaminotetrazolo[1,5-a]-[1,3,5]triazin-7-one (2). Hexane was evaporated leaving 0.12 g of 6-allyloxy-4-azido-N,N-dimethyl-1,3,5-triazin-2-amine (3). The aqueous wash was evaporated, and the residue separated by column chromatography (eluent EtOAc) to give 0.27 g of 2-(1-allyl-1H-tetrazol-5-yl)-1,1-dimethylguanidine (4) and additional compound 2 (0.06 g).
3-Allyl-5-dimethylaminotetrazolo[1,5- a ][1,3,5]triazin-7-one (2). Yield 1.23 g (56%), white crystals, mp 144–146°С. IR spectrum, ν, cm−1: 3091, 3027, 2987, 2960, 2933, 2877, 1724, 1631, 1560, 1475, 1394, 1378, 1303, 1284, 1226, 1149, 1126, 1087, 958, 946, 829, 767, 703. 1H NMR spectrum, δ, ppm (J, Hz): 3.13 (3Н, s) and 3.17 (3Н, s, N(CH3)2); 4.91 (2Н, dt, J = 5.6, J = 1.5, NCH2); 5.32–5.40 (2Н, m, =СH2); 6.02 (1Н, ddt, J = 16.0, J = 10.9, J = 5.6, CН=CH2). 13C NMR spectrum, δ, ppm: 36.7, 37.2 (N(CH3)2); 48.1 (NCH2); 119.7 (=CH2); 130.1 (СН=CH2); 145.6 (C=O); 150.3 (C-3а); 163.3 (C-5). Found, %: С 43.52; H 5.15; N 44.19. C8H11N7O. Calculated, %: С 43.44; H 5.01; N 44.32.
6-Allyloxy-4-azido- N , N -dimethyl-1,3,5-triazin-2-amine (3). Yield 0.12 g (5%), white crystals, mp 67–69°С. IR spectrum, ν, cm−1: 3097, 3016, 2935, 2852, 2121, 1591, 1519, 1461, 1396, 1336, 1249, 1187, 1087, 1037, 995, 937, 885, 798, 763. 1H NMR spectrum, δ, ppm (J, Hz): 3.09 (3Н, s) and 3.11 (3Н, s, N(CH3)2); 4.82 (2Н, dt, J = 5.6, J = 1.4, OCH2); 5.26 (1Н, dq, J = 10.5, J = 1.3) and 5.37 (1Н, dq, J = 17.2, J = 1.6, =СH2); 6.02 (1Н, ddt, J = 17.2, J = 10.5, J = 5.6, CН=СH2). 13C NMR spectrum, δ, ppm: 36.0 (N(CH3)2); 67.4 (ОCH2); 118.2 (=CH2); 132.6 (CH=CH2); 166.0 (C–NMe2); 169.5 (C–N3); 170.3 (C–O). Found, %: С 43.60; H 5.12; N 44.23. C8H11N7O. Calculated, %: С 43.44; H 5.01; N 44.32.
2-(1-Allyl-1 Н -tetrazol-5-yl)-1,1-dimethylguanidine (4). Yield 0.27 g (18%), white crystals, mp 68–70°С. IR spectrum, ν, cm−1: 3467, 3303, 3025, 2935, 2875, 1629, 1573, 1529, 1506, 1421, 1400, 1328, 1261, 1174, 1091, 1024, 989, 939, 767, 750. 1H NMR spectrum, δ, ppm (J, Hz): 2.99 (6Н, s, NCH3); 4.72 (2Н, d, J = 5.4, NCH2); 5.07 (1Н, d, J = 17.5) and 5.18 (1Н, d, J = 10.2, =СH2); 5.94 (1Н, ddt, J = 16.0, J = 10.6, J = 5.6, CН=СH2); 7.55 (2Н, br. s, NH2). 13C NMR spectrum, δ, ppm: 37.0 (N(CH3)2); 46.6 (NCH2); 118.0 (=CH2); 132.3 (CH=CH2); 157.1 (С–NH2); 157.3 (C-5 tetrazole). Found, %: С 42.98; H 6.67; N 50.35. C7H13N7. Calculated, %: С 43.07; H 6.71; N 50.22.
Hydrolysis of 3-allyl-5-dimethylaminotetrazolo[1,5- a ]-[1,3,5]triazin-7-one (2). Н2О (0.1 ml, 5.6 mmol) was added to a solution of compound 2 (0.44 g, 2 mmol) in MeCN (10 ml), and the mixture stirred at room temperature for 72 h (control by TLC). Acetonitrile was evaporated under reduced pressure leaving a residue of pure 2-(1-allyl-1H-tetrazol-5-yl)-1,1-dimethylguanidine (4). Yield 0.38 g (98%).
X-ray structural analysis of compounds 2, 4. Crystals of compound 2 suitable for X-ray structural analysis were grown from EtOAc by slow evaporation. X-ray structural analysis of a single crystal of compound 2 was performed on a Bruker APEX-II CCD automated diffractometer using λMoKα beam.
Crystals of compound 4 suitable for X-ray structural analysis were grown from MeOH by slow evaporation. X-ray structural analysis of a single crystal of compound 4 was performed on a Xcalibur S automated diffractometer using λMoKα beam.
Collection of data, solving and refinement of the unit cell parameters were performed using the program CrysAlis CCD,8 processing of the diffraction data was done using the program CrysAlis RED.8 The structure was solved with the direct method using program SHELXL9, refinement was performed by full-matrix least-squares technique on F 2 in anisotropic (isotropic for hydrogen atoms) approximation. Representation of the molecular structure in crystal was rendered using the program Mercury.10 The full set of X-ray structural data was deposited at the Cambridge Crystallographic Data Center (deposits CCDC 1421214 (compound 2) and CCDC 1421215 (compound 4)).
This work was supported by funding from the Ministry of Education and Science of the Russian Federation within the framework of the Project part of the State Assignment to Samara State Technical University (project № 4.813.2014/K).
References
(a) De Clercq, E. Nucleosides, Nucleotides Nucleic Acids 2012, 31, 339. (b) Ojwang, J. O.; Ali, S.; Smee, D. F.; Morrey, J. D.; Shimasaki, C. D.; Sidwell, R. W. Antiviral Res. 2005, 68, 49. (c) Krečmerová, M.; Otmar, M. Future Med. Chem. 2012, 4, 991.
Guo, H.-M.; Wu, S.; Niu, H.-Y.; Song, G.; Qu, G.-R. In Chemical Synthesis of Nucleoside Analogues; John Wiley & Sons, Inc., 2013, p. 103.
(a) Khalymbadzha, I. A.; Shestakova, T. S.; Subbotina, J. O.; Eltsov, O. S.; Musikhina, A. A.; Rusinov, V. L.; Chupakhin, O. N.; Karpenko, I. L.; Jasko, M. V.; Kukhanova, M. K.; Deev, S. L. Tetrahedron 2014, 70, 1298. (b) Kjellberg, J.; Johansson, N. G. Nucleosides Nucleotides 1989, 8, 225.
(a) Bakharev, V. V.; Gidaspov, A. A. Chem. Heterocycl. Compd. 2006, 42, 417. [Khim. Geterotsikl. Soedin. 2006, 466.] (b) Fedorov, B. S.; Utienyshev, A. N.; Ghidaspov, A. A.; Kachanovskaya, E. V.; Bakharev, V. V.; Fadeev, M. A. Chem. Heterocycl. Compd. 2005, 41, 496. [Khim. Geterotsikl. Soedin. 2005, 582.]
Parfenov, V. E.; Bakharev, V. V.; Zavodskaya, A. V.; Selezneva, E. V.; Gidaspov, A. A.; Suponitsky, K. Yu. Tetrahedron Lett. 2014, 55, 7072.
(a) Golankiewicz, B.; Januszczyk, P.; Zeidler, J.; Popenda, M. Nucleosides Nucleotides Nucleic Acids 2004, 23, 127. (b) Gelotte, K. O.; Mason, D. N.; Meckler, H.; Shieh, W.-C.; Starkey, C. M. J. Heterocycl. Chem. 1990, 27, 1549. (c) Francis, J. E.; Cash, W. D.; Psychoyos, S.; Ghai, G.; Wenk, P.; Friedmann, R. C.; Atkins, C.; Warren, V.; Furness, P.; Hyun, J. L.; Stone, G. A.; Desai, M.; Williams, M. J. Med. Chem. 1988, 31, 1014. (d) Rusinov, V. L.; Ulomskii, E. N.; Kozhevnikov, D. N.; Chupakhin, O. N.; Aleksandrov, G. G. Zh. Org. Khim. 1996, 32, 770. (e) Rusinov, V. L.; Ulomskii, E. N.; Aleksandrov, G. G.; Parshin, V. E.; Chupakhin, O. N. Chem. Heterocycl. Compd. 1991, 27, 561. [Khim. Geterotsikl. Soedin. 1991, 700.] (f) Orihuela, S.; Sánchez, M. P. ; Quirós, M.; Molina, J.; Faure, R. J. Mol. Struct. 1997, 415, 285. (g) Schroeter, G.; Finck, E. Ber. Dtsch. Chem. Ges. 1938, 71, 671.hybridised
(a) Moriarty, R. M.; Bailey III, B. R.; Prakash, I.; Miller, R. S. J. Org. Chem. 1985, 50, 3710. (b) Peet, N. P. J. Heterocycl. Chem. 1987, 24, 223.
Oxford Diffraction (2006). CrysAlis CCD (Version 1.171.29.9) and CrysAlis RED (Version 1.171.29.9). Oxford Diffraction Ltd., Abingdon.
Sheldrick, G. M. Acta Crystallogr., Sect. A: Found. Crystallogr. 2008, 64A, 112.
Macrae, C. F.; Bruno, I. J.; Chisholm, J. A.; Edgington, P. R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P. A. J. Appl. Crystallogr. 2008, 41, 466.
Author information
Authors and Affiliations
Corresponding author
Additional information
Translated from Khimiya Geterotsiklicheskikh Soedinenii, 2015, 51(8), 745–748
Rights and permissions
About this article

Cite this article
Bakharev, V.V., Parfenov, V.E., Gidaspov, A.A. et al. Alkylation of 5-dimethylaminotetrazolo-[1,5-a][1,3,5]triazin-7-one. Chem Heterocycl Comp 51, 745–748 (2015). https://doi.org/10.1007/s10593-015-1768-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10593-015-1768-4