
Abstract
We have examined transcription loops on lampbrush chromosomes of the newt Notophthalmus by superresolution microscopy. Because of the favorable, essentially two-dimensional morphology of these loops, an average optical resolution in the x–y plane of about 50 nm was achieved. We analyzed the distribution of the multifunctional RNA-binding protein CELF1 on specific loops. CELF1 distribution is consistent with a model in which individual transcripts are tightly folded and hence closely packed against the loop axis.
Similar content being viewed by others


Introduction
The physical organization of chromatin at intermediate levels of resolution is poorly understood—that is, between the level of the nucleosome and what can be resolved by conventional light microscopy. Because of the extreme level of chromatin compaction in mitotic chromosomes and interphase nuclei, giant chromosomes with their more extended organization have provided much useful information, particularly the polytene chromosomes of Diptera and the lampbrush chromosomes (LBCs) of various vertebrates. LBCs are the largest known chromosomes, reaching lengths of up to 1 mm in oocytes of some amphibians. Their major structural features have been known for a long time and are well summarized in Callan’s classic monograph (Callan 1986). In brief, the longitudinal axis of each chromosome consists of multiple regions of condensed inactive chromatin, the chromomeres, from which paired loops of active chromatin extend laterally (Gaginskaya et al. 2009; Gall et al. 2004; Morgan 2002). Each loop consists of one or more transcription units (TUs) readily visible by conventional light microscopy (reviewed in Gall et al. 1983). LBCs can be centrifuged onto a standard microscope slide or coverslip, which reduces their complex three-dimensional organization to two dimensions.
To gain new insight into the organization of the TUs, we used spectral position determination microscopy (SPDM) (Lemmer et al. 2008). SPDM is a method of localization microscopy (Betzig et al. 2006; Hess et al. 2006; Rust et al. 2006) that uses standard fluorophores; it is also termed direct optical reconstruction microscopy (Heilemann et al. 2008) or ground-state depletion imaging microscopy (Fölling et al. 2008). SPDM is based on the precise determination of the position of each individually detected fluorescent molecule. From this information, an image of the underlying biological structure can be reconstructed with an effective resolution far below the diffraction limit of light. Localization microscopy and other superresolution methods, in combination with fluorescent immunostaining and in situ hybridization, have already given new insights into several aspects of nuclear structure and gene activity, including details of the nuclear envelope, the organization of chromatin domains, and the relationship between transcription and chromatin structure (Bohn et al. 2010; Markaki et al. 2010; Schermelleh et al. 2008; Tykocinski et al. 2010)
For the analysis shown below, we made use of both the high structural resolution (~50 nm) and the single-molecule information provided by the detection of individual fluorophores. This unique combination, which is currently provided only by localization microscopy, allowed us to gain insight into the folding of mRNA on LBC loops with a previously unmatched level of detail.
Materials and methods
Setup for SPDM
Superresolution microscopy measurements were performed with a wide-field fluorescence microscopy setup according to Lemmer et al. (2008). This technique detects single molecules using a light-induced, long-lived dark state of the fluorophore (Dickson et al. 1997; McAnaney et al. 2005; Peterman et al. 1999; Schuster et al. 2005; Sinnecker et al. 2005). Illumination of the sample with an excitation intensity in the 10 kW/cm² range pushes the fluorophores into a long-lived dark state within a few seconds. The stochastic recovery of the molecules to the fluorescent state (followed by their transition to another long-lived dark state) is then used for their detection.
For image acquisition, we used diode-pumped solid-state lasers with wavelengths of 488 and 568 nm (Sapphire HP 488 and Sapphire 568, Coherent, Dieburg, Germany). No additional laser was needed for the switching/bleaching of the molecules. Fluorescent light was detected with a 1.4 NA oil-immersion objective (HCX PL APO, 63X, Leica, Wetzlar, Germany) and a high quantum efficiency CCD camera (SensiCam QE, PCO Imaging, Kehlheim, Germany). Bandpass filters (XF3003 520DF40, Laser Components GmbH, Olching, Germany; F37-609, AHF, Analysentechnik, Tübingen, Germany) were used to select the emitted fluorescence.
For the present experiments, data stacks consisting of several thousand images were recorded with an exposure time of 150 ms each. Measurements for the two different channels were performed sequentially. Determination of the fluorophore positions was performed as described previously (Gunkel et al. 2009; Kaufmann et al. 2009; Lemmer et al. 2008). The chromatic shift between the images from the two different channels was globally corrected via cross-correlation. Local shifts were considerably smaller than the achieved structural resolution and therefore have been neglected in further analyses.
LBC preparations
Adult female newts, Notophthalmus viridescens, were purchased from Charles D. Sullivan, Nashville TN and maintained in aquaria at ~8 °C with weekly feeding. Ovaries were removed and LBC preparations made as described (Gall and Wu 2010) with the following modifications. LBCs were centrifuged onto 22 mm coverslips instead of microscope slides. They were stained with two antibodies: mouse mAb 3B1 (IgG) against CELF1 (CUG-BP1) from Abcam and mouse mAb H14 (IgM) against RNA polymerase II phosphorylated on serine 5 of the C-terminal domain, a gift from Jeffry Corden (Patturajan et al. 1998). After immunostaining, the coverslips were mounted on standard 3″ × 1″ microscope slides with Prolong Gold antifade reagent (Invitrogen, Grand Island, NY). TetraSpeck Fluorescent Microspheres (0.5 μm; Invitrogen) were included for microscope calibration.
Distance analysis of CELF1 molecules
As the first step of the distance analysis, a polygon chain (white line in Fig. 2c, f) was manually fitted to the DNA strand using the image of the green channel (Alexa 488, polymerase II (pol II)). The start point of CELF1 binding was identified from the red channel (Alexa 594, CELF1). An algorithm implemented in MATLAB (MathWorks, Natick, MA) determined the shortest distance between each CELF1 molecule and the polygon chain as well as its distance along the DNA strand from the start point of CELF1 binding. In this manner, the distance of each CELF1 molecule from the DNA strand was determined relative to the distance that molecule had travelled since it attached to the nascent RNA chain (Fig. 1). The distance analysis was performed not only for each detected CELF1 molecule but also for clusters formed by these molecules. Detection and characterization of the CELF1 clusters was done using an algorithm described previously (Kaufmann et al. 2011).

Interpretation of LBC loop morphology based on folding of nascent RNA chains. In the absence of processing, the length of nascent RNA chains (in bases) will be equal to the length of the DNA template from which they are transcribed. Left, fully extended RNA chains. This condition is approximated in E.M. images of LBCs isolated in distilled water (Hamkalo and Miller 1973; Miller and Hamkalo 1972). Right, folded RNA chains as postulated for LBCs isolated in isotonic saline. Horizontal lines represent the DNA template (LBC loop axis). Arrows indicate direction of transcription. P promoter region. Green and red circles represent Pol II and CELF1 molecules, respectively
Results
Superresolution microscopy of CELF1 and polymerase II
The results reported here extend observations originally made by Morgan on LBCs of the axolotl, Ambystoma mexicanum (Morgan 2007). Morgan analyzed the distribution of the multifunctional RNA-binding protein CELF1 (Barreau et al. 2006) on specific LBC loops, showing three features clearly. First, CELF1 protein is strongly concentrated on a small subset of transcription loops. Second, it is limited to a restricted region of the TUs on which it occurs. And third, within that region, it has a characteristic distribution: proximally (toward the start of the TU) it is closely associated with pol II on the loop axis, whereas distally (away from the start) it becomes more and more displaced from the axis. Morgan interpreted these features as follows. Because CELF1 is an RNA-binding protein, it binds to the nascent transcript when pol II transcribes a specific binding sequence, and it remains attached to this sequence as the nascent transcript elongates. Consequently, the binding sequence and its attached CELF1 become progressively displaced laterally from the loop axis (Fig. 1; Morgan’s Fig. 6).
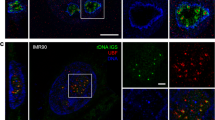
We performed SPDM measurements on single TUs that had been immunostained with two antibodies, one against CELF1 labeled red with Alexa 594 and one against phosphorylated pol II labeled green with Alexa 488 (Fig. 2). CELF1 provides information about the nascent RNA chains, whereas Pol II allows this information to be related to the DNA that forms the axis of the TU. In our images, it was possible to determine the positions of individual molecules in the x–y plane to a mean accuracy of ~23 nm, corresponding to a structural resolution of ~50 nm. Figure 2 illustrates the difference in resolution between confocal fluorescence microscopy images (a, d) and the superresolution microscopy images (b, e) of the same structure. Further examples of superresolution microscopy images of CELF1 molecules on LBCs are shown in Fig. 3.

Images of a LBC loop and analysis of CELF1 distribution. a Confocal fluorescence image of a single transcription unit immunostained for CELF1 (red, Alexa 594) and polymerase II (green, Alexa 488). b Superresolution microscopy (SPDM) image of the same region. c Analysis of CELF1 molecules (red). Large yellow error mark, the start point of CELF 1 attachment to the nascent transcripts and also the start point for determining distances along the loop axis (DNA strand). The loop axis is represented by the white line. d–f Enlarged images of the regions marked by white boxes in (a–c). Yellow arrows in (f) indicate vertical distances of CELF1 molecules from the loop axis

Superresolution images of LBC loops. Each panel shows one or more LBC loops immunostained for CELF1 (red, Alexa 594) and polymerase II (green, Alexa 488). In these images, the average lateral resolution in the x–y plane is about 50 nm in both channels
Distance analysis of CELF1 molecules along the DNA strand
If CELF1 binds to a specific splice site within a nascent transcript, then the spatial distribution of CELF1 staining should give insight into the behavior of that site as the transcript proceeds around the loop. The easiest parameter to measure is the perpendicular distance of the CELF1 stain from the loop axis as a function of distance along the loop. If both the DNA in the loop axis and the nascent RNA transcript were fully extended, these two distances would be roughly the same (Fig. 1). Obviously, this is not the case. Folding of the nascent chain will greatly reduce the ratio and is the starting assumption for our analysis. For individual CELF1 signals, we made two measurements. First, we determined the vertical distance between the CELF1 signal and the loop axis (Fig. 2f). Because the pol II signal is discontinuous, we determined a theoretical loop axis by plotting a polygon chain (white line in Fig. 2c, f). Second, we determined the distance (along the loop axis) between each CELF1 signal and the point at which CELF1 first appeared on the loop axis (Fig. 2c, yellow error mark). The distance analysis was performed using an algorithm developed especially for this purpose (“Materials and methods”). The results of the analyses of eight measurements are shown in Fig. 4a. Here, the vertical distance of individual CELF1 molecules to the loop axis (Fig. 2f) is plotted vs. the distance along the axis from the start of CELF1 staining (Fig. 2c, yellow error mark). A similar analysis is shown in Fig. 4b, but here we measured the distance from the axis of clusters formed by the CELF1 molecules. The two most obvious features of this analysis, evident from simple inspection of the micrographs, are (1) the distance between an individual CELF1 signal and the loop axis increases in the direction of transcription and (2) each signal is much closer to the axis than its distance from the point at which CELF1 first appears on the loop (Fig. 1).

Analysis of CELF1 molecules. a Vertical distances of single CELF1 molecules from the DNA strand are plotted versus the distances of those molecules from the start point of CELF1 attachment to the loop axis (yellow error mark in Fig. 2c). Values are averaged in intervals of 500 nm along the abscissa. Error bars indicate the error of the means. A model function of the type c + N α was fitted to the data. b Distances of CELF1 clusters from the DNA strand are plotted as in (a). Values are averaged in intervals of 600 nm. A model function of the same kind as in (a) was fitted to the data
Fang et al. (2011) describe a model for the folding of single-stranded RNA (ssRNA) based on the formation of double-stranded duplexes by contiguous base pairs. According to their simulations, the lengths of a folded ssRNA molecule and its fully extended chain are related by a power law. Based on their analysis, we fitted model functions of the type c + N α to the experimental data, where N is the length of the chain. The yellow curves in the diagrams in Fig. 4 represent the best fit to the data. The curve shapes in Fig. 4a, b are very similar. The simulations of Fang et al. yielded a value of ~0.7, whereas the fit to our experimental data gives a value closer to ~0.5. This corresponds to a secondary structure of the mRNA that is more condensed relative to the ssRNA than predicted by the theoretical simulation.
Discussion
Several features make LBCs particularly favorable objects for the study of chromosome organization. First is their extraordinary size, which results from extreme decondensation at the molecular level. Second, they are easy to isolate freehand from the giant germinal vesicle, thereby physically separating them from each other and from other nuclear components. Third, they are transcriptionally active, permitting analysis not only of general chromosome structure but also of individual TUs. Finally, they can be centrifuged onto a microscope slide or coverslip, thus reducing the three-dimensional complexity of the intact nucleus to a two-dimensional problem. These features have been used to their advantage here and in numerous earlier studies.
Our superresolution analysis of CELF1 staining provides quantitative data about the TUs in LBC loops. The generally accepted model of LBC structure assumes that each loop represents one or more TUs. In the simplest case, a loop corresponds to a single TU with transcription starting at or near the “thin” end of the loop and terminating at the “thick” end. This model is strongly supported by electron microscopic images of GV contents prepared as “Miller spreads.” Such images show closely spaced nascent transcripts extending laterally from a linear axis (Hamkalo and Miller 1973; Miller and Hamkalo 1972). The width of a LBC loop as viewed by phase contrast or confocal microscopy, however, is very much less than the length of the attached nascent transcripts seen in these electron micrographs. Furthermore, the increase in width from thin to thick end is not proportional to the distance from the thin end. These two features are reflected in the data for CELF1 staining shown in Fig. 4a, b.
We presume that folding of the nascent RNA chains is the major factor responsible for the small width of a loop relative to the length of its attached RNA fibers. In addition to intrinsic folding of the RNA molecule, additional compaction probably results from association of RNA binding proteins with the nascent transcripts. An indication of this tighter folding is the smaller value of our measured α compared with the value provided by the theoretical simulations of Fang et al. (2011). In their model, folding of the ssRNA depends only on the development of local double strands. Splicing is a third factor that could influence loop morphology. The splicing machinery definitely associates with nascent LBC loops (Patel and Bellini 2008; Patel et al. 2007), but it is not known whether spliced introns are removed from the loops during transcription or after release of the transcripts into the nucleoplasm. Early release of introns would presumably influence the width of a loop, preventing the width from increasing in proportion to the distance from transcription initiation.
Superresolution images should help to clarify additional aspects of loop structure, especially if combined with a variety of molecular markers. Particularly useful will be analysis of additional splicing components, the exon-junction complex, and hnRNP proteins that may be involved in targeting transcripts to specific pathways or locations in the cell.
Abbreviations
- CCD:
-
Charge coupled device camera
- CELF1:
-
CUG-BP1 and ETR-3 Like Factor 1
- CUG-BP1:
-
CUG Binding Protein 1
- CUG:
-
oligoribonucleotide 5´CUG3´
- ETR-3:
-
Embryonic lethal abnormal vision Type RNA-binding protein 3
- dSTORM:
-
digital optical reconstruction microscopy
- EM:
-
Electron microscopy
- GSDIM:
-
Ground state depletion imaging microscopy
- LBC:
-
Lampbrush chromosome
- Pol II:
-
RNA polymerase 2
- SPDM:
-
Spectral position determination microscopy
- ssRNA:
-
single stranded RNA
- TU:
-
Transcription unit
References
Barreau C, Paillard L, Mereau A, Osborne HB (2006) Mammalian CELF/Bruno-like RNA-binding proteins: molecular characteristics and biological functions. Biochimie 88:515–525
Betzig E, Patterson GH, Sougrat R et al (2006) Imaging intracellular fluorescent proteins at nanometer resolution. Science 313:1642–1645
Bohn M, Diesinger P, Kaufmann R et al (2010) Localization microscopy reveals expression-dependent parameters of chromatin nanostructure. Biophys J 99:1358–1367
Callan HG (1986) Lampbrush chromosomes. Springer, Berlin
Dickson RM, Cubitt AB, Tsien RY, Moerner WE (1997) On/off blinking and switching behaviour of single molecules of green fluorescent protein. Nature 388:355–358
Fang LT, Yoffe AM, Gelbart WM, Ben-Shaul A (2011) A sequential folding model predicts length-independent secondary structure properties of long ssRNA. J Phys Chem B 115:3193–3199
Fölling J, Bossi M, Bock H et al (2008) Fluorescence nanoscopy by ground-state depletion and single-molecule return. Nature Methods 5:943–945
Gaginskaya E, Kulikova T, Krasikova A (2009) Avian lampbrush chromosomes: a powerful tool for exploration of genome expression. Cytogen Gen Res 124:251–267
Gall JG, Diaz MO, Stephenson EC, Mahon KA (1983) The transcription unit of lampbrush chromosomes. In: Subtelny S, Kafatos F (eds) Gene structure and regulation in development, vol. 41. Alan R. Liss, New York, pp 137–146, Symp Soc Dev Biol
Gall JG, Wu Z (2010) Examining the contents of isolated Xenopus germinal vesicles. Methods 51:45–51
Gall JG, Wu Z, Murphy C, Gao H (2004) Structure in the amphibian germinal vesicle. Exp Cell Res 296:28–34
Gunkel M, Erdel F, Rippe K et al (2009) Dual color localization microscopy of cellular nanostructures. Biotech J 4:927–938
Hamkalo BA, Miller OL Jr (1973) Electronmicroscopy of genetic activity. Annu Rev Biochem 42:379–396
Heilemann M, van de Linde S, Schuttpelz M et al (2008) Subdiffraction-resolution fluorescence imaging with conventional fluorescent probes. Angew Chem Int Ed Engl 47:6172–6176
Hess ST, Girirajan TP, Mason MD (2006) Ultra-high resolution imaging by fluorescence photoactivation localization microscopy. Biophys J 91:4258–4272
Kaufmann R, Lemmer P, Gunkel M et al (2009) SPDM: single molecule superresolution of cellular nanostructures. Proc Soc Photo-Opt Instr Eng 7185:1–19
Kaufmann R, Muller P, Hildenbrand G, Hausmann M, Cremer C (2011) Analysis of Her2/neu membrane protein clusters in different types of breast cancer cells using localization microscopy. J Microsc 242:46–54
Lemmer P, Gunkel M, Baddeley D et al (2008) SPDM: light microscopy with single-molecule resolution at the nanoscale. Appl Phys B 93:1–12
Markaki Y, Gunkel M, Schermelleh L et al (2010) Functional nuclear organization of transcription and DNA replication: a topographical marriage between chromatin domains and the interchromatin compartment. Cold Spring Harb Symp Quant Biol 75:475–492
McAnaney TB, Zeng W, Doe CF et al (2005) Protonation, photobleaching, and photoactivation of yellow fluorescent protein (YFP 10C): a unifying mechanism. Biochem 44:5510–5524
Miller OL Jr, Hamkalo BA (1972) Visualization of RNA synthesis on chromosomes. Int Rev Cytol 33:1–25
Morgan GT (2002) Lampbrush chromosomes and associated bodies: new insights into principles of nuclear structure and function. Chromosome Res 10:177–200
Morgan GT (2007) Localized co-transcriptional recruitment of the multifunctional RNA-binding protein CELF1 by lampbrush chromosome transcription units. Chromosome Res 15:985–1000
Patel SB, Bellini M (2008) The assembly of a spliceosomal small nuclear ribonucleoprotein particle. Nucleic Acids Res 36:6482–6493
Patel SB, Novikova N, Bellini M (2007) Splicing-independent recruitment of spliceosomal small nuclear RNPs to nascent RNA polymerase II transcripts. J Cell Biol 178:937–949
Patturajan M, Schulte RJ, Sefton BM et al (1998) Growth-related changes in phosphorylation of yeast RNA polymerase II. J Biol Chem 273:4689–4694
Peterman EJG, Brasselet S, Moerner WE (1999) The fluorescence dynamics of single molecules of green fluorescent protein. J Phys Chem A103:10553–10560
Rust MJ, Bates M, Zhuang X (2006) Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat Methods 3:793–795
Schermelleh L, Carlton PM, Haase S et al (2008) Subdiffraction multicolor imaging of the nuclear periphery with 3D structured illumination microscopy. Science 320:1332–1336
Schuster J, Cichos F, von Borczyskowski C (2005) Blinking of single dye molecules in various environments. Opt Spectr 98:712–717
Sinnecker D, Voigt P, Hellwig N, Schaefer M (2005) Reversible photobleaching of enhanced green fluorescent proteins. Biochem 44:7085–7094
Tykocinski LO, Sinemus A, Rezavandy E et al (2010) Epigenetic regulation of promiscuous gene expression in thymic medullary epithelial cells. Proc Natl Acad Sci USA 107:19426–19431
Acknowledgments
CC and RK thank the Frontier Project of Heidelberg University and the Deutsche Forschungsgemeinschaft (DFG) for support. Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under award number R01GM33397 to JGG. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. JGG is an American Cancer Society Professor of Developmental Genetics.
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible Editor: Herbert Macgregor.
Rights and permissions
About this article
Cite this article
Kaufmann, R., Cremer, C. & Gall, J.G. Superresolution imaging of transcription units on newt lampbrush chromosomes. Chromosome Res 20, 1009–1015 (2012). https://doi.org/10.1007/s10577-012-9306-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10577-012-9306-z