
Abstract
When rare plants are distributed across a range of habitats, ecotypic differentiation may arise requiring customized conservation measures. The rate of local adaptation may be accelerated in complex landscapes with numerous physical barriers to gene flow. In such cases, examining the distribution of genetic diversity is essential in determining conservation management units. We investigated the distribution of genetic diversity in the federally threatened Camissonia benitensis (Onagraceae), which grows in two distinct serpentine habitats across several watersheds in San Benito, Fresno, and Monterey Cos., CA, USA. We compared genetic diversity with that of its two widespread relatives, C. contorta and C. strigulosa, and examined the potential for hybridization with the latter species. Genotyping results using seven heterospecific microsatellite markers indicate that differentiation between habitat types was weak (F ST = 0.0433) and in an AMOVA analysis, there was no significant partitioning of molecular variation between habitats. Watersheds accounted for 11.6 % of the molecular variation (pairwise F ST = 0.1823–0.4275). Three cryptic genetic clusters were identified by InStruct and STRUCTURE that do not correlate with habitat or watershed. C. benitensis exhibits 5–11× higher inbreeding levels and 0.54× lower genetic diversity in comparison to its close relatives. We found no evidence of hybridization between C. benitensis and C. strigulosa. To maximize conservation of the limited amount of genetic diversity in C. benitensis, we recommend mixing seed representing the three cryptic genetic clusters across the species’ geographic range when establishing new populations.
Similar content being viewed by others


Introduction
Habitat heterogeneity can lead to the development of phenotypically and genetically distinct ecotypes via local adaptation (Kawecki and Ebert 2004; Kossover et al. 2009; Kruckeberg 1986, 1991; Lesica and Shelly 1995; Moyle et al. 2012; Sambatti and Rice 2006). Local adaptation can proceed more rapidly in complex landscapes because geographic barriers can physically separate nascent ecotypes providing partial reproductive isolation through microallopatry (Grossenbacher and Whittall 2011). Reduced gene flow between ecotypes can ultimately lead to speciation (Abbott and Comes 2007; Coyne and Orr 2004; McNeilly and Antonovics 1968). Thus, the combined forces of habitat heterogeneity and geographic isolation can create ecotypes and spur plant diversification. When a species of conservation concern is distributed across diverse habitat types and in a complex landscape, an examination of gene flow and genetic subdivision can aid in preserving the maximum amount of genetic variation (Kramer and Havens 2009; McKay et al. 2005). Understanding how these factors have influenced the amount and structuring of genetic diversity is an important first step in characterizing populations and developing effective in situ and ex situ management strategies (Frankham 2005; Rao and Hodgkin 2002). Microsatellites have been used to estimate barriers to gene flow (Arif et al. 2010; Selkoe and Toonen 2006) and analytical methods such as those for determining genetic structure allow for rigorous hypothesis testing (e.g., habitat divergence vs. geographic barriers) and can even reveal unexpected genetic partitioning (Gao et al. 2006; Pritchard et al. 2000).
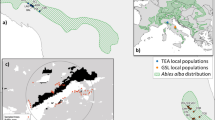
Camissonia benitensis P. H. Raven (Onagraceae) occurs on two distinct serpentine habitat types in a complex landscape in the Bureau of Land Management’s Clear Creek Management Area of southern San Benito County, CA, USA (Fig. 1). The species is a strict serpentine endemic (Safford et al. 2005). Serpentine is an ultramafic rock that weathers to produce extremely chemically adverse soils that are deficient in nitrogen, phosphorus, potassium, and calcium and have potentially phytotoxic concentrations of magnesium and nickel (Kruckeberg 1984). Camissonia benitensis is a diminutive annual herb with very small flowers (Fig. 2) that are primarily self-pollinating (Raven 1969; Taylor 1990) since the pollen dehisces in bud where it is deposited on receptive stigmas 1 h before the flower opens (O’Dell unpublished data). Historically, the species was only known from alluvial stream terraces adjacent to creeks or rivers of several distinct watersheds within or in close proximity to the New Idria serpentine mass. The species was federally listed as threatened in 1985, largely due to adverse impacts to its stream terrace habitat from off-highway vehicles (USFWS 2006). Recent field surveys located additional populations on upland geologic transition zones along the margins of serpentine outcrops (USFWS 2006). The discovery of C. benitensis in geologic transition zone habitat has increased the total number of known occurrences from 64 in 2009 to approximately 426 by 2012 (BLM 2010, 2011, 2012), yet it remains unknown whether stream terrace populations are genetically differentiated from transition zone populations.

Location of Camissonia benitensis study populations. Camissonia benitensis is a local serpentine endemic near San Benito Mountain, San Benito County, CA, USA. Map denotes serpentine soils (green), major streams and rivers (blue lines), known C. benitensis stream terrace populations (red), and known geologic transition zone populations (orange). A total of 12 populations were sampled from stream terrace habitat (ST) and 17 populations were sampled from transition zone habitat (TZ). Pie charts next to each population name indicate the percentage of individuals within that population that were assigned to each of the three InStruct clusters. Samples that were not consistently and confidently placed into a single cluster were assigned based on the highest assignment probability

Camissonia benitensis is a diminutive annual plant that typically reaches a height of only 5 cm or less under natural conditions. Exceptionally large, multi-branched specimens have a decumbent growth form and may reach 20 cm in diameter. The species has very small flowers with petals 3.5–4 mm long
We tested three hypotheses that may affect the distribution of genetic diversity in C. benitensis. First, habitat differences between the stream terrace and transition zones may reduce gene flow in C. benitensis populations occupying the two habitat types and create local adaptation (ecotypic differentiation). Although there are no obvious differences in flowering time or morphology in plants from either habitat type, there are substantial differences in slope steepness and the associated plant community. Stream terrace populations occur on gentle slopes <15° with leather oak (Quercus durata), manzanita (Arctostaphylos visicida and A. pungens), and several conifers (Pinus sabiniana, P. coulteri, and P. jeffreyi). Transition zone populations are found much further from water in uplands on slopes as steep as 60° and can be associated with blue oak (Quercus douglasii) or California juniper (Juniperus californicus) woodland and scrub oak (Quercus berberidifolia) (BLM 2010, 2011). Second, physical distance between populations could be positively correlated with genetic distance. Isolation by distance is predicted when gene flow is low, and with C. benitensis being primarily self-pollinating, gene flow will be mainly dependent on the rate of seed dispersal. Although the range of C. benitensis is fairly small (435 km2), other studies have detected significant isolation-by-distance at comparably small geographic scales (Furches et al. 2009; Peterson et al. 2002). Third, there is substantial topographic variation within the small range of C. benitensis, extending from 595 m at the western limit of its range to 1,284 m at the eastern range limit. Six major watersheds divide the species range, which may lead to genetic subdivision between plants from different watercourses that may not be detected by raw geographic distances (Whittall et al. 2006; Whittall et al. 2004). Due to the small range of C. benitensis, we can test the three potential barriers to gene flow (habitat type, distance, and watershed) in this study by including samples from populations distributed throughout the entire geographical range of the species.
The distributional range of C. benitensis overlaps with two of its presumed closest relatives—C. contorta and C. strigulosa (Raven 1969; Taylor 1990). These two species could provide a useful comparison of the genetic diversity between a strict serpentine endemic (C. benitensis), a serpentine tolerator (C. strigulosa; found on both serpentine and non-serpentine soils), and a non-tolerator (C. contorta; found only on non-serpentine soils) (Anacker et al. 2011; Safford et al. 2005). Camissonia strigulosa grows sympatrically with several C. benitensis populations and is also tetraploid with identical chromosome counts (2n = 28; Raven 1969), so there is potential for hybridization. Camissonia contorta is more widespread and grows in the vicinity of C. benitensis, but occupies non-serpentine habitats, and is a hexaploid (2n = 42; Raven 1969), thereby reducing the chances of hybridization with C. benitensis.
Determining the number of conservation units and therefore the proper source material to use in reintroductions of C. benitensis depends on the distribution of genetic variation within and among populations and potential hybridization/introgression with close relatives. Microsatellites have recently been developed for Camissoniopsis cheiranthifolia (formerly Camissonia cheiranthifolia) and successfully amplified in six species in the former genus Camissonia (Camissoniopsis bistorta, C. micrantha, C. lewisii, Eulobus crassifolius, E. californica, and E. angelorum; Lopez-Villalobos, Samis and Eckert unpublished data). In this study, we used these heterospecific microsatellite loci to address three main goals to aid in the conservation of C. benitensis: (1) test for genetic differentiation between habitat types, among watersheds, and across geographic distance of C. benitensis, (2) determine the distribution of genetic diversity in C. benitensis compared to its close relatives, and (3) analyze the hybridization potential between C. benitensis and C. strigulosa.
Materials and methods
Sample collection and DNA extraction
For all samples, a population was defined as a group of individuals separated from other groups by unoccupied habitat and at least 0.40 km (0.25 miles) as stated from the California Natural Diversity Database. In this study, populations were separated by an average of 10.5 km ± 0.31 SE (range 0.43–31.0 km). Fresh leaf or flower tissue from C. benitensis individuals (n = 213) was collected from the extent of the species’ range during spring 2011 (Fig. 1). On average, we sampled 19 individuals per population (range 11–23) separated by at least 1 m from six locations [four stream terrace (ST), two transition zone (TZ)]. Sample sizes for field populations are as follows: 01TZ: n = 17, 04ST: n = 17, 07ST: n = 20, 20ST: n = 23, 28TZ: n = 11, 29ST: n = 23. Samples were also collected from plants grown from soil seed bank collected from 23 additional populations (eight stream terrace, 15 transition zone). For the seed bank samples, 20 soil aliquots of 225 mL were collected at least 1 m apart from within a single population, sieved to <2 mm, and homogenized to create a composite sample. Approximately 475 mL of each composite soil sample was thinly spread on plastic flats (Anderson Die-Deep Propagation Flat, Stuewe and Sons, Inc., Corvallis, OR, USA) filled with potting soil (MiracleGro Moisture Control potting mix) and seeds in the seed bank were germinated at ambient climate at the BLM Hollister Field Office (Hollister, CA, USA; (BLM 2011). Plants were cultivated to maturity and tissue from four individuals on average (range 1–8) representing each population was randomly sampled from the flats. Sample sizes for seed bank populations are as follows: 02TZ: n = 8, 03TZ: n = 4, 05TZ: n = 4, 06TZ: n = 4, 08TZ: n = 6, 09TZ: n = 4, 10TZ: n = 4, 11ST: n = 4, 12ST: n = 4, 13TZ: n = 4, 14ST: n = 3, 15TZ: n = 8, 16TZ: n = 4, 17TZ: n = 5, 18ST: n = 4, 19ST: n = 1, 21ST: n = 4, 22TZ: n = 5, 23ST: n = 4, 24TZ: n = 5, 25ST: n = 4, 26TZ: n = 4, 27TZ: n = 5.
To compare genetic diversity of C. benitensis to close relatives, tissue was also collected at two C. contorta (Bear Valley: n = 19, Pinnacles Bench Trail: n = 23) and three C. strigulosa locations (Oat Canyon: n = 24, White Creek: n = 19, Coalinga Road: n = 19) within or near (within 56 km) a C. benitensis population. DNA from ~15 mg of each tissue sample was extracted with the NucleoSpin Plant II kit (Macherey–Nagel, Bethlehem, PA, USA) using lysis buffer PL2.
Microsatellite amplification
Microsatellite loci were amplified with primers developed for Camissoniopsis cheiranthifolia (Lopez-Villalobos, Samis and Eckert unpublished data). Of the 23 loci tested, five were intact based on direct sequencing of one sample per species on an ABI 3730xl DNA Analyzer (Sequetech, Santa Clara, CA, USA) following the BigDye protocol (Life Technologies, Foster City, CA, USA). Two additional loci with interrupted microsatellites determined by Sanger sequencing) were included in the study because they exhibited useful variation in fragment length within and among species (Table 1; Online Resources 1 and 2). PCR was performed in 25 μL reaction volumes using the following reagents (and their final concentrations): 1× Buffer B, 2.5 mM MgCl2, 0.25 mM dNTPs, 0.6–0.8 μM forward and reverse primer, and 1.5 U Taq DNA polymerase (New England Biolabs, Massachusetts, USA). Reverse primers were labeled on the 5′ end with the fluorophore 6-FAM or NED. Thermal cycling conditions were: 94 °C for 5 min, followed by 40 cycles of 94 °C for 25 s, 47–59 °C (Table 1) for 15 s, and 72 °C for 40 s. A final 5 min extension at 72 °C was followed by a 4 °C hold.
Fragments were separated and sized on an ABI 3730xl DNA Analyzer (Cornell University Core Laboratories, Ithaca, NY, USA) using a GeneScan 500 LIZ size standard (Life Technologies) following Cornell’s recommended reaction conditions published online. Samples were genotyped with GeneMapper software (v4.0, Life Technologies) using the default microsatellite analysis settings. Alleles were manually scored and binned based on size similarity. Overall, there did not seem to be a large effect of null alleles that can be caused by priming site mutations or large allele dropout (Selkoe and Toonen 2006). Out of 2,219 samples (317 individuals × 7 loci), only 30 or 1.4 % failed to amplify the first time, which could have been due to priming site variation. Re-amplification at or below the original annealing temperature always succeeded so that each sample was amplified for all seven loci. In addition, we were able to detect an average of 12 bps difference in allele sizes within an individual (range 6–27). The average difference between the smallest and largest allele sizes among species and loci was 14 (range 0–53), so there did not seem to be a problem with large allele dropout.
Analysis of microsatellite data
For all analyses, loci were only used if they were variable across the species tested and did not exhibit signs of duplication (>2 alleles/locus; for C. contorta only). The C. benitensis population differentiation, within-species inbreeding, and C. benitensis/C. strigulosa hybridization analyses included five, four, and seven loci, respectively. Population structuring within C. benitensis was determined with STRUCTURE (version 2.3.3; Pritchard et al. 2000). An admixture model was used and the assumed number of population clusters (k) was tested from 1 to 10 for five independent runs using a burn-in and MCMC sampling length of 1 × 106 generations each and λ = 0.5920 (empirically determined). Since deviations from Hardy–Weinberg equilibrium due to self-fertilization can lead to overestimation of admixture in STRUCTURE (Gao et al. 2007), the program InStruct (version 1; Gao et al. 2006), which does not assume Hardy–Weinberg equilibrium, was run for comparison using identical burn-in lengths, MCMC repetitions, and chain numbers. We are not aware of any simulations looking at the effect of null alleles on InStruct sample assignment. However, sample assignment by STRUCTURE is robust to null alleles (Carlsson 2008) and other studies have found very similar results between the programs (Gunn et al. 2011; Tatarenkov et al. 2007), even when there is some evidence of null alleles (Niu et al. 2012). For both analyses, the actual number of population clusters that best fit the data was calculated using the Δk method of Evanno et al. (2005) in STRUCTURE Harvester (Earl and vonHoldt 2012) or by hand using the likelihood values. Both analyses converged on the same k and STRUCTURE did not appear to overestimate admixture as only 38 % of samples showed significant admixture (<0.95 assignment probability) compared to 66 % of samples in the InStruct analysis. For simplicity and model accuracy, only the InStruct assignments are reported here.
Population differentiation between C. benitensis stream terrace and transition zone habitat types was tested using F ST in GenePop (version 4.1; (Raymond and Rousset 1995). Significance was determined with a Markov chain algorithm using a burn-in (dememorization) of 1 × 106 batches followed by 50,000 batches, and 5,000 iterations per batch. Hierarchical analysis of molecular variance (AMOVA) was also performed in Arlequin (version 3.5; Excoffier and Lischer 2010) using 10,000 permutations and samples were partitioned as follows: between habitat types, among populations within habitat types, and within populations.
Differentiation among watersheds was examined by AMOVA and F ST as described above. In the AMOVA, variation was partitioned among watersheds, among populations within watersheds, and within populations. Populations were grouped into watersheds as follows: Group 1-Laguna Creek (02TZ, 03TZ), Group 2-Clear Creek (04ST, 10TZ, 11ST, 12ST, 13TZ, 14ST, 18ST, 19ST, 21ST), Group 3-Larious Creek (05TZ, 06TZ, 07ST, 08TZ, 09TZ), Group 4-Sampson Creek (16TZ, 17TZ), Group 5-Upper San Benito River (22TZ, 23ST, 24TZ, 25ST), and Group 6-White Creek (27TZ, 28TZ, 29ST). Four populations (01TZ, 15TZ, 20ST, and 26TZ) were not included in the analysis since each population was the only one in its watershed.
An isolation-by-distance analysis was conducted to test for geographic structure in the distribution of genetic variation. Two-dimensional Cartesian coordinates for the analysis were acquired for all 29 C. benitensis populations. Mantel tests with 1 × 106 permutations were used to estimate significance. The analysis was conducted on all populations combined and separately on populations within each habitat type. The average number of migrants per generation, Nm, was calculated for all populations following the method of Barton and Slatkin (1986).
Levels of inbreeding within C. benitensis, C. contorta, and C. strigulosa populations were estimated with Wright’s inbreeding coefficient (F IS) in GenePop. Since null alleles can overestimate F IS, we also calculated inbreeding coefficients for each species and population using INEst, which can account for the presence of null alleles (Chybicki and Burczyk 2009). The distribution of genetic variation within and among species was examined with an analysis of molecular variance (AMOVA) in Arlequin using 10,000 permutations to assess the significance of the components of molecular variance.
To compare allelic diversity across species (C. benitensis and C. strigulosa) with drastically different sample sizes, we used Simpson’s Diversity Index (1-D). A C++ script (source code available upon request) was used to bootstrap 100,000 replicates, with replicates being drawn from a pool of three randomly selected populations per species. The script was run twice using small and large sample sizes. For the small sample size, we sampled 12 individuals from populations with at least four individuals. For the large sample size, we sampled 45 individuals from populations with at least 10 individuals. The ranges of the magnitude differences among all loci were calculated for both sample size numbers. The Simpson’s Diversity Index of C. contorta is not reported as the value ranged inconsistently due to its polyploid nature.
Hybridization between C. benitensis and its closely related and often sympatric C. strigulosa was examined with STRUCTURE and InStruct as described above, except λ = 0.5962 (empirically determined). Again, both analyses converged on the same k using the Δk method, although InStruct had less admixture in this analysis (4 % of InStruct versus 9 % of STRUCTURE samples had less than 0.95 assignment probability). Only the InStruct assignments are reported below.
Results
Habitat, watershed and population differentiation
In the InStruct (and STRUCTURE) analysis, the data best fit a model with three genetic clusters of individuals (Fig. 3; Online Resource 3). Post-hoc pairwise F ST among the three clusters identified by InStruct ranged from 0.1948 to 0.3080 (P < 0.0001). Additionally, almost 21 % of all genetic variation was partitioned among clusters (Table 2). Samples were largely clustered according to population. For 62 % of the populations, all individuals sampled from that location were assigned to the same cluster. The remaining 38 % of the populations had individuals assigned to two or even all three different clusters (Fig. 1). Within these populations, the second or third clusters usually had individuals assigned to them with admixture.

Genetic differentiation between Camissonia benitensis stream terrace (n = 111) and transition zone (n = 102) habitat types. Plot shows the InStruct analysis indicating the probability an individual belongs to each of three assumed clusters. Assignment probabilities represent an average after aligning the probability values from five independent chains
Although C. benitensis stream terrace and transition zone habitats are spatially distinct, the pairwise F ST for differentiation between habitats was weak, yet significant (F ST = 0.0433; P < 0.0001). In the AMOVA analysis, none of the molecular variation in C. benitensis was partitioned between the two habitat types (Table 2). Among the six watersheds, genetic subdivision was larger than for habitat types (pairwise F ST ranged from 0.1823 to 0.4275; P < 0.0001) and 11.6 % of the AMOVA variation was found among watersheds (Table 2). Additionally, there were no significant correlations between spatial proximity and genetic similarity from the isolation-by-distance analysis (all populations: P = 0.78; stream terrace: P = 0.06; transition zone: P = 0.47). Gene flow between populations based on the average number of migrants was low (Nm = 0.306).
Within population inbreeding levels
Camissonia benitensis had exceptionally high levels of inbreeding in comparison to C. contorta and C. strigulosa, regardless of whether we accounted for null alleles or not (Table 3; Online Resource 4). The inbreeding coefficient for C. benitensis was 0.813 or 0.279 from GenePop and INEst, respectively. On average, this value was 5× that of C. contorta and 11× that of C. strigulosa.
Genetic variation among species
AMOVA indicated that only 29 % of the total variation could be explained by genetic differences among the three species (Table 4). Depending on which locus was examined and whether we used small or large sample sizes, C. benitensis genetic diversity was 0.18–0.98× that of C. strigulosa (mean = 0.54×).
Hybridization potential
InStruct repeatedly found two genetic clusters that generally differentiated C. benitensis from C. strigulosa (Fig. 4). Most of the C. benitensis samples were grouped in the same cluster, but 3.3 % were consistently assigned to both clusters with low probability (individuals from C. benitensis populations 07ST, 15TZ, and 28TZ in Fig. 1). The C. strigulosa individuals that grouped together primarily came from the two non-serpentine populations. However, 24.2 % of C. strigulosa were more genetically similar to C. benitensis and came from a single serpentine population. It is unlikely that these individuals were misidentified because they do not co-occur with C. benitensis and all of their alleles at three loci are otherwise unique compared to C. benitensis. The 29ST population at White Creek where both species co-occur had no alleles in common, except at one locus where C. benitensis was invariant.

Genetic differentiation and hybridization potential between Camissonia benitensis (n = 213 individuals, 29 populations) and C. strigulosa (n = 62 individuals, 3 populations). InStruct analysis plotting the probability an individual belongs to each of two assumed clusters. Assignment probabilities represent an average after aligning the probability values from five independent chains
Discussion
Habitat, watershed and population differentiation
Populations of C. benitensis show evidence of cryptic genetic subdivision that does not correlate with habitat type, watershed, or physical distance between populations. Although the F ST between habitats was highly significant, none of the genetic variation was partitioned among habitats in an AMOVA analysis. Based on the isolation-by-distance analysis, physical distance among all populations could not account for these genetic differences. There was marginally insignificant isolation-by-distance among just the stream terrace populations, which is consistent with seeds likely being moved short distances within watersheds. A landscape genetic approach would be interesting to compare with physical distances, now that we have identified some potential resistance barriers to gene flow such as watershed (McRae 2006). Watershed represented a slightly better subdivision than habitat based on F ST and AMOVA results; however, the InStruct results indicated the genetic data were best grouped into three clusters of individuals that were independent of habitat or watershed. Almost one quarter of all genetic variation was partitioned among these three cryptic genetic clusters. Null alleles could have affected the accuracy of InStruct cluster assignment, but STRUCTURE, which is robust to null alleles (Carlsson 2008), also found three cryptic genetic clusters. The biological basis for the clustering remains unexplained as populations belonging to each of the three clusters show no apparent phenotypic, ecological, or geographic similarity and populations from the same cryptic genetic cluster can be up to 29 km distant from one another (the approximate width of the entire species range). Other studies have also detected evidence of cryptic genetic structuring (Brown et al. 2012; Xu et al. 2006). In our study, it is possible that the number of microsatellite markers examined was not large enough to detect differentiation between habitats or watersheds. Increasing the number of markers could sample more of the genome and determine whether the cryptic subdivision persists.
Within local populations, there were differences in the preservation of genetic diversity. Some populations (38 %) in the InStruct analysis contain individuals belonging to more than one assignment, while the rest of the populations contain individuals that cluster together. These populations may still harbor undetected genetic variation. For example, additional alleles may be hidden in the seed bank (Honnay et al. 2008), which has been conservatively estimated as averaging 519× the size of the standing populations (BLM 2011). Our sampling included field collected plants and plants germinated from the seed bank, and there were slight differences in the distribution of genetic diversity (field collected F IS = 0.868 ± 0.06; seed bank F IS = 0.780 ± 0.10) and significant population differentiation between field and seed bank samples (F ST = 0.072; P < 0.0001). Most of the 11 populations with individuals assigned to more than one cluster were collected from the seed bank (seed bank: 7/11; field: 4/11), so it may act as an additional source of variation (Ellner and Hairston 1994).
Gene flow between C. benitensis populations is not enough to reduce inter-population differentiation as most genetic variation occurred among populations. The observed low gene flow could be due to short seed dispersal distances (Colling et al. 2010; England et al. 2002; Finger et al. 2011; Furches et al. 2009; Schaal 1980). Seed dispersal is most likely the primary gene flow mechanism in self-pollinated plants where pollen flow is rare. Gene flow via seed dispersal in C. benitensis is thought to occur by water (USFWS 2006). Long-distance seed transport may be infrequent because the average number of migrants is low and may not counteract divergence between distant populations (Mills and Allendorf 1996). Fragmented habitat distribution could also inhibit gene flow (England et al. 2002; Furches et al. 2009; Provan et al. 2008) by limiting the species’ ability to colonize new populations once dispersed. Populations of C. benitensis are found on a narrow range of habitats consisting of relatively stable soils, sparse woody overstory, and little competition from other native or invasive species (BLM 2010). The presence of non-suitable habitat separating populations generates a patchy distribution, which could further increase genetic isolation.
Within population inbreeding levels
Camissonia benitensis exhibits high levels of inbreeding (GenePop F IS = 0.813; INEst F IS = 0.279). The small sample sizes of some C. benitensis populations likely did not inflate our estimate of inbreeding. The average inbreeding level of just the field-collected samples, which had larger sample sizes, was very similar to the full dataset (field collected F IS = 0.869, all populations F IS = 0.813). This level of inbreeding in C. benitensis is indicative of predominantly self-pollinating species like Arabidopsis thaliana (F IS = 0.92–1.0; Stenoien et al. 2005), Mimulus laciniatus (F IS = 0.80; Awadalla and Ritland 1997), Medicago lupulina (F IS = 0.92; Yan et al. 2009), and Triticum aestivum (F IS = 0.84–0.98; Rousselle et al. 2011). It is unclear why C. contorta and C. strigulosa would have much lower levels of inbreeding. They have comparably small flowers and are inferred to have a similar self-pollinating mating system since pollen was observed dehiscing in bud (Raven 1969). It is possible there could still be variation in rates of self-pollination within species as this value can differ greatly among groups (Herlihy and Eckert 2004; Kalisz et al. 2012; Routley et al. 1999). If C. benitensis had higher self-pollination rates than the two other species, its F IS value would be higher. In addition, C. benitensis could have smaller population or seed bank size than the two other species. Although this information is not yet available, future studies could examine this potential difference.
There was a large discrepancy in inbreeding values depending on whether we estimated inbreeding with GenePop or INEst. We believe the number of null alleles calculated by INEst was overestimated. The null allele frequency was 0.25 on average (range 0.022–0.543) depending on the species or locus (Online Resource 5). Based on the null allele frequencies, we calculated the expected number of homozygotes with null alleles for the four loci included in the analysis, which should represent the number of initial PCR failures. We should have expected 58 PCR failures, but we only had 22 failures at those loci. Note that 58 is an underestimate, as we assumed Hardy–Weinberg equilibrium to calculate the expected number of failures. Even if there were some null alleles, we were mainly interested in relative differences between species rather than the absolute value to see whether C. benitensis showed comparatively high levels of inbreeding. In comparison to the other species, C. benitensis had higher levels of inbreeding regardless of the method used to calculate F IS.
Genetic variation among species
When comparing the distribution of genetic diversity for C. benitensis, C. strigulosa, and C. contorta, there was 1.5× more variation within species than among species. The lack of differentiation is most likely not due to gene flow through hybridization. If an individual represented a recent hybridization event, some of its microsatellite loci would be genetically similar to each parent species. This is unlikely since there was very low genetic variation within populations of C. benitensis to start with, which would not be expected if a population contained alleles from multiple species. Three possibilities could explain the lack of variation among species. First, an identical sized allele shared among two or three of the species could have arisen from independent mutations. If this occurred, there would be divergence between the species that went undetected. Second, both C. benitensis and C. contorta were thought to be derived from C. strigulosa or from hybridization between C. strigulosa and another species, respectively (Raven 1969). The shared and relatively recent ancestry between all three species could explain the lack of differentiation. Third, it is also likely that variation present in the ancestral species underwent incomplete allele sorting as the species diverged (Avise 1994; Cooper et al. 2010; Funk and Omland 2003). In this case, when different populations of the three species became fixed for alleles found in the more variable common ancestor, alleles could have sorted in such a way that they became shared across species.
Hybridization potential
Hybridization with more widespread plants negatively impacts rare species and could lead to extinction (Soltis and Gitzendanner 1999). If hybrids were viable, introgression would dilute the gene pool of the rare species through genetic assimilation and hybrids could further compete for resources or habitat (Levin et al. 1996). This is especially problematic for endemic species because local extinction at populations where both parents co-occur could critically reduce the already diminished population numbers of the species at risk (Francisco-Ortega et al. 2000; Liston et al. 1990; Rieseberg and Gerber 1995).
Based on our sampling, no hybridization events were detected between C. benitensis and C. strigulosa. Three conditions would need to be met for us to conclude hybridization between these species from our data: (1) in the InStruct analysis, some of the individuals would not have a definite assignment probability to one of the two clusters because their microsatellite loci would be a mixture of both parents, (2) each species would need to be found in the same geographic area, and (3) there would likely be shared alleles between co-occurring or geographically adjacent populations. Of the three C. benitensis populations containing individuals not confidently assigned to either C. benitensis or C. strigulosa clusters, only one population (07ST) is within 100 meters of C. strigulosa, but it is most genetically identical to other C. benitensis individuals.
Reintroduction implications
When features of a heterogeneous landscape do not correlate with genetic structure, the ideal way to preserve genetic variation is not as straightforward as if genetic structure was associated with landscape features. In C. benitensis, careful management during reintroductions is of utmost importance to preserve the small amount of genetic diversity that exists. The results from this study can provide some recommendations to help ex situ recovery. First, habitat type (stream terrace vs. transition zone) and watershed do not represent the best subdivision of the genetic data so it might be possible to mix seeds without regard to these groupings. We did not see evidence of ecotypic differentiation nor incipient speciation, despite substantial habitat differences among populations. However, there could still be divergence at traits under selection between both habitat types even though we detected low microsatellite divergence (Leinonen et al. 2008; McKay and Latta 2002; Reed and Frankham 2001). Future studies should test for local adaptation between habitat types and perform crosses to test for reproductive isolation. In addition, the conclusion about the lack of habitat/watershed differentiation is based on five microsatellite loci with admixture (66 %). Since accurate assignment of admixed samples may require greater than five loci (Pritchard et al. 2000), the inclusion of several more loci could generate different results. Second, a special emphasis should be placed on collecting seed from the populations with individuals belonging to multiple InStruct-based clusters. These populations are: 01TZ, 02TZ, 04ST, 10TZ, 15TZ, 17TZ, 18ST, 20ST, 25ST, 26TZ, and 29ST. The 01TZ population is especially important because it contains individuals assigned to all three of the InStruct clusters. Mixing seed from populations representing all three clusters when establishing new introduced populations will maximize conservation of genetic diversity in C. benitensis (Godefroid et al. 2011).
References
Abbott RJ, Comes HP (2007) Blowin in the wind: the transition from ecotype to species. New Phytol 175(2):197–200. doi:10.1111/j.1469-8137.2007.02127.x
Anacker BL, Whittall JB, Goldberg EE, Harrison SP (2011) Origins and consequences of serpentine endemism in the California flora. Evolution 65(2):365–376. doi:10.1111/j.1558-5646.2010.01114.x
Arif IA, Bakir MA, Khan HA, Al Farhan AH, Al Homaidan AA, Bahkali AH, Al Sadoon M, Shobrak M (2010) A brief review of molecular techniques to assess plant diversity. Int J Mol Sci 11(5):2079–2096. doi:10.3390/ijms11052079
Avise JC (1994) Molecular markers, natural history and evolution. Chapman & Hall, New York
Awadalla P, Ritland K (1997) Microsatellite variation and evolution in the Mimulus guttatus species complex with contrasting mating systems. Mol Biol Evol 14(10):1023–1034
Barton NH, Slatkin M (1986) A quasi-equilibrium theory of the distribution or rare alleles in a subdivided population. Heredity 56:409–415
BLM (2010) Annual summary report for the 2009–2010 season monitoring and status of Camissonia benitensis and implementation of the 2006 record of decision. Hollister, CA
BLM (2011) Annual report for the 2010–2011 season monitoring and status of Camissonia benitensis and implementation of the 2006 record of decision. Hollister, CA
BLM (2012) Annual report for the 2012 season. Monitoring and status of Camissonia benitensis and implementation of the 2006 record of decision. Hollister, CA
Brown JE, Bauman JM, Lawrie JF, Rocha OJ, Moore RC (2012) The structure of morphological and genetic diversity in natural populations of Carica papaya (Caricaceae) in Costa Rica. Biotropica 44(2):179–188. doi:10.1111/j.1744-7429.2011.00779.x
Carlsson J (2008) Effects of microsatellite null alleles on assignment testing. J Hered 99(6):616–623. doi:10.1093/jhered/esn048
Chybicki IJ, Burczyk J (2009) Simultaneous estimation of null alleles and inbreeding coefficients. J Hered 100(1):106–113. doi:10.1093/jhered/esn088
Colling G, Hemmer P, Bonniot A, Hermant S, Matthies D (2010) Population genetic structure of wild daffodils (Narcissus pseudonarcissus L.) at different spatial scales. Plant Syst Evol 287(3–4):99–111. doi:10.1007/s00606-010-0298-x
Cooper EA, Whittall JB, Hodges SA, Nordborg M (2010) Genetic variation at nuclear loci fails to distinguish two morphologically distinct species of Aquilegia. PLoS One 5(1):e8655. doi:e865510.1371/journal.pone.0008655
Coyne JA, Orr AH (2004) Speciation. Sinauer Associates, Sunderland
Earl DA, vonHoldt BM (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4(2):359–361. doi:10.1007/s12686-011-9548-7
Ellner S, Hairston NG (1994) Role of overlapping generations in maintaining genetic variation in a fluctuating environment. Am Nat 143(3):403–417. doi:10.1086/285610
England PR, Usher AV, Whelan RJ, Ayre DJ (2002) Microsatellite diversity and genetic structure of fragmented populations of the rare, fire-dependent shrub Grevillea macleayana. Mol Ecol 11(6):967–977. doi:10.1046/j.1365-294X.2002.01500.x
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14(8):2611–2620. doi:10.1111/j.1365-294X.2005.02553.x
Excoffier L, Lischer HEL (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10(3):564–567. doi:10.1111/j.1755-0998.2010.02847.x
Finger A, Kettle CJ, Kaiser-Bunbury CN, Valentin T, Doudee D, Matatiken D, Ghazoul J (2011) Back from the brink: potential for genetic rescue in a critically endangered tree. Mol Ecol 20(18):3773–3784. doi:10.1111/j.1365-294X.2011.05228.x
Francisco-Ortega J, Santos-Guerra A, Kim SC, Crawford DJ (2000) Plant genetic diversity in the Canary Islands: a conservation perspective. Am J Bot 87(7):909–919. doi:10.2307/2656988
Frankham R (2005) Genetics and extinction. Biol Conserv 126(2):131–140. doi:10.1016/j.biocon.2005.05.002
Funk DJ, Omland KE (2003) Species-level paraphyly and polyphyly: frequency, causes, and consequences, with insights from animal mitochondrial DNA. Annu Rev Ecol Evol Syst 34:397–423. doi:10.1146/annurev.ecolsys.34.011802.132421
Furches MS, Wallace LE, Helenurm K (2009) High genetic divergence characterizes populations of the endemic plant Lithophragma maximum (Saxifragaceae) on San Clemente Island. Conserv Genet 10(1):115–126. doi:10.1007/s10592-008-9531-3
Gao H, Williamson S, Bustamante CD (2006) InStruct (version 1) [Computer Program]. Available at http://cbsuapps.tc.cornell.edu/InStruct.aspx. Accessed 03 August 2011
Gao H, Williamson S, Bustamante CD (2007) A Markov chain Monte Carlo approach for joint inference of population structure and inbreeding rates from multilocus genotype data. Genetics 176(3):1635–1651. doi:10.1534/genetics.107.072371
Godefroid S, Piazza C, Rossi G, Buord S, Stevens AD, Aguraiuja R, Cowell C, Weekley CW, Vogg G, Iriondo JM, Johnson I, Dixon B, Gordon D, Magnanon S, Valentin B, Bjureke K, Koopman R, Vicens M, Virevaire M, Vanderborght T (2011) How successful are plant species reintroductions? Biol Conserv 144(2):672–682. doi:10.1016/j.biocon.2010.10.003
Grossenbacher DL, Whittall JB (2011) Increased floral divergence in sympatric monkey flowers. Evolution 65(9):2712–2718
Gunn BF, Baudouin L, Olsen KM (2011) Independent origins of cultivated coconut (Cocos nucifera L.) in the old world tropics. PLoS One. doi:10.1371/journal.pone.0021143
Herlihy CR, Eckert CG (2004) Experimental dissection of inbreeding and its adaptive significance in a flowering plant, Aquilegia canadensis (Ranunculaceae). Evolution 58(12):2693–2703. doi:10.1554/04-439
Honnay O, Bossuyt B, Jacquemyn H, Shimono A, Uchiyama K (2008) Can a seed bank maintain the genetic variation in the above ground plant population? Oikos 117(1):1–5. doi:10.1111/j.2007.0030-1299.16188.x
Kalisz S, Randle A, Chaiffetz D, Faigeles M, Butera A, Beight C (2012) Dichogamy correlates with outcrossing rate and defines the selfing syndrome in the mixed-mating genus Collinsia. Ann Bot 109(3):571–582. doi:10.1093/aob/mcr237
Kawecki TJ, Ebert D (2004) Conceptual issues in local adaptation. Ecol Lett 7(12):1225–1241. doi:10.1111/j.1461-0248.2004.00684.x
Kossover O, Frenkel Z, Korol A, Nevo E (2009) Genetic diversity and stress of Ricotia lunaria in “Evolution Canyon,” Israel. J Hered 100(4):432–440. doi:10.1093/jhered/esp014
Kramer AT, Havens K (2009) Plant conservation genetics in a changing world. Trends Plant Sci 14(11):599–607. doi:10.1016/j.tplants.2009.08.005
Kruckeberg AR (1984) California serpentines: flora, vegetation, geology, soils, and management problems. University of California Press, California
Kruckeberg AR (1986) The stimulus of unusual geologies for plant speciation: an essay. Syst Bot 11(3):455–463. doi:10.2307/2419082
Kruckeberg AR (1991) Geoedaphics and island biogeography for vascular plants: an essay. Aliso 13:225–238
Leinonen T, O’Hara RB, Cano JM, Merila J (2008) Comparative studies of quantitative trait and neutral marker divergence: a meta-analysis. J Evol Biol 21(1):1–17. doi:10.1111/j.1420-9101.2007.01445.x
Lesica P, Shelly JS (1995) Effects of reproductive mode on demography and life-history in Arabis fecunda (Brassicaceae). Am J Bot 82(6):752–762. doi:10.2307/2445615
Levin DA, FranciscoOrtega J, Jansen RK (1996) Hybridization and the extinction of rare plant species. Conserv Biol 10(1):10–16. doi:10.1046/j.1523-1739.1996.10010010.x
Liston A, Rieseberg LH, Mistretta O (1990) Ribosomal DNA evidence for hybridization between island endemic species of Lotus. Biochem Syst Ecol 18(4):239–244. doi:10.1016/0305-1978(90)90067-p
McKay JK, Latta RG (2002) Adaptive population divergence: markers, QTL and traits. Trends Ecol Evol 17(6):285–291. doi:10.1016/s0169-5347(02)02478-3
McKay JK, Christian CE, Harrison S, Rice KJ (2005) “How local is local?” A review of practical and conceptual issues in the genetics of restoration. Restor Ecol 13(3):432–440. doi:10.1111/j.1526-100X.2005.00058.x
McNeilly T, Antonovics J (1968) Evolution in closely adjacent plant populations IV. Barriers to gene flow. Heredity 23:205–218
McRae BH (2006) Isolation by resistance. Evolution 60(8):1551–1561. doi:10.1554/05-321.1
Mills LS, Allendorf FW (1996) The one-migrant-per-generation rule in conservation and management. Conserv Biol 10(6):1509–1518. doi:10.1046/j.1523-1739.1996.10061509.x
Moyle LC, Levine M, Stanton ML, Wright JW (2012) Hybrid sterility over tens of meters between ecotypes adapted to serpentine and non-serpentine soils. Evol Biol 39(2):207–218. doi:10.1007/s11692-012-9180-9
Niu HY, Hong L, Wang ZF, Shen H, Ye WH, Mu HP, Cao HL, Wang ZM, Bradshaw CJA (2012) Inferring the invasion history of coral berry Ardisia crenata from China to the USA using molecular markers. Ecol Res 27(4):809–818. doi:10.1007/s11284-012-0957-1
Peterson A, Bartish IV, Peterson J (2002) Genetic structure detected in a small population of the endangered plant Anthericum liliago (Anthericaceae) by RAPD analysis. Ecography 25(6):677–684. doi:10.1034/j.1600-0587.2002.250604.x
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155(2):945–959
Provan J, Beatty GE, Hunter AM, McDonald RA, McLaughlin E, Preston SJ, Wilson S (2008) Restricted gene flow in fragmented populations of a wind-pollinated tree. Conserv Genet 9(6):1521–1532. doi:10.1007/s10592-007-9484-y
Rao VR, Hodgkin T (2002) Genetic diversity and conservation and utilization of plant genetic resources. Plant Cell, Tissue Organ Cult 68(1):1–19
Raven PH (1969) A revision of the genus Camissonia (Onagraceae). Contributions to the US National Herbarium 37:155–396
Raymond M, Rousset F (1995) Genepop (version 1.2): population genetics software for exact tests and ecumenicism. J Hered 86(3):248–249
Reed DH, Frankham R (2001) How closely correlated are molecular and quantitative measures of genetic variation? A meta-analysis. Evolution 55(6):1095–1103. doi:10.1111/j.0014-3820.2001.tb00629.x
Rieseberg LH, Gerber D (1995) Hybridization in the Catalina-Island mountain mahogany (Cercocarpus traskiae): RAPD evidence. Conserv Biol 9(1):199–203. doi:10.1046/j.1523-1739.1995.09010199.x
Rousselle Y, Thomas M, Galic N, Bonnin I, Goldringer I (2011) Inbreeding depression and low between-population heterosis in recently diverged experimental populations of a selfing species. Heredity 106(2):289–299. doi:10.1038/hdy.2010.72
Routley MB, Mavraganis K, Eckert CG (1999) Effect of population size on the mating system in a self-compatible, autogamous plant, Aquilegia canadensis (Ranunculaceae). Heredity 82:518–528. doi:10.1038/sj.hdy.6885220
Safford HD, Viers JH, Harrison SP (2005) Serpentine endemism in the California flora: a database of serpentine affinity. Madro 52:222–257
Sambatti JBM, Rice KJ (2006) Local adaptation, patterns of selection, and gene flow in the Californian serpentine sunflower (Helianthus exilis). Evolution 60(4):696–710. doi:10.1554/05-479.1
Schaal B (1980) Measurement of gene flow in Lupinus texensis. Nature 284:450–451
Selkoe KA, Toonen RJ (2006) Microsatellites for ecologists: a practical guide to using and evaluating microsatellite markers. Ecol Lett 9(5):615–629. doi:10.1111/j.1461-0248.2006.00889.x
Soltis PS, Gitzendanner MA (1999) Molecular systematics and the conservation of rare species. Conserv Biol 13(3):471–483. doi:10.1046/j.1523-1739.1999.97286.x
Stenoien HK, Fenster CB, Tonteri A, Savolainen O (2005) Genetic variability in natural populations of Arabidopsis thaliana in northern Europe. Mol Ecol 14(1):137–148. doi:10.1111/j.1365-294X.2004.02359.x
Tatarenkov A, Gao H, Mackiewicz M, Taylor DS, Turner BJ, Avise JC (2007) Strong population structure despite evidence of recent migration in a selfing hermaphroditic vertebrate, the mangrove killifish (Kryptolebias marmoratus). Mol Ecol 16(13):2701–2711. doi:10.1111/j.1365-294X.2007.03349.x
Taylor DW (1990) Ecology and life history of the San Benito evening primrose (Camissonia benitensis). Santa Cruz, CA
USFWS (2006) Recovery plan for Camissonia benitensis (San Benito evening-primrose). US Fish and Wildlife Service, Sacramento
Whittall JB, Hellquist CB, Schneider EL, Hodges SA (2004) Cryptic species in an endangered pondweed community (Potamogeton Potamogetonaceae) revealed by AFLP markers. Am J Bot 91(12):2022–2029. doi:10.3732/ajb.91.12.2022
Whittall JB, Carlson ML, Beardsley PM, Meinke RJ, Liston A (2006) The Mimulus moschatus alliance (Phrymaceae): molecular and morphological phylogenetics and their conservation implications. Syst Bot 31(2):380–397. doi:10.1600/036364406777585810
Xu X, Lu BR, Chen YH, Xu M, Rong J, Ye PY, Chen JK, Song ZP (2006) Inferring population history from fine-scale spatial genetic analysis in Oryza rufipogon (Poaceae). Mol Ecol 15(6):1535–1544. doi:10.1111/j.1365-294X.2006.02870.x
Yan J, Chu HJ, Wang HC, Li JQ, Sang T (2009) Population genetic structure of two Medicago species shaped by distinct life form, mating system and seed dispersal. Ann Bot 103(6):825–834. doi:10.1093/aob/mcp006
Acknowledgments
Aaron Thom assisted with tissue collection and Stephanie Saffouri helped with the DNA extractions. This research was supported by a Bureau of Land Management grant awarded to JBW.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Dick, C.A., Herman, J.A., O’Dell, R.E. et al. Cryptic genetic subdivision in the San Benito evening primrose (Camissonia benitensis). Conserv Genet 15, 165–175 (2014). https://doi.org/10.1007/s10592-013-0533-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-013-0533-4