
Abstract
Submersed aquatic macrophyte beds provide important ecosystem services, yet their distribution and extent has declined worldwide in aquatic ecosystems. Effective restoration of these habitats will require, among other factors, reintroduction of genetically diverse source material that can withstand short- and long-term environmental fluctuations in environmental conditions. We examined patterns of genetic diversity in Vallisneria americana because it is a cosmopolitan freshwater submersed aquatic macrophyte and is commonly used for restoring freshwater habitats. We sampled 26 naturally occurring populations of V. americana in the Chesapeake Bay estuary and its tributaries and found that the majority of populations have high genotypic diversity and are not highly inbred. Fourteen of the populations had high allelic and genotypic diversity and could serve as source sites for restoration material. However, substantial geographic structuring of genetic diversity suggests that caution should be used in moving propagules to locations distant from their source. In particular, we suggest that propagules at least be limited within four primary geographic areas that correspond to freshwater tidal and non-tidal, oligohaline, and seasonally mesohaline areas of the Chesapeake Bay.
Similar content being viewed by others
Introduction
Beds of submersed aquatic vegetation (SAV) provide habitat for fish and aquatic invertebrates (Rozas and Odum 1987, 1988; Wyda et al. 2002; Rozas and Minello 2006) and food resources for migratory waterfowl (Krull 1970; Korschgen and Green 1988). SAV also provides critical ecosystem services in that it improves water quality by stabilizing sediments (Sand-Jensen 1998; Madsen et al. 2001) and buffering nutrient levels (Brix and Schierup 1989; Takamura et al. 2003; Moore 2004). Unfortunately, the abundance, distribution, and diversity of SAV beds in coastal aquatic habitats have declined world-wide owing to extensive agricultural, industrial, and urban development in coastal zones (Cooper 1995; Short and Wyllie-Echeverria 1996; Orth et al. 2006; Procaccini et al. 2007). Such is the case in the Chesapeake Bay estuary (Costanza and Greer 1995; Boesch et al. 2001; Kemp et al. 2005), where current SAV coverage is <15% of the 250,000 ha estimated to have existed historically (Stevenson and Confer 1978; Dennison et al. 1993; Orth et al. 2008).
Programs to restore SAV acreage to the Chesapeake Bay and its tributaries have been implemented to mitigate declines. However, these programs have resulted in minimal increases in SAV extent. Poor water and habitat quality at many restoration sites are likely the primary reasons for disappointing results (van Katwijk et al. 2009). Our goal in this paper is to assess the amounts and patterns of genetic diversity in the submersed aquatic plant species Vallisneria americana Michx. (Hydrocharitaceae) to begin to investigate the possibility that genetic factors are contributing to low restoration success rates (Frankel 1974; Frankham 1995a; Hughes et al. 2008). Genetic diversity can affect population persistence in dynamic environments (Lande and Shannon 1996) and the chances for successful establishment of restored populations (Williams 2001). Unfortunately, assessments of this type of diversity often are not directly included in management and restoration plans because it is hard to quantify without sophisticated equipment and substantial expense. Our intent is to provide a description of spatial patterns of genetic variation within and among populations of V. americana that can contribute to the design of restoration efforts.
Amongst SAV species, V. americana has suffered substantial population size declines in the northern freshwater reaches of the Chesapeake Bay and its tributaries (Kemp et al. 1983). V. americana is a cosmopolitan, dioecious, perennial macrophyte that is native to eastern North American freshwater and oligohaline habitats (Korschgen and Green 1988; Catling et al. 1994). The species reproduces sexually and vegetatively (Wilder 1974) and the relative frequency of the two reproductive modes is unknown. Distribution of V. americana is limited to habitats characterized by a maximum water depth of 7 m in clear water, substrates ranging from gravel to hard clay, water temperatures between 20 and 40°C, and salinity below 18ppt (Korschgen and Green 1988). It is further limited by turbidity, nutrient content in the water column, water pH, gas exchange, water current, and competition with other plant species and grazing by animals (Hunt 1963; Barko et al. 1982; Titus and Stephens 1983; Korschgen and Green 1988; Doering et al. 2001; Kemp et al. 2004; Jarvis and Moore 2008).
Full restoration of V. americana within the Chesapeake Bay will depend on linking both physical and biological factors (Allendorf and Luikart 2007). Previous investigations across a wide range of habitats have examined the abiotic growth requirements and ecology of V. americana. These include salinity (Doering et al. 2001; Kreiling et al. 2007; Boustany et al. 2010), light attenuation (Titus and Adams 1979; Korschgen et al. 1997; Kreiling et al. 2007; Boustany et al. 2010), temperature (Titus and Adams 1979), suspended nitrogen (Kreiling et al. 2007), germination requirements (Jarvis and Moore 2008), effects of competition (Titus and Stephens 1983), and sex-ratios and natural fecundity (Doust and Laporte 1991; Titus and Hoover 1991). Here we build on this previous knowledge and quantify the levels and patterns of genetic diversity within and indirect measures of gene flow among naturally occurring sites supporting V. americana in the Chesapeake Bay.
Given the magnitude of decline in V. americana population size and extent in the Bay, we wanted to quantify the levels of genetic diversity and inbreeding overall and within remaining populations (Williams and Davis 1996; Williams 2001; Hufford and Mazer 2003) to know if levels were low enough to cause concern for survival and reproduction (Dudash 1990; Frankham 1995a; Gigord et al. 1998; Saccheri et al. 1998; Westemeier et al. 1998; Reed and Frankham 2003). We also wanted to know what amounts of genetic diversity are available because this diversity can affect probability of persistence of remaining populations, potential for unaided recovery, and selection of source material for propagation and planting. Unfortunately, there is no way to know how much genetic diversity there was prior to population size declines, nor exactly how much is enough to be safe from genetic concerns. We compare current levels of genetic diversity with those in other SAV species to understand if amounts of genetic diversity are substantially lower than expected such that they would cause concern for elevated levels of risk. We also wanted to understand patterns of differentiation because they provide insight into ecological and evolutionary processes that are relevant to restoration. For example, if populations are naturally highly differentiated, moving material among locations could have negative consequences due to outbreeding depression resulting from moving locally adapted individuals to less suitable locations (Montalvo and Ellstrand 2001). On the other hand, if historically high connectivity among populations of V. americana had been reduced or eliminated (Young et al. 1996), effective population size within habitat patches would be reduced, and the rate of inbreeding and genetic drift increased relative to historical conditions (Frankham 1995b, 1996). In this circumstance, knowledge of long-term patterns of gene flow can focus restoration efforts on locations that have potential for reestablishing natural movement among anthropogenically isolated sites. In total, the genetic data we present here provide useful guidance for the restoration community actively working with V. americana in the Chesapeake Bay.
Methods
Sampling localities and protocol
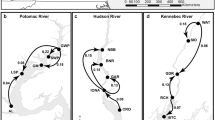
In 2007, 2008, and 2010, we sampled from 26 naturally occurring sites of V. americana present in tidal and non-tidal reaches of Chesapeake Bay tributaries (Table 1) to quantify patterns of allelic and genotypic diversity and historic gene flow. Collection sites were identified with the help of managers and scientists working within the Mid-Atlantic region of the USA. Sampling represented the geographical and ecological extent of the species in the Bay (Fig. 1). Other regions of the Bay are too deep or too saline to support this species. We sampled the Potomac River extensively because plant material from the river has been harvested in the past for use in restoration projects.

Structure results (bottom; colored bars) for Vallisneria americana collection sites (top; colored symbols) visited in 2007, 2008, and 2010. Coloring of bars corresponds to coloring of symbols. When K = 4, collection sites from the upper Potomac, lower Potomac, central Bay, and northern Bay form four distinct groupings. PL was excluded from the analysis due to low genotypic diversity. Sites not shown are CON (near WSP), and CBH/CHC (near DC). Dark blue hashed areas represent general and isolated areas where Vallisneria occurs in the Bay (Moore et al. 2000)
From each site, we collected ~30 shoots, each approximately 5–10 m apart. Samples were often taken blindly as the water was generally too turbid to see shoots, but the distances among samples were kept as consistent as possible given the natural variation in densities at sites. Latitude and longitude coordinates were recorded for each sampled shoot using global positioning systems in all but three sites (CBH, CBC, CON). Shoot tissue was placed on ice and frozen at −80°C until DNA extraction and genotyping.
DNA extraction and genotyping
Genomic DNA was isolated and purified using methods described in (Burnett et al. 2009). We genotyped 11 microsatellite loci representing tri-nucleotide repeats from each sample using robust primers with specific amplification that were developed for the species (Burnett et al. 2009). Polymerase chain reactions (PCR) were performed on an MJ Research PTC-200 Peltier Thermal Cycler using proprietary reagents in the TopTaq DNA Polymerase Kit (QIAGEN). Reaction conditions for all loci followed Burnett et al. (2009) with the exception of the locus Vaam_AAG004, for which we added dimethyl sulfoxide and Q-Solution (QIAGEN) to each reaction for optimal specificity. PCR products were separated and measured on an ABI 3730xl DNA Analyzer with GeneScan™-500 ROX™ or 500 LIZ™ Size Standard (Applied Biosystems) after tagging the PCR product with fluorescent labeled forward primers (Applied Biosystems). Peak data were then analyzed using Genemapper v3.7 (Applied Biosystems) and all allele calls were also visually inspected.
Ambiguity in calls resulting from human or PCR error can result in individuals being misclassified and cascading errors in subsequent analyses. For quality control purposes we reran every ambiguous call up to three times (as necessary). If after three attempts the sample was still ambiguous, the alleles were coded as missing data. In addition, we confirmed genotype calls by re-extracting DNA from 32 samples, rerunning all PCRs and re-genotyping at all loci. These samples were chosen because together they were present across all eight 96 well plates used in the initial fragment analysis. This confirmatory process was completed several months after the initial analysis of the raw data and scoring was done without looking at the initial scores. We detected no allele scoring differences in any of these samples.
Genotypic diversity
We detected clones within and across sites by identifying identical multilocus genotypes using the program GenClone v2.0 (Arnaud-Haond and Belkhir 2007). Because mutation and scoring errors can lead to individuals originating from the same sexual reproductive event having different genotypes we used Genodive v2.0b17 (Meirmans and Van Tienderen 2004) to quantify pairwise differences in alleles among all individuals. Genodive calculates a distance matrix based on the minimum number of mutation steps that are needed to transform the genotype of one individual into the genotype of the other, summed over all loci. Individuals with distances below a threshold in the distance matrix (threshold = 11) were considered to represent the same genet (Rogstad et al. 2002; Meirmans and Van Tienderen 2004). This threshold represents the minimum number of mutation steps that is needed to transform the genotype of one individual into the genotype of another and was chosen because it was it was prior to the point of inflection in the distribution number of clones. Beyond this threshold, genotypes that were different at multiple loci would be identified as one genet, which we considered inappropriate. We compared genets identified using this method with those that would be identified using complete multilocus matches and found 66 individuals differed due to 3–6 base pair mutation at a single locus and 25 individuals were missing data at one locus but matched exactly at all nine other loci. Thus, everything we identified as a clone was also identified when exact multilocus matches were required, but we lumped 91 ramets with another genotype that would be identified as unique if missing data or the mutations were coded separately.
We assessed the probability that shoots with identical genotypes were members of the same clone rather than occurring by chance by using Pgen (Parks and Werth 1993) to estimate the probability of the occurrence of each genotype based on allele frequencies in each population. We then calculated the probability of sampling a second occurrence of each genotype given the number of genets sampled using Psec (Parks and Werth 1993). These calculations were done using the program GenClone. For each site, the proportion of unique genotypes was calculated as (G − 1)/(N − 1), where G is the number of unique genotypes and N is the total number of shoots sampled (Pleasants and Wendel 1989; Arnaud-Haond et al. 2007). For subsequent analyses, each genet within a population was represented by only one shoot (ramet).
The dispersal of vegetative tissues across long distances has been documented in other submersed aquatics (Langeland 1996; Fér and Hroudová 2008), providing the possibility for sharing of V. americana genotypes among sites. To assess the extent of such sharing we pooled all samples, and quantified shared genotypes among sites in Genodive. As with the within-population comparisons, everything we determined to be a clone was an exact multilocus match.
Measures of genetic diversity
For all loci, observed number of alleles (A n), expected (H e) and observed (H o) heterozygosity, proportion of polymorphic loci (P), and private alleles (A p) within each of the 26 collection sites and across all sites combined were calculated using GDA v1.1 (Lewis and Zaykin 2001). To compare allelic diversity among collection sites and regions, we controlled for varying sample size by conducting a rarefaction analysis using the program HP-Rare v1.0 (Kalinowski 2004, 2005); rarefied estimates were not used in other analyses. Shannon’s information index (I) was calculated using PopGene v1.32 (Yeh et al. 1997).
Wright’s F is was calculated for the global dataset using the estimator f (Weir and Cockerham 1984) in GDA to test for site-level deviations from Hardy–Weinberg equilibrium. Significance of F is was tested by obtaining confidence limits around each estimate generated by 1000 bootstraps in GDA. Significant departures from Hardy–Weinberg equilibrium can indicate a departure from random breeding.
We examined each site that had more than two genotypes for presence of a recent genetic bottleneck using a test for heterozygote excess in the program Bottleneck v 1.2.02 (Cornuet and Luikart 1996). Bottleneck computes heterozygote excess as the difference between expected heterozygosity (H e) and heterozygosity expected at equilibrium (H eq) for each site from the number of alleles given the sample size (Cornuet and Luikart 1996). Significance of the difference between H e and H eq was tested using a one-tailed Wilcoxon’s sign rank test under a two-phase mutation model which provides results intermediate between an infinite allele model and a stepwise mutation model that are considered to be most appropriate for microsatellites (Di Rienzo et al. 1994).
Population differentiation
We assessed patterns of genetic differentiation in three complementary ways. First we used the program Structurama v1.0 (Huelsenbeck and Andolfatto 2007) to identify theoretical a posteriori ‘populations’ from our collection of sites based on minimal deviations from both Hardy–Weinberg and linkage equilibrium as in Pritchard et al. (2000). Structurama differs from the program Structure (Pritchard et al. 2000) in that the number of theoretical populations is included as a parameter in the model and a posterior distribution of the probabilities of each number is generated. Prior number of populations and expected number of populations were set as random variables. The sampler was run for 1,000,000 generations and sampled every 25 generations for a total of 40,000 samples. Four heated chains (temperature = 0.1) were used in the analysis. Data were summarized after discarding 10,000 burn-in samples. We chose the mean partition value as the number of theoretical populations (K) containing the highest posterior probability. Because Structurama lacks clearly interpretable visualization of individual assignments we used Structure v2.3.2 (Pritchard et al. 2000) to assess distinctiveness of theoretical populations (Berryman 2002) by assigning individuals to the number of populations inferred by Structurama. Structure was run assuming prior admixture, with 1,000,000 steps in the Bayesian sampler, using a burn-in of 50,000 steps. The analysis was run 10 times, and the best run was selected based on the highest likelihood score.
To provide a general overview of site-level differentiation, we calculated global and pairwise estimates of Wright’s F st, using Weir and Cockerham’s (1984) estimate θ as calculated in GDA. Significance was assessed by generating confidence limits derived from 1000 bootstrap samples. All θ values were normalized to account for the theoretical maximum value and thus allow for future comparison across studies (Hedrick 2005; Meirmans 2006) using the program Genodive (Meirmans and Van Tienderen 2004). There is no significance test for these normalized values (Meirmans 2006). To account for potential limitations of F st in quantifying differentiation (Hedrick 2005; Jost 2008), we also calculated pairwise and global values of Jost’s (2008) measure of genetic differentiation, D, using Chao et al.’s (2008) estimate D est_Chao in SMOGD v 1.2.5 (Crawford 2009). Significance was assessed by generating confidence limits derived from 1000 bootstrap samples in SMOGD.
We tested for relationships between linearized pairwise F st (F st /(1 − F st ) (Slatkin 1995) among sites and two different geographic distances using a Mantel test as implemented by the program IBDWS v3.16 (Jensen et al. 2005). Significance was assessed using 1,000 randomizations in IBDWS. We used pairwise Euclidean geographic distances calculated from the GPS coordinates collected in the field, and the shortest distance over water among paired sites using Pathmatrix v1.1 (Ray 2005). Euclidian distance is potentially realistic for seed dispersal by waterfowl that can fly over land whereas the weighted geographic distances are more realistic for water-dispersed pollen.
We used principal components analysis (PCA) on the variance–covariance matrix of allele frequencies, using Genodive, to understand the distribution of variance among sampled locations that is a function of variation in allelic composition. PCA provides a different perspective from the Structurama/Structure analyses because it represents the relative degree of genetic similarity among sites in a continuous rather than categorical framework.
Estimates of gene flow among populations
Because coalescent-based methods can provide more accurate and powerful estimates of migration than classical frequentist estimates (Rosenberg and Nordborg 2002; Holsinger and Weir 2009), we quantified migration among population groupings using Migrate-n v3.2.6 (Beerli and Felsenstein 1999, 2001; Beerli 2006). Migrate-n employs a likelihood method of parameter estimation utilizing coalescent theory to estimate asymmetric migration among populations under an equilibrium model that assumes migration has been constant over time (Beerli and Felsenstein 1999). Estimating migration among all sites would require estimating 462 parameters. To estimate a reasonable number of parameters given our data, we limited migration to four groupings based on results from the Structurama/Structure analyses and geographic proximity of sites. The HL locality was difficult to assign to a group in Structure (Fig. 1) due to assignment probabilities being split between groupings and geographic distance from other sites; it therefore was excluded from this analysis.
Migrate-n was run with the following parameters. Data were treated under a Brownian motion mutational model where mutation rate was calculated as a random variable from the data and missing alleles were discarded. The Bayesian sampler started from a random genealogy with a full migration model, where both migration rate (M) and population size (θ) were free to vary. The sampler utilized uniform priors for both M and θ. To reduce the size of the tree-space explored by the samples, the priors were constrained based on exploratory analyses between 0 and 4.5 with delta = 0.01 for θ, and 0–150 with delta = 30 for M each with 500 bins. Four parallel chains with a swap interval of 1.0 were run with heating values of 10, 7, 4, and 1. One long chain of 80,000 recorded steps was sampled every 20 steps, for a total of 1,600,000 sampled parameters values. Subsequent posterior distributions were summarized after a burn-in of 10,000 steps. The burnin value was selected following examination of exploratory data analyses. Convergence of the run was assessed using effective sample size calculated in migrate-n.
The number of immigrants per generation (Nm) was estimated as 4Nm j = M ij × θj, where θ j is the effective population size of the recipient population and M ij is the migration rate from population i to population j.
Results
Genetic diversity
We sampled a total of 675 shoots, representing 427 unique genotypes. Within each of 26 locations, we sampled an average of 26.0 shoots (Table 1). A median of 68% of sampled shoots within sites represented unique genets, but the proportion of shoots representing multiple genets varied from 0.00 to 1.000 (Table 1). Eight of nine sites upstream from and including PL in the Potomac River and site HL in the Mattaponi River were particularly low in genotypic diversity, with genotypic diversity ranging between 0 and 0.38 of sampled shoots being unique genets (Table 1). Site PL was the most extreme, with all 30 samples representing a single genotype. Two exceptions to the trend of low genotypic diversity upstream of PL in the Potomac River were WF and WSP that had clonal diversity values of 0.58 and 0.76, respectively.
Five genotypes were shared among sites within the upper Potomac River (Table 2). Two of these genotypes dominated multiple sites, often comprising 53–100% of sampled shoots. Those two genotypes spanned large geographic distances; one genotype covered approximately 160 river km and the other was present across 132 river km. We found no genotypes shared among other sites within the Chesapeake Bay.
The probability of recovering any given genotype by chance ranged from 5.63 × 10−16 to 5.75 × 10−7 (SD = 3.97 × 10−8). The probability of finding a second occurrence of each genotype, given the number of genets sampled, ranged from 2.37 × 10−13 to 2.45 × 10−4 (SD = 1.70 × 10−5). The genotypes that spanned large geographic distances in the Potomac River ranged in the probability of occurrence from 6.5 × 10−11 to 1.5 × 10−7 and in the probability of re-sampling one of those genotypes from 2.75 × 10−8 to 6.57 × 10−5 (Table 2). Thus we consider these identical genotypes to be clones that resulted from the same sexual reproduction even.
Many loci showed departure from Hardy–Weinberg equilibrium; however, the degree of deviation was often minimal (Table 3). The locus AAGX013 showed significant departure from HWE, and also had a large amount of missing data (31.92%); therefore, it was excluded from subsequent analyses. The amount of missing data in the remaining 10 loci was negligible, averaging 0.84% and ranging from 0.23 to 2.35%.
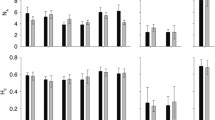
The proportion of polymorphic loci within sites was 0.854 (SD = 0.139). The average number of alleles per locus across all sites combined was 8.70 (SD = 4.08) and within sites was 3.91 (SD = 1.40). When we standardized by number of genets, the number of alleles among sites was similar indicating that genotypic diversity largely controlled allelic diversity. Between one and five private alleles were found in nine populations. Seven of the sites with private alleles were in the main stem of the Chesapeake Bay (Table 1). Sites with private alleles were also relatively high in genotypic diversity (>18 genets). None of the sites with low genotypic diversity in the Potomac River had private alleles.
Observed heterozygosity was high at all sites (avg H o = 0.535; SD = 0.086). Nine sites departed significantly from Hardy–Weinberg equilibrium (Table 1); six sites had more heterozygotes than expected (EN, Tour1, Tour2, CON, WF, and HL) and three had fewer heterozygotes (GWP, AL, LSP; Table 1). Shannon’s information index was similar among all sites except the HL site, and those sampled in the Potomac River above Great Falls, MD (Table 1).
Based on analysis with the program Bottleneck (Cornuet and Luikart 1996), 3 of the 24 sites we could analyze (MP, SCN, and POR) showed evidence that H e significantly exceeds H eq, which suggests that they have undergone recent genetic bottlenecks (Table 1). Of the sites in the lower Potomac with significant F is, two of these sites supported only two genotypes and thus did not have the minimum number of samples to run Bottleneck; the third only met the minimum requirement of three genotypes. Lack of a significant bottleneck for this site could easily have been due to the small sample size.
Population differentiation
Bayesian clustering analysis as implemented by Structurama indicated that there are four genetic subdivisions in the 26 sampled locations of V. americana in the Chesapeake Bay (Pr[K = 4|X] = 0.9993). When Structure was run assuming K = 4 to visualize individual clusters three primary divisions were noted: northern Bay localities, central Bay localities, and Potomac River localities (Fig. 1). A further subdivision between the upper and lower Potomac River was identified. Mixed population assignments of individuals provide evidence of similarity among all members of the upper Potomac and several lower Potomac sites (GWP, SWP, GM). The sites LSP and AL had low probability of assignment into the upper Potomac localities (Fig. 1). The Potomac River sites also have a very small degree of admixture with the central Bay sites, which is most evident in LSP (Fig. 1). Site HL from the Mattaponi River was difficult to assign, with assignment probabilities being split between the Potomac group and the central Bay group.
Overall, we observed moderate levels of global genetic differentiation among all sites combined (θ = 0.114, 95% CI = 0.081–0.152). The PL location was excluded from these analyses because it is not possible to calculate F st or D for a site with only one sample. Within regions identified in Structure, the median pairwise values of θ among sites ranged from ~0.020 in the upper and central Bay, to 0.043 among sites in the lower Potomac, to 0.10 in the upper Potomac. The median pairwise θ value of sites from different regions was 0.114 and the range was from 0.013 to 0.32. Thus, the pairwise differences among sites from the upper Potomac (range was from −0.02 to 0.31) were similar to differences among other sites from different regions. The global D est_Chao (0.124, 95% CI = 0.008–0.352) was slightly higher than θ. The median pairwise D est_Chao among regions was 0.07. Within region median values of D est_Chao were lower than those observed with θ (northern Bay = 0.02; central Bay = 0.01; upper Potomac = 0.01; lower Potomac = 0.009), and indicate that differentiation within regions was substantially lower than among regions.
There were significant relationships between genetic distance and both straight-line (r = 0.39; P < 0.001) and weighted (r = 0.59; P < 0.001) distances (Fig. 2) for all sites combined. Relationships with both geographic distances were also significant in the upper (straight-line: r = 0.41; P < 0.001; weighted: r = 0.47; P < 0.001) and lower Potomac River (straight-line: r = 0.69; P < 0.001; weighted: r = 0.93; P < 0.001) groups. In the northern Chesapeake Bay, neither measure of geographic distance provided a significant correlation. The central Chesapeake Bay tended to have larger genetic distances among sites relative to the northern Chesapeake Bay (distance table not shown); however, the correlation was not significant for either distance measure.

Linearized F st (F st /(1 − F st ) (Slatkin 1995) genetic distance regressed against a Euclidean geographic distance and b the shortest distance over water among collection sites (weighted geographic distance)
The PCA on the variance–covariance matrix of allele frequencies showed that allelic composition was generally more similar within than among the four geographic regions within the Chesapeake Bay identified in the Structure analysis (Fig. 3). The first axis explained 27.58% of the variance in allele frequencies and captured differences among the regions. The second axis explained 18.65% of the variance and was driven primarily by two sites with extremely low genotypic diversity (G = 2 in CON and G = 1 in PL). Both populations were distinct due to chance fixation of some alleles and the fact that given small number of genets present in each site, allele frequencies are by necessity limited to a small range of values, and those values happened to be higher than those in other populations. The alleles that were fixed in these sites were also present in other sites but the resulting large differences in allele frequency placed CON and PL away from all other sites, and compressed the remaining sites into a small portion of Axis 2 (Fig. 3).

Principal components analysis of the covariance matrix of allele frequencies. Axis 1: Eigenvalue = 0.29, percent of variation explained = 27.58; and axis 2: Eigen value = 0.19, percent of variation explained = 18.65. Symbols represent the four genetic regions within the Chesapeake Bay
Migration
Effective sample size, a measure of convergence, exceeded 1000 samples for all parameters. The number of migrants per generation (4Nm) among the four groups identified using Structure and geographic proximity varied from 7.69 to 29.91 (Fig. 4). The upper Potomac River population grouping was largely isolated from all other populations. The lower Potomac River population grouping had apparent migrant exchange with both the northern and central population groupings with relatively equal frequency (4Nm = 25.41–29.91). The northern Chesapeake Bay received nearly the same number of migrants from (4Nm = 28.14; CI = 23.21–32.96) as it contributed to (4Nm = 21.29; CI = 17.06–26.24; Fig. 4) the central Chesapeake Bay. In contrast, the upper Potomac River appeared to share more migrants with the lower Potomac (4Nm = 17.39; CI = 12.44–21.62) than the lower Potomac shared with the upper Potomac (4Nm = 9.91; CI = 7.67–13.61), but the confidence intervals in these estimates overlapped to a small degree.

Per generation bidirectional migration rates (4Nm) among the four population grouping recovered from analysis in Migrate-n
Discussion
Overall, most sites of V. americana in the Chesapeake Bay support a diversity of genotypes and alleles, and most are not highly inbred. This is good news for the future of the species in the Bay because high genetic diversity increases a population’s capacity to persist under variable environmental conditions (Frankham 1995a; Procaccini and Piazzi 2001; Williams 2001; Reed and Frankham 2003) and to adapt to novel conditions (Frankham 2005; Lavergne and Molofsky 2007; Barrett and Schluter 2008). The genotypically diverse sites can also serve as sources of material for restoring V. americana to currently unoccupied sites. The geographic structuring of genetic diversity we documented is important to consider if movement of propagules around the Bay is proposed. Despite the relatively positive general outlook, evidence for recent bottlenecks in three sites, signs of inbreeding at three sites, and low genotypic diversity in the upper Potomac River raise concern for long-term effects of the previous population declines.
Genetic diversity
Species level allelic richness in the Chesapeake Bay and its tributaries was on par with what has been found in other SAV species from throughout the world, which ranges from 2 to 18 alleles per locus (Reusch et al. 1999b, 2000; Rhode and Duffy 2004; Pollux et al. 2007; van Dijk et al. 2009; Campanella et al. 2010). Our site-level allele richness was also mostly within the typical ranges of values found in these same studies of other SAV species (2.3–10.5 alleles per locus). The three exceptions that had particularly low allelic richness (1.5–1.7 alleles/locus) supported only 1 or 2 unique genotypes each (Table 1). Beyond these extreme cases, lower allelic diversity was associated with lower genotypic diversity, typically with <30% of sampled shoots in low allelic diversity sites being unique genets.
Evidence of recent bottlenecks based on heterozyote excess in three sites (MP, SCN, and POR) and the significant inbreeding coefficients in three sites in the lower Potomac River (GWP, LSP, AL; Table 1) cause some concern. However, widespread inbreeding was not observed despite low levels of genotypic diversity (and therefore effective population size). The dioecious mating system of V. americana enforces outcrossing and may explain why inbreeding was not more prevalent. Determining the full implications of apparent bottlenecks and inbreeding requires understanding their fitness consequences, which is beyond the scope of this study.
One of our more striking results is that genotypic diversity ranged from 0 to 1.0, meaning that sites ranged from being monoclonal to every sampled shoot being distinct. It also means sites range from having no detectable sexual reproduction to no detectable asexual reproduction. Such variation in mating structure across this same spatial scale is not common in aquatic species but has been documented in Typha minima Hoppe (Till-Bottraud et al. 2010) and in Posidonia oceanica Delile (0.1–0.97; Arnaud-Haond et al. 2010). The general paradigm that Vallisneria populations are maintained primarily by vegetative reproduction (e.g., McFarland and Shafer 2008) is not supported by our data.
The sites with low genotypic diversity relative to other V. americana locations in the Bay are those in the upper Potomac River, site HL in the Mattaponi River, and sites SCN and SFP in the central Chesapeake Bay. Variation in levels of genotypic diversity among sites is interesting because of the advantages typically associated with high genotypic diversity and for the insights into the potential mechanisms that might have caused these sites to have fewer, more extensive clones than other sites in the Bay. Higher genotypic diversity has been correlated with increased resistance to periodic stressors and more resilience after climatic extremes in experimental settings (Hughes and Stachowicz 2004; Reusch et al. 2005; Hughes and Stachowicz 2009) and with increased survival of transplants (Procaccini and Piazzi 2001). Thus, although sites in the upper Potomac River support extensive cover, the few highly successful genotypes may not provide the genetic variation necessary to withstand novel perturbations or adapt to future conditions. It is important to note that the effect of genotypic diversity on the stability of SAV beds is still unclear. At least some field observations indicated higher mortality in more genetically diverse populations of P. oceanica (Arnaud-Haond et al. 2010). Further, sedimentation rate was a stronger predictor of shoot mortality in P. oceanica than were genetic diversity or even demographic parameters (Arnaud-Haond et al. 2010).
Clearly, at extreme levels of disturbance that exceed physiological tolerances, no amount of genetic diversity will be sufficient to withstand or overcome perturbations, and environmental factors become more important. Short of such extremes, it is plausible that a limited number of genotypes will be sufficiently resistant to survive perturbations, which would result in less genotypically diverse populations in high disturbance sites. Conversely, low genotypic diversity in more stable sites has been explained as resulting from one genotype becoming dominant. Periodic or fluctuating disturbance could foster more genotypic diversity if survival and fitness of genotypes differed across conditions (Hammerli and Reusch 2003). The patterns observed in any particular case will depend on the magnitude and frequency of disturbance and the interaction between that disturbance and genotypic or phenotypic abilities to withstand it. Without monitoring over time, it is not possible to know if low genotypic diversity is a signature of past environmental perturbations that have left only tolerant genotypes or the result of stochastic losses.
In addition to having low genotypic diversity, multiple sites along the upper Potomac River shared the same genotype (Table 2). The geographic extent of the five shared genotypes is remarkable: two of them extended a distance of 130 and 160 river km, and the remaining three genotypes covered distances of 50 river km. The probability of recovering the specific genotypes by chance if they were not identical by descent given global allele frequencies is astronomically small 10−7 to 10−11 (Parks and Werth 1993), and the probability of finding a second occurrence of each genotype, given the number of genets sampled, is 10−5 to 10−8 (Parks and Werth 1993). A typical mutation rate of microsatellite loci (~10−3 to 10−4 per allele per generation; Thuillet et al. 2002; Vigouroux et al. 2002) does provide the possibility that these genotypes are merely identical in state (Mank and Avise 2003); however, it is highly unlikely that mutation events simultaneously produced identical individuals across such a large geographic range. Although a large proportion of studied angiosperm species exhibit clonality that extends across more than one location (Ellstrand and Roose 1987), extremely large clonal extent is rare. Examples of the larger known clonal extents include a single Populus tremuloides Michx. clone that covers an area of roughly 43 ha (Mitton and Grant 1996), and several submersed aquatic species that are known to have clones that extend >5 km (Reusch et al. 1999a; Ruggiero et al. 2002). Most studies of other SAV species indicate that clones are primarily limited to within individual sites (Titus and Hoover 1991; Campanella et al. 2010) with extents typically limited to the scale of ~18 m (Becheler et al. 2010), to 78 m (Arnaud-Haond et al. 2010), to ~250 m (Zipperle et al. 2009).
Vegetative expansion of V. americana through rhizomes is generally limited to within a few meters of the parent plant (Titus and Hoover 1991). Maximum seasonal lateral growth of V. americana from the upper Potomac River genotypes is 60 cm under greenhouse conditions (Engelhardt, unpublished data). At this ideal growth rate it would take roughly 260,000 years to grow 130–160 km, and even supposing growth occurred from a central location outward, it would take 130,000 years to traverse that distance. It is unlikely that habitat necessary to allow this vegetative growth would have been sufficiently continuous and stable throughout the stretch of the river for such a long period of time. Thus, although lateral vegetative growth within sites could potentially lead to local dominance by one or a few genotypes, it is highly improbable that lateral growth alone is responsible for genotypes extending 50–160 km along the Potomac River.
The question, then, is how did these few genotypes come to extend and dominate over such large areas? Specific mechanisms could include passive stochastic loss and colonization, deterministic processes based on competitive ability, selective advantages due to environmental tolerance of particular genotypes, or a combination of passive and deterministic processes. Passive processes could include initial chance colonization by few genotypes that expanded in place, or stochastic loss of genotypes within sites followed by repeated recolonization by a small number of genotypes. More deterministic processes include selection in response to abiotic factors or competition. If particular genotypes were resistant to abiotic stressors, they would become dominant as other genotypes were eliminated. Dominance by a few clones could also result if downstream sites were colonized by a small number of competitively superior vegetative propagules from upstream populations, widespread dominance of a limited number of genotypes would result. We offer these mechanisms as possible explanations; our current data are not sufficient to infer mechanism but are more consistent with some possibilities than others, and clearly point to the need for further experiments.
Tubers of V. americana are generally negatively buoyant, but they can become positively buoyant if attached to shoot fragments (Titus and Hoover 1991). The extensive clones we observed in the Upper Potomac River could have originated from dislodged shoots and tubers that were carried downstream in floods (Fér and Hroudová 2008). Flooding events sufficiently extreme to cause scouring are common in the Potomac River and removal of individuals from suitable habitat would create opportunities for expansion of chance colonists. It is likely that upstream populations have either had low diversity due to founder events, or that diversity has been lost from small, isolated sites. Once upstream populations have low genotypic diversity, opportunities to gain new diversity would be limited due to unidirectional water flow from headwaters to mouth. Large distances from other major bodies of water yield small chances of recolonization from sources other than nearby low diversity sites (Chen et al. 2007). The process could generate a positive feedback loop in that as particular genotypes become more dominant, they become more likely to be source material for additional colonizations. An additional consequence of low genotypic diversity that may in turn facilitate dominance of a few genotypes is the reduced probability of having both males and females, which limits sexual reproduction. Existing clones could have higher potential to spread and occupy larger areas than they might in populations that also had sexually produced propagules. We have no quantitative data on sex ratios but we have observed fertile fruits at all sites, indicating some sexual reproduction is occurring. However, for the same level of search effort, we found substantially fewer fruits at many of the upper Potomac River sites than we found in other locations throughout the Bay.
Another explanation that we considered to possibly explain widespread dominance was the introduction of competitively superior genotypes into the Potomac River via restoration or other activities, or through natural mechanisms such as ingestion and dispersal of tubers via waterfowl. We know of no restoration activities within any of these regions. Additionally, many of the sites visited were not easily accessible, which would hinder the inadvertent introduction by humans through recreational activities such as boating or through activities such as dumping of aquaria.
It is most likely that the unprecedented size of the large V. americana clones in the Potomac River has resulted from a combination of local spread via rhizomes and repeated longer distance dispersal of tubers during storm events. Clearly, much still needs to be learned regarding dispersal of vegetative propagules from parent populations (Titus and Hoover 1991). Regardless of the mechanisms, lower genotypic and allelic diversity in the upper Potomac River sites compared to other localities in the Bay suggests that they should be considered cautiously as source material for restoration plantings. Sampling shoots from even widespread locations is highly likely to yield the same genotype. If the upper Potomac River were used as a source for restoration, using seed rather than vegetative material would improve chances of representing more genetic diversity and of including both male and females in restoration plantings.
Genetic differentiation and migration
The overall patterns of genetic differentiation among sites in the Bay related strongly to geographic distance (both straight line and weighted and is indicative of equilibrium between genetic drift and gene flow (Hutchison and Templeton 1999). Beyond coarse geographic trends, Structure analysis indicated the Chesapeake Bay can be broken into four genetic regions. These subdivisions roughly correspond to regions of differing salinity. The northern Chesapeake Bay is oligohaline and the central Chesapeake Bay is oligohaline to seasonally mesohaline (Pritchard 1952). Sites in the lower Potomac River are oligohaline and are strongly tidally influenced while the upper Potomac River is entirely freshwater. Such environmental differences can increase isolation among populations (Keeley 1979; Stanton et al. 1997; Doebeli and Dieckmann 2003), influence patterns of occurrence and hybridization (Crain et al. 2004; Blum et al. 2010), and drive adaptation to local conditions (Clausen et al. 1941; Antonovics and Bradshaw 1970; Linhart and Grant 1996; Antonovics 2006).
The admixture among the regions implies at least historic gene flow among sites, and results from the full Migrate-n analysis show evidence of some exchange between the two regions within the Potomac River (Fig. 4). Even with this admixture, the level of substructuring we detected is surprising given the potential for the Bay to represent one large, hydrologically connected unit (e.g., van Dijk et al. 2009). The degree of substructuring is greater than has been found in other studies at similar scales (Campanella et al. 2010).
The level of differentiation we observed among sites within each region is similar to levels documented from hydrologically connected populations of several Vallisneria species (G st = 0.02–0.06; Lokker et al. 1994; Chen et al. 2007) and other seagrass populations sampled from similar spatial scales (Campanella et al. 2010). When sites are pooled, the degree of genetic differentiation between the north and central Chesapeake Bay (D est_Chao = 0.060) is at the upper range of the levels documented among connected sites. Levels of differentiation among sample sites in different regions are more similar to those found in isolated water bodies: F st = 0.132–0.202 and G st = 0.457 (Laushman 1993; Wang et al. 2010). Interestingly, the amount of gene flow between the north and central localities estimated by Migrate-n is theoretically enough (4Nm = 21.29–28.14) to swamp out genetic differentiation among populations. If successful migration among populations is sufficiently common (e.g., >1 migrant per generation), genetic subdivision is not likely to occur (Wright 1931; Slatkin 1981, 1985, 1987). Several factors could be influencing the observed patterns of gene flow among the populations. Coalescent-based analyses integrate estimates of migration and effective population size over 4Ne generations (Kingman 1982a, b). A disconnect between current patterns of genetic differentiation and the amount of historic gene flow among populations could exist (Sork et al. 1999). In addition, genetic differentiation can occur in presence of substantial gene flow (Morrell et al. 2003). In cases where extreme environmental heterogeneity exists among sites, reproductive isolation can develop and be sustained even in the face of genetic exchange among populations (Caisse and Antonovics 1978; Antonovics 2006).
We interpret the inferred regions cautiously because sampling from a continuous population with local mating structure can yield ‘populations’ using the program Structure (Schwartz and McKelvey 2008). However, most sites we sampled in the northern and central Bay were from discrete beds that are isolated from other beds by depth and salinity beyond the limits of tolerance for Vallisneria. Thus, although they would have been more extensive historically, it is not likely that many of the now isolated beds would ever have been continuous. In contrast, the upper Potomac River is probably best considered one extensive relatively continuous population with a combination of extensive vegetative dispersal and of sexual reproduction among spatially proximal individuals. Within the upper Potomac, F st and Jost’s D values (Table 1) reflect local mating structure while the extensive distribution of some genotypes (Table 2) indicate connectivity over large distances that is not reflected in other statistics calculated including only one representative of each genotype. There are no extensive natural physical barriers along this part of the river, and there is no abrupt environmental change. There are several small dams that cause 1–2 km breaks in the distribution of Vallisneria by increasing sediment deposition immediately upstream and causing extensive scouring immediately below. In contrast, differences in F st and Jost’s D between the upper and lower Potomac are more similar to those in between other regions, and no genotypes are shared. The major environmental difference between two parts of the river is the tidal influence in the lower reaches of the river that is absent above Great Falls, MD. More intensive sampling between our existing sampling locations is needed to elucidate finer scale patterns of population structure, clonal diversity, and clonal extent, which are necessary to understand spatial mating and dispersal structure.
Implications for restoration
Goals for ‘restoration’ can range from simply returning vegetation to a site, to full-scale ecological restoration. Ecological restoration is defined as, “an intentional activity that initiates or accelerates the recovery of an ecosystem with respect to its health, integrity and sustainability” (Society for Ecological Restoration International Science and Policy Working Group 2004). This definition requires, the restored ecosystem to be self-sustaining and be sufficiently resilient to endure the normal periodic stress events in the local environment. (http://www.ser.org/content/ecological_restoration_primer.asp#5). There are three main paradigms for selecting material for revegetation efforts.
1. Select a few particularly well performing genotypes for a particular set of criteria and propagate those genotypes in a manner similar to development of cultivars in agriculture and horticulture. This approach lends itself to efficient commercial production of source material and development of material with resistance to known pests or pathogens or with characteristics that meet specific needs. Planting one or a few genotypes over broad areas may be successful in the short-term but provides no raw material for evolution to changing abiotic conditions or novel pathogens. Although it is sometime applied in revegetation project, it is generally not considered acceptable in ecological restoration.
2. Select propagules such that amounts and types of genetic diversity in restored populations reflect those found in surrounding natural populations. This approach recognizes the importance of local adaptation and uses local genetic stock. A major goal is to prevent founder events in the restoration process that can occur during collection, cultivation or planting so that future evolutionary potential is maintained. At the same time, propagule sources can be selected based on spatial proximity or habitat similarity (van Katwijk et al. 2009) between the source and reference site that are deemed to be sufficiently local. This approach can be problematic if individual sites are genetically depauperate and or inbred, but prevents planting maladapted stock or causing genetic pollution of local populations (McKay et al. 2005). However, the presence of local adaptation is not documented for most species and the spatial scale at which such adaptations may occur is likely to be idiosyncratic. Unnecessarily restricting source material for widespread species with little or no local adaptation can severely hamper restoration efforts (Broadhurst et al. 2008).
3. Use large numbers of propagules of diverse origin, letting natural selection sort out appropriate genotypes for a particular site (Broadhurst et al. 2008). This approach is suggested for relatively common, widespread species that have long-distance dispersal abilities but that are now fragmented and in which individual remnants do not support much remaining diversity or in which inbreeding depression may be causing reduced fitness. Such an approach is also suggested for large-scale regional restoration efforts in which sufficient propagules may not exist within small isolated fragments. Advocates of this approach suggest that the genetic diversity of the source material is as important as or more important than being ‘local.’ Inappropriate use of genetic stocks in environments to which they are not adapted can substantially impact the success of restored populations (Montalvo et al. 1997; Hufford and Mazer 2003). Restoration failure may result when the foreign genetic stock provisions resources at inappropriate times (Jones et al. 2001), is maladapted to local conditions (McKay et al. 2005), or contributes to outbreeding depression (Templeton 1997; Montalvo and Ellstrand 2001; Potts et al. 2003).
Although they provide insight into only the one aspect of genetic diversity, our results inform aspects of each of these potential approaches. We found that levels of genotypic and allelic diversity at most sites are high and can serve as source populations for restoration material. Exceptions include upper Potomac River sites (e.g., HCK, POR, WF), and two sites in the central Bay (SCN, SFP). Low diversity in sites and presence of shared genotypes among sites in the upper Potomac River also cautions against the use of that region for source material without prior thought and understanding of the potential implications of low diversity collections. On the other hand, the widespread genotypes in the low diversity sites could be candidates for intensive propagation if their dominance was shown to relate to superior competitive ability that confers resistance to environmental stressors affecting the Potomac River. We do not advocate approaches that reduce genetic diversity, but as part of a comprehensive restoration program, having genotypes that can withstand and even flourish under stressful conditions could be beneficial. Our current data only provide a starting point for investigation of such possibilities.
Based on the diversity we observed, we found no compelling evidence for the need for genetic rescue of any population through introduction of genotypes or the need to mix genotypes in restoration plantings (Hedrick and Fredrickson 2010). We have no way of knowing the original levels of genetic diversity in the Bay, but, despite extensive population size declines, there is no evidence of catastrophic losses in that most remaining sites are not genetically depauperate or homogeneous. Confirmation of this assertion requires comparing fitness in apparently bottlenecked populations with populations that have no indication of severe reduction.
The spatial substructuring we detected among sites in the northern and central Bay suggests that caution should be used in moving propagules to locations distant from their source. It is also necessary to more thoroughly understand the population structure within the Potomac River to determine the scales at which there is genetic interaction from dispersal of vegetative propagules, pollen, and seed. Specifically, we suggest that movement of propagules for restoration activities be limited to within each of the four primary geographic areas that are related to environmental factors, in particular salinity. We find no strong evidence against moving propagules within regions. Our data do not allow us to assess the degree to which the genetic differences we detected indicate adaptation to local environmental conditions. We are just beginning to conduct experiments to determine whether there is evidence for local adaptation within these regions and if there are fitness consequences of crossing individuals from different regions. Until more investigations relating these patterns with fitness are completed, it is prudent to be cautious and carefully select plant material from within one of the genetic regions.
References
Allendorf FW, Luikart G (2007) Conservation and the genetics of populations. Blackwell Publishing, Malden
Antonovics J (2006) Evolution in closely adjacent plant populations X: long-term persistence of prereproductive isolation at a mine boundary. Heredity 97:33–37
Antonovics J, Bradshaw AD (1970) Evolution in closely adjacent plant populations. VIII. Clinal patterns at a mine boundary. Heredity 25:349–362
Arnaud-Haond S, Belkhir K (2007) GENCLONE: a computer program to analyse genotypic data, test for clonality and describe spatial clonal organization. Mol Ecol Notes 7:15–17
Arnaud-Haond S, Duarte CM, Alberto F, Serrao EA (2007) Standardizing methods to address clonality in population studies. Mol Ecol 16:5115–5139
Arnaud-Haond S, Marba N, Diaz-Almela E, Serrao EA, Duarte CM (2010) Comparative analysis of stability-genetic diversity in seagrass (Posidonia oceanica) meadows yields unexpected results. Estuar Coast 33:878–889
Barko J, Hardin D, Matthews M (1982) Growth and morphology of submersed freshwater macrophytes in relation to light and temperature. Can J Bot 60:877–887
Barrett RDH, Schluter D (2008) Adaptation from standing genetic variation. Trends Ecol Evol 23:38–44
Becheler R, Diekmann O, Hily C, Moalic Y, Arnaud-Haond S (2010) The concept of population in clonal organisms: mosaics of temporally colonized patches are forming highly diverse meadows of Zostera marina in Brittany. Mol Ecol 19:2394–2407
Beerli P (2006) Comparison of Bayesian and maximum-likelihood inference of population genetic parameters. Bioinformatics 22:341–345
Beerli P, Felsenstein J (1999) Maximum-likelihood estimation of migration rates and effective population numbers in two populations using a coalescent approach. Genetics 152:763–773
Beerli P, Felsenstein J (2001) Maximum likelihood estimation of a migration matrix and effective population sizes in n subpopulations by using a coalescent approach. Proc Natl Acad Sci USA 98:4563–4568
Berryman AA (2002) Population: a central concept for ecology? Oikos 97:439–442
Blum MJ, Knapke E, McLachlan JS, Snider SB, Saunders CJ (2010) Hybridization between Schoenoplectus sedges across Chesapeake Bay marshes. Conserv Genet 11:1885–1898
Boesch DF, Brinsfield RB, Magnien RE (2001) Chesapeake Bay eutrophication: scientific understanding, ecosystem restoration, and challenges for agriculture. J Environ Qual 30:303–320
Boustany RG, Thomas CM, Rebecca FM (2010) Effects of salinity and light on biomass and growth of Vallisneria americana from Lower St. Johns River, FL, USA. Wetl Ecol Manag 18:203–217
Brix H, Schierup HH (1989) The use of aquatic macrophytes in water-pollution control. Ambio 18:100–107
Broadhurst LM, Lowe A, Coates DJ, Cunningham SA, McDonald M, Vesk PA, Yates C (2008) Seed supply for broadscale restoration: maximizing evolutionary potential. Evol Appl 1:587–597
Burnett RK, Lloyd MW, Engelhardt KAM, Neel MC (2009) Development of 11 polymorphic microsatellite markers in a macrophyte of conservation concern, Vallisneria americana Michaux (Hydrocharitaceae). Mol Ecol Resour 9:1427–1429
Caisse M, Antonovics J (1978) Evolution in closely adjacent plant populations IX. Evolution of reproductive isolation in clinal populations. Heredity 40:371–384
Campanella JJ, Bologna PAX, Smith SM, Rosenzweig EB, Smalley JV (2010) Zostera marina population genetics in Barnegat Bay, New Jersey, and implications for grass bed restoration. Popul Ecol 52:181–190
Catling PM, Spicer KW, Biernacki M, Doust JL (1994) The biology of Canadian weeds.103. Vallisneria americana Michx. Can J Plant Sci 74:883–897
Chao A, Jost L, Chiang SC, Jiang YH, Chazdon RL (2008) A two-stage probabilistic approach to multiple-community similarity indices. Biometrics 64:1178–1186
Chen L, Xu LM, Huang HW (2007) Genetic diversity and population structure in Vallisneria spinulosa (Hydrocharitaceae). Aquat Bot 86:46–52
Clausen J, Keck DD, Hiesey WM (1941) Regional differentiation in plant species. Am Nat 75:231–250
Cooper SR (1995) Chesapeake Bay watershed historical land-use-impact on water-quality and diatom communities. Ecol Appl 5:703–723
Cornuet JM, Luikart G (1996) Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 144:2001–2014
Costanza R, Greer J (1995) The Chesapeake Bay and its watershed: a model for sustainable ecosystem management? In: Gunderson LH, Holling CS, Light SS (eds) Barriers and bridges to the renewal of ecosystems and institutions. Columbia University Press, New York, pp 169–213
Crain CM, Silliman BR, Bertness SL, Bertness MD (2004) Physical and biotic drivers of plant distribution across estuarine salinity gradients. Ecology 85:2539–2549
Crawford NG (2009) SMOGD: software for the measurement of genetic diversity. Mol Ecol Resour. doi:10.1111/j.1755-0998.2009.02801.x
Dennison WC, Orth RJ, Moore KA, Stevenson JC, Carter V, Kollar S, Bergstrom PW, Batiuk RA (1993) Assessing water-quality with submersed aquatic vegetation. Bioscience 43:86–94
Di Rienzo A, Peterson AC, Garza JC, Valdes AM, Slatkin M, Freimer NB (1994) Mutational processes of simple-sequence repeat loci in human populations. Proc Natl Acad Sci USA 91:3166–3170
Doebeli M, Dieckmann U (2003) Speciation along environmental gradients. Nature 421:259–264
Doering PH, Chamberlain RH, McMunigal JM (2001) Effects of simulated saltwater intrusions on the growth and survival of wild celery, Vallisneria americana, from the Caloosahatchee estuary (south Florida). Estuaries 24:894–903
Doust JL, Laporte G (1991) Population sex-ratios, population mixtures and fecundity in a clonal dioecious macrophyte, Vallisneria americana. J Ecol 79:477–489
Dudash M (1990) Relative fitness of selfed and outcrossed progeny in a self-compatible, protandrous species, Sabatia angularis L. (Gentianaceae): a comparison in three environments. Evolution 44:1129–1139
Ellstrand NC, Roose ML (1987) Patterns of genotypic diversity in clonal plant-species. Am J Bot 74:123–131
Fér T, Hroudová Z (2008) Detecting dispersal of Nuphar lutea in river corridors using microsatellite markers. Freshw Biol 53:1409–1422
Frankel OH (1974) Genetic conservation—our evolutionary responsibility. Genetics 78:53–65
Frankham R (1995a) Conservation genetics. Annu Rev Genet 29:305–327
Frankham R (1995b) Inbreeding and extinction—a threshold effect. Conserv Biol 9:792–799
Frankham R (1996) Relationship of genetic variation to population size in wildlife. Conserv Biol 10:1500–1508
Frankham R (2005) Stress and adaptation in conservation genetics. J Evol Biol 18:750–755
Gigord L, Lavigne C, Shykoff JA (1998) Partial self-incompatibility and inbreeding depression in a native tree species of La Réunion (Indian Ocean). Oecologia 117:342–352
Hammerli A, Reusch TBH (2003) Inbreeding depression influences genet size distribution in a marine angiosperm. Mol Ecol 12:619–629
Hedrick PW (2005) A standardized genetic differentiation measure. Evolution 59:1633–1638
Hedrick PW, Fredrickson R (2010) Genetic rescue guidelines with examples from Mexican wolves and Florida panthers. Conserv Genet 11:615–626
Holsinger KE, Weir BS (2009) Genetics in geographically structured populations: defining, estimating and interpreting Fst. Nat Rev Genet 10:639–650
Huelsenbeck JP, Andolfatto P (2007) Inference of population structure under a Dirichlet process model. Genetics 175:1787–1802
Hufford KM, Mazer SJ (2003) Plant ecotypes: genetic differentiation in the age of ecological restoration. Trends Ecol Evol 18:147–155
Hughes AR, Stachowicz JJ (2004) Genetic diversity enhances the resistance of a seagrass ecosystem to disturbance. Proc Natl Acad Sci USA 101:8998–9002
Hughes AR, Stachowicz JJ (2009) Ecological impacts of genotypic diversity in the clonal seagrass Zostera marina. Ecology 90:1412–1419
Hughes AR, Inouye BD, Johnson MTJ, Underwood N, Vellend M (2008) Ecological consequences of genetic diversity. Ecol Lett 11:609–623
Hunt G (1963) Wild celery in the lower Detroit River. Ecology 44:360–370
Hutchison DW, Templeton AR (1999) Correlation of pairwise genetic and geographic distance measures: inferring the relative influences of gene flow and drift on the distribution of genetic variability. Evolution 53:1898–1914
Jarvis JC, Moore KA (2008) Influence of environmental factors on Vallisneria americana seed germination. Aquat Bot 88:283–294
Jensen JL, Bohonak AJ, Kelley ST (2005) Isolation by distance, web service. BMC Genet 6:13
Jones AT, Hayes MJ, Hamilton NRS (2001) The effect of provenance on the performance of Crataegus monogyna in hedges. J Appl Ecol 38:952–962
Jost L (2008) G st and its relatives do not measure differentiation. Mol Ecol 17:4015–4026
Kalinowski ST (2004) Counting alleles with rarefaction: private alleles and hierarchical sampling designs. Conserv Genet 5:539–543
Kalinowski ST (2005) HP-RARE 1.0: a computer program for performing rarefaction on measures of allelic richness. Mol Ecol Notes 5:187–189
Keeley JE (1979) Population differentiation along a flood frequency gradient: physiological adaptations to flooding in Nyssa sylvatica. Ecol Monogr 49:89–108
Kemp WM, Boynton WR, Stevenson JC, Twilley RR, Means JC (1983) The decline of submerged vascular plants in upper Chesapeake Bay: summary of results concerning possible causes. Mar Technol Soc J 17:78–89
Kemp WM, Batiuk R, Bartleson R, Bergstrom P, Carter V, Gallegos CL, Hunley W, Karrh L, Koch EW, Landwehr JM, Moore KA, Murray L, Naylor M, Rybicki NB, Stevenson JC, Wilcox DJ (2004) Habitat requirements for submerged aquatic vegetation in Chesapeake Bay: water quality, light regime, and physical-chemical factors. Estuaries 27:363–377
Kemp WM, Boynton WR, Adolf JE, Boesch DF, Boicourt WC, Brush G, Cornwell JC, Fisher TR, Glibert PM, Hagy JD, Harding LW, Houde ED, Kimmel DG, Miller WD, Newell RIE, Roman MR, Smith EM, Stevenson JC (2005) Eutrophication of Chesapeake Bay: historical trends and ecological interactions. Mar Ecol Prog Ser 303:1–29
Kingman JFC (1982a) The coalescent. Stoch Process Appl 13:235–248
Kingman JFC (1982b) On the genealogy of large populations. J Appl Probab 19A:27–43
Korschgen C, Green W (1988) American wild celery (Vallisneria americana): ecological considerations for restoration. Technical report 19. US Fish and Wildlife Service, Washington, DC
Korschgen CE, Green WL, Kenow KP (1997) Effects of irradiance on growth and winter bud production by Vallisneria americana and consequences to its abundance and distribution. Aquat Bot 58:1–9
Kreiling RM, Yin Y, Gerber DT (2007) Abiotic influences on the biomass of Vallisneria americana Michx. in the Upper Mississippi River. River Res Appl 23:343–349
Krull JN (1970) Aquatic plant-macroinvertebrate associations and waterfowl. J Wildl Manag 34:707–718
Lande R, Shannon S (1996) The role of genetic variation in adaptation and population persistence in a changing environment. Evolution 50:434–437
Langeland KA (1996) Hydrilla verticillata (L.F.) Royle (Hydrocharitaceae), “The perfect aquatic weed”. Castanea 61:293–304
Laushman RH (1993) Population genetics of hydrophilous angiosperms. Aquat Bot 44:147–158
Lavergne S, Molofsky J (2007) Increased genetic variation and evolutionary potential drive the success of an invasive grass. Proc Natl Acad Sci USA 104:3883–3888
Lewis PO, Zaykin D (2001) Genetic data analysis: computer program for the analysis of alleleic data. Free program distributed by the authors over the internet from http://ewis.eeb.unconn.edu/lewishome/software.html
Lokker C, Susko D, Lovett-Doust L, Lovett-Doust J (1994) Population genetic structure of Vallisneria americana, a dioecious clonal macrophyte. Am J Bot 81:1004–1012
Linhart YB, Grant MC (1996) Evolutionary significance of local genetic differentiation in plants. Annu Rev Ecol Syst 27:237–277
Madsen JD, Chambers PA, James WF, Koch EW, Westlake DF (2001) The interaction between water movement, sediment dynamics and submersed macrophytes. Hydrobiologia 444:71–84
Mank JE, Avise JC (2003) Microsatellite variation and differentiation in North Atlantic eels. J Hered 94:310–314
McFarland DG, Shafer DJ (2008) Factors influencing reproduction in American wild celery: a synthesis. J Aquat Plant Manage 46:129–144
McKay JK, Christian CE, Harrison S, Rice KJ (2005) “How local is local?”—a review of practical and conceptual issues in the genetics of restoration. Restor Ecol 13:432–440
Meirmans PG (2006) Using the AMOVA framework to estimate a standardized genetic differentiation measure. Evolution 60:2399–2402
Meirmans PG, Van Tienderen PH (2004) GENOTYPE and GENODIVE: two programs for the analysis of genetic diversity of asexual organisms. Mol Ecol Notes 4:792–794
Mitton JB, Grant MC (1996) Genetic variation and the natural history of quaking aspen. Bioscience 46:25–31
Montalvo A, Ellstrand N (2001) Nonlocal transplantation and outbreeding depression in the subshrub Lotus scoparius (Fabaceae). Am J Bot 88:258–269
Montalvo AM, Williams SL, Rice KJ, Buchmann SL, Cory C, Handel SN, Nabhan GP, Primack R, Robichaux RH (1997) Restoration biology: a population biology perspective. Restor Ecol 5:277–290
Moore KA (2004) Influence of seagrasses on water quality in shallow regions of the lower Chesapeake Bay. J Coastal Res 45:162–178
Moore KA, Wilcox DJ, Orth RJ (2000) Analysis of the abundance of submersed aquatic vegetation communities in the Chesapeake Bay. Estuaries 23:115–127
Morrell PL, Lundy KE, Clegg MT (2003) Distinct geographic patterns of genetic diversity are maintained in wild barley (Hordeum vulgare ssp spontaneum) despite migration. Proc Natl Acad Sci USA 100:10812–10817
Orth RJ, Carruthers TJB, Dennison WC, Duarte CM, Fourqurean JW, Heck KL, Hughes AR, Kendrick GA, Kenworthy WJ, Olyarnik S, Short FT, Waycott M, Williams SL (2006) A global crisis for seagrass ecosystems. Bioscience 56:987–996
Orth RJ, Wilcox DJ, Whiting JR, Nagey LS, Owens AL, Kenne AK (2008) 2007 Distribution of submerged aquatic vegetation in the Chesapeake Bay and coastal bays. VIMS special scientific report number 151. Final report to US EPA, Chesapeake Bay Program, Annapolis, MD. http://webvimsedu/bio/sav/sav07/indexhtml
Parks JC, Werth CR (1993) A study of spatial features of clones in a population of bracken fern, Pteridium aquilinum (Dennstaedtiaceae). Am J Bot 80:537–544
Pleasants JM, Wendel JF (1989) Genetic diversity in a clonal narrow endemic, Erythronium propullans, and in its widespread progenitor, Erythronium albidum. Am J Bot 76:1136–1151
Pollux BJA, Jong MDE, Steegh A, Verbruggen E, Van Groenendael JM, Ouborg NJ (2007) Reproductive strategy, clonal structure and genetic diversity in populations of the aquatic macrophyte Sparganium emersum in river systems. Mol Ecol 16:313–325
Potts BM, Barbour RC, Hingston AB, Vaillancourt RE (2003) Genetic pollution of native eucalypt gene pools—identifying the risks. Aust J Bot 51:1–25
Pritchard DW (1952) Salinity distribution and circulation in the Chesapeake Bay estuarine system. J Mar Res 11:106–123
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Procaccini G, Piazzi L (2001) Genetic polymorphism and transplantation success in the mediterranean seagrass Posidonia oceanica. Restor Ecol 9:332–338
Procaccini G, Olsen JL, Reusch TBH (2007) Contribution of genetics and genomics to seagrass biology and conservation. J Exp Mar Biol Ecol 350:234–259
Ray N (2005) PATHMATRIX: a GIS tool to compute effective distances among samples. Mol Ecol Notes 5:177–180
Reed DH, Frankham R (2003) Correlation between fitness and genetic diversity. Conserv Biol 17:230–237
Reusch TBH, Bostrom C, Stam WT, Olsen JL (1999a) An ancient eelgrass clone in the Baltic. Mar Ecol 183:301–304
Reusch TBH, Stam WT, Olsen JL (1999b) Microsatellite loci in eelgrass Zostera marina reveal marked polymorphism within and among populations. Mol Ecol 8:317–321
Reusch TBH, Stam WT, Olsen JL (2000) A microsatellite-based estimation of clonal diversity and population subdivision in Zostera marina, a marine flowering plant. Mol Ecol 9:127–140
Reusch TBH, Ehlers A, Hammerli A, Worm B (2005) Ecosystem recovery after climatic extremes enhanced by genotypic diversity. Proc Natl Acad Sci USA 102:2826–2831
Rhode JM, Duffy JE (2004) Relationships between bed age, bed size, and genetic structure in Chesapeake Bay (Virginia, USA) eelgrass (Zostera marina L.). Conserv Genet 5:661–671
Rogstad SH, Keane B, Beresh J (2002) Genetic variation across VNTR loci in central North American Taraxacum surveyed at different spatial scales. Plant Ecol 161:111–121
Rosenberg NA, Nordborg M (2002) Genealogical trees, coalescent theory and the analysis of genetic polymorphisms. Nat Rev Genet 3:380–390
Rozas LP, Minello TJ (2006) Nekton use of Vallisneria americana Michx. (wild celery) beds and adjacent habitats in coastal Louisiana. Estuar Coast 29:297–310
Rozas LP, Odum WE (1987) Fish and macrocrustacean use of submerged plant beds in tidal fresh-water marsh creeks. Mar Ecol 38:101–108
Rozas LP, Odum WE (1988) Occupation of submerged aquatic vegetation by fishes—testing the roles of food and refuge. Oecologia 77:101–106
Ruggiero MV, Turk R, Procaccini G (2002) Genetic identity and homozygosity in North-Adriatic populations of Posidonia oceanica: an ancient, post-glacial clone? Conserv Genet 3:71–74
Saccheri I, Kuussaari M, Kankare M, Vikman P, Fortelius W, Hanski I (1998) Inbreeding and extinction in a butterfly metapopulation. Nature 392:491–494
Sand-Jensen K (1998) Influence of submerged macrophytes on sediment composition and near-bed flow in lowland streams. Freshw Biol 39:663–679
Schwartz MK, McKelvey KS (2009) Why sampling scheme matters: the effect of sampling scheme on landscape genetic results. Conserv Gent 10:441–452
Short FT, Wyllie-Echeverria S (1996) Natural and human-induced disturbance of seagrasses. Environ Conserv 23:17–27
Slatkin M (1981) Estimating levels of gene flow in natural populations. Genetics 99:323–335
Slatkin M (1985) Gene flow in natural populations. Annu Rev Ecol Syst 16:393–430
Slatkin M (1987) Gene flow and the geographic structure of natural populations. Science 236:787–792
Slatkin M (1995) A measure of population subdivision based on microsatellite allele frequencies. Genetics 139:457–462
Society for Ecological Restoration International Science & Policy Working Group (2004) The SER international primer on ecological restoration. Society for Ecological Restoration International, Tucson. www.ser.org
Sork VL, Nason J, Campbell DR, Fernandez JF (1999) Landscape approaches to historical and contemporary gene flow in plants. Trends Ecol Evol 14:219–224
Stanton ML, Galen C, Shore J (1997) Population structure along a steep environmental gradient: consequences of flowering time and habitat variation in the snow buttercup, Ranunculus adoneus. Evolution 51:79–94
Stevenson JC, Confer NM (1978) Summary of available information on Chesapeake Bay submerged vegetation. Fish and Wildlife Service, Washington, DC, pp 283–335
Takamura N, Kadono Y, Fukushima M, Nakagawa M, Kim BHO (2003) Effects of aquatic macrophytes on water quality and phytoplankton communities in shallow lakes. Ecol Res 18:381–395
Templeton AR (1997) Coadaptation and outbreeding depression. In: Soule M (ed) Conservation biology the science of scarcity and diversity. Sinauer Associates, Inc., Sunderland, pp 105–116
Thuillet AC, Bru D, David J, Roumet P, Santomi S, Sourdille P, Bataillon T (2002) Direct estimation of mutation rate for 10 microsatellite loci in durum wheat, Triticum turgidum (L.) Thell. ssp durum desf. Mol Biol Evol 19:122–125
Till-Bottraud I, Poncet BN, Rioux D, Girel J (2010) Spatial structure and clonal distribution of genotypes in the rare Typha minima Hoppe (Typhaceae) along a river system. Bot Helv 120:53–62
Titus JE, Adams MS (1979) Coexistence and the comparative light relations of the submersed macrophytes Myriophyllum spicatum L. and Vallisneria americana Michx. Oecologia 40:273–286
Titus JE, Hoover DT (1991) Toward predicting reproductive success in submersed freshwater angiosperms. Aquat Bot 41:111–136
Titus JE, Stephens MD (1983) Neighbor influences and seasonal growth-patterns for Vallisneria americana in a mesotrophic lake. Oecologia 56:23–29
van Dijk JK, van Tussenbroek BI, Jimenez-Duran K, Marquez-Guzman GJ, Ouborg J (2009) High levels of gene flow and low population genetic structure related to high dispersal potential of a tropical marine angiosperm. Mar Ecol 390:67–77
van Katwijk M, Bos A, de Jonge V, Hanssen L, Hermus D, de Jong D (2009) Guidelines for seagrass restoration: Importance of habitat selection and donor population, spreading of risks, and ecosystem engineering effects. Mar Pollut Bull 58:179–188
Vigouroux Y, Jaqueth JS, Matsuoka Y, Smith OS, Beavis WF, Smith JSC, Doebley J (2002) Rate and pattern of mutation at microsatellite loci in maize. Mol Biol Evol 19:1251–1260
Wang B, Song Z, Liu G, Lu F, Li W (2010) Comparison of the extent of genetic variation of Vallisneria natans and its sympatric congener V. spinulosa in lakes of the middle-lower reaches of the Yangtze River. Aquat Bot. doi:10.1016/j.aquabot.2009.1012.1006
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolution 38:1358–1370
Westemeier RL, Brawn JD, Simpson SA, Esker TL, Jansen RW, Walk JW, Kershner EL, Bouzat JL, Paige KN (1998) Tracking the long-term decline and recovery of an isolated population. Science 282:1695–1698
Wilder GJ (1974) Symmetry and development of pistillate Vallisneria americana (Hydrocharitaceae). Am J Bot 61:846–866
Williams SL (2001) Reduced genetic diversity in eelgrass transplantations affects both population growth and individual fitness. Ecol Appl 11:1472–1488
Williams SL, Davis CA (1996) Population genetic analyses of transplanted eelgrass (Zostera marina) beds reveal reduced genetic diversity in southern California. Restor Ecol 4:163–180
Wright S (1931) Evolution in Mendelian populations. Genetics 16:97–159
Wyda JC, Deegan LA, Hughes JE, Weaver MJ (2002) The response of fishes to submerged aquatic vegetation complexity in two ecoregions of the mid-atlantic bight: Buzzards Bay and Chesapeake Bay. Estuaries 25:86–100
Yeh FC, Yang R-C, Boyle TBJ, Ye Z-H, Mao JX (1997) POPGENE, the user-friendly shareware for population genetic analysis. Molecular Biology and Biotechnology Centre, University of Alberta, Canada
Young A, Boyle T, Brown T (1996) The population genetic consequences of habitat fragmentation for plants. Trends Ecol Evol 11:413–418
Zipperle AM, Coyer JA, Reise K, Gitz E, Stam WT, Olsen JL (2009) Clonal architecture in an intertidal bed of the dwarf eelgrass Zostera noltii in the Northern Wadden Sea: persistence through extreme physical perturbation and the importance of a seed bank. Mar Biol 156:2139–2148
Acknowledgments
The authors gratefully acknowledge the assistance of a number of organizations and individuals that were instrumental in assistance with location of sites and collection of samples: Maryland Department of Natural Resources, North Bay Camp, Jason Granberg, Peter Bergstrom, Lee Karrh, Mark Lewandowski, Stan Kollar, Nancy Rybicki, Todd Beser, and Jason Jullian. We thank Paul Widmeyer for creating the study area map. Our funding was provided through NOAA Sea Grant Maryland, and University of Maryland Department of Plant Science and Landscape Architecture and Maryland Agricultural Experiment Station.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Lloyd, M.W., Burnett, R.K., Engelhardt, K.A.M. et al. The structure of population genetic diversity in Vallisneria americana in the Chesapeake Bay: implications for restoration. Conserv Genet 12, 1269–1285 (2011). https://doi.org/10.1007/s10592-011-0228-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-011-0228-7