
Abstract
Episomal vectors assembled from defined genetic components are a promising alternative to traditional gene therapy vectors that integrate in the host genome and may cause insertional mutations. The vector pEPI-eGFP is stably retained in the episomal state in cultured mammalian cells at low copy number for many generations without integration into the host genome. Although pEPI-eGFP is a fully engineered vector, little is known about how it interacts with the host genome and about the molecular mechanisms that are responsible for its transcriptional activity. We have analyzed the expression of the episomal reporter gene eGFP under conditions that affect the chromatin state of the genome. We have also constructed pEPI derivatives carrying a tandem array of lac operator sequences, which allows in vivo visualization and manipulation of the chromatin state of the episome. We show that changes in chromatin state of both the host and pEPI-eGFP induces changes in episomal gene activity and influences the episome’s nuclear distributions. We conclude that episomal genes are subject to control systems of the host, similarly to their counterparts in the host genome.
Similar content being viewed by others

Introduction
The stable and safe transfer of genes to mammalian and in particular human cells is of great interest for biomedical and biotechnological research. A number of transformation techniques have been developed (reviewed in Conese et al. 2004; Glover et al. 2005; Jackson et al. 2006; Van Craenenbroeck et al. 2000), most of which employ vectors that assure stable expression by integration in the host genome. However, integration can disrupt host genes and position effects make expression of the transgene unpredictable (Hacein-Bey-Abina et al. 2003a; Hacein-Bey-Abina et al. 2003b). As an alternative, non-integrating episomal vectors have been developed. Such episomes are often based on viral sequences (Conese et al. 2004; Van Craenenbroeck et al. 2000). They can be retained in the episomal state in the host cell during cell division, may have a stable level of gene expression and are assumed not to be subject to position effects because of their non-integrated state. However, since viral gene products can be detrimental due to immunological complications or undesired interactions with host cell components, their use in humans is restricted (reviewed in Glover et al. 2005; Lipps et al. 2003).
Recently, a fully engineered non-viral episomal vector (pEPI-eGFP) has been developed that may be a safe alternative for gene transfer in mammals (Stehle et al. 2007; Jenke et al. 2004b; Baiker et al. 2000; Piechaczek et al. 1999). After transfection pEPI-eGFP becomes a persistent episome in mammalian cells. This property is attributed to the combination of a 2 kb S/MAR (scaffold/matrix attachment region) sequence, which has been isolated from the human β-interferon locus, and read-through transcription of a gene into the S/MAR sequence (Jackson et al. 2006; Jenke et al. 2004b; Stehle et al. 2003). Episomal pEPI-eGFP is claimed to be present in cells at low copy numbers, typically 2–10 per cell, based on fluorescence in situ hybridization (FISH) labeling (Baiker et al. 2000; Jenke et al. 2004b; Stehle et al. 2007). Interestingly, pEPI-eGFP not only remains episomal in cultured cells, it was also found to persist in various tissues of pig fetuses after sperm-mediated transfer into the embryo, illustrating the robustness of the S/MAR-based episomal system (Manzini et al. 2006). These properties make pEPI-eGFP a promising vector for applications that aim at permanent genetic transformation of mammals, including gene therapy in man.
Despite the potential of the pEPI-GFP-type episomes, we have only limited understanding of the molecular mechanisms that ensure their extra-chromosomal persistence and regulate expression of their genes. Issues such as episomal replication, faithful segregation during mitosis, and control of transcription have hardly been addressed yet. It has been shown that persistent pEPI-eGFP replicates once per cell cycle in early S-phase (Schaarschmidt et al. 2004) and acquires a nucleosomal organization enriched in marks for transcriptionally active chromatin, such as trimethylated histone H3K4 (Stehle et al. 2007). Interestingly, these marks are particularly enriched at the S/MAR sequence in pEPI-eGFP in a cell-cycle-dependent manner (Rupprecht and Lipps 2009). Thus, although pEPI-eGFP is a stable extra-chromosomal replicon, its chromatin state is at least in part controlled by the host. For genes integrated in the host genome it is well documented that the local nuclear environment plays an important role in gene expression, often by affecting the chromatin state (Finlan et al. 2008, reviewed in Fraser and Bickmore 2007; Kumaran et al. 2008; Zhao et al. 2009). Whether such processes also act in trans, affecting episomes, is unknown.
To get insight into in trans-regulatory mechanisms between host and episomal chromatin we have combined in vivo 3D live cell microscopy with molecular analysis of the episome by chromatin immunoprecipitation (ChIP). Using the lac operator/lac repressor (lacO/lacR) system (Robinett et al. 1996; Verschure et al. 2005), we have visualized individual episomes in living cells and manipulated their functional state. Results show that transcriptional activation is accompanied by a moderate relocation of the episomes towards nuclear center. We provide evidence that episomes can be transcriptionally activated by changing the chromatin state of the host and likely also of the episome, in particular by inhibiting DNA methylation, decreasing histone H3K9 tri-methylation and inhibiting histone deacetylation. We show that an increase in acetylated episomal histone H3 is correlated with enhanced episomal transcription. Taken together, our data suggest that episomal genes behave remarkably similar to host genes.
Materials and methods
Vectors
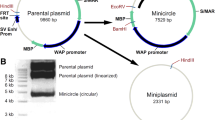
Episomal vectors used were pEPI-eGFP (Baiker et al. 2000; Jenke et al. 2004b; Piechaczek et al. 1999) and pELO64 (Fig. 1a, b). Plasmid pELO64 was constructed by restriction with BamHI/SalI of pEPI-eGFP and insertion of a BamHI/SalI fragment containing 64 lac operator binding sites from plasmid pPs-8.8 (Robinett et al. 1996). For visualization of pELO64 in vivo, a plasmid expressing mCherry-lac repressor based on eGFP-lac repressor-AscI-NLS (Verschure et al. 2005) was used after eGFP was replaced by mCherry by BsrGI/AgeI cloning (Fig. 1c). Targeted epigenetic manipulation of pELO64 was achieved by using a plasmid expressing mCherry-lac repressor-VP16 acidic activation domain. The VP16 acidic activation domain was inserted into the mCherry-lac repressor plasmid by EcoRV/MunI cloning downstream of the lac repressor sequence (Fig. 1c).

Plasmids used in this study and episomal status. a Map of pEPI-eGFP. b Map of pELO64-eGFP. An array of 64 lac operator sites were cloned downstream of the S/MAR sequence of pEPI-eGFP. c Map of pmCherry-lac repressor. d Southern blot analysis of DNA from CHO-K1 cells carrying pEPI-eGFP and pELO64 after restriction with BamHI. The linearized episomes pEPI-eGFP and pELO64 are detected as a single band at 6.7 kb (lane 1) and 9.2 kb (lane 2), respectively. Lane L, size ladder
Cultured cells and transfection
Chinese Hamster Ovary (CHO-K1) cells were grown in Ham’s F12 medium (Invitrogen, Carlsbad, CA, USA) supplemented with 10% heat-inactivated fetal bovine serum (FBS; Invitrogen), 100 units/ml penicillin and 100 units/ml streptomycin (Invitrogen). Suv39h1 -/- cells and immortalized mouse embryonic fibroblasts (IMEFs) were grown as described elsewhere (Peters et al. 2001). HCT116 cells were cultured in McCoy’s 5A medium (Invitrogen) supplemented with 10% heat-inactivated FBS (Invitrogen), 100 units/ml penicillin and 100 units/ml streptomycin (Invitrogen). All cells were cultured at 37°C with 5% CO2. Transfection of all cells was carried out with Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. For the establishment of cell lines that persistently carry the episomal vector, 24 h after transfection with the episomal construct, CHO-K1, Suv39h1 −/− and IMEF cells were selected on culture medium supplemented with respectively 500 μg/ml G418 (CHO-K1 cells) or 400 μg/ml G418 (Suv39h1 −/− and IMEF cells) for 3 weeks. The selected cells were subsequently cultured with a 2-fold lower concentration of G418 (Invitrogen). Transient transfection with mCherry-lac repressor for in vivo observations was carried out following the same protocol and cells were analyzed 48 to 72 h after transfection.
DNA isolation and Southern blot analysis
Total genomic DNA was isolated from 106 CHO-K1 cells and digested with the appropriate restriction enzyme; 5 μg DNA of each sample were subsequently separated on 0.8% agarose gel and blotted overnight to a nylon membrane (Fluka, Buchs SG, Switzerland). A polymerase chain reaction (PCR)-amplified 609-bp fragment, synthesized with the primers 5′-GACGTAAACGGCCACAAGTT-3′ and 5′-GAACTCCAGCAGGACCATGT-3′ on the eGFP sequence, was labeled with 32P and used as a probe.
Fluorescence in situ hybridization
FISH analysis was carried out essentially as described elsewhere (Goetze et al. 2007). Prior to the RNAse treatment a 5-min incubation in 0.1 mg/ml pepsin in 0.01 M HCl was added. pELO64 DNA was used as a probe and labeled with either Biotin- or Digoxigenin-Nick Translation kits (Roche, Mannheim, Germany). Prior to microscopy samples were counterstained with 4′, 6′-diamidino-2-phenylindole (DAPI; 1 μg/ml in Vectashield, Vector Laboratories, Burlingame, CA, USA).
Confocal scanning laser microscopy for in vivo and FISH observation
Images were taken with a Zeiss LSM 510 (Zeiss, Oberkochen, Germany) confocal laser scanning microscope using a 63X Plan-A (1.4 NA) oil immersion objective (Zeiss). For imaging eGFP, Alexa-488 and fluorescein an argon 488-nm laser was used for excitation. Emitted fluorescence was detected using a 505–530-nm band pass filter. For imaging mCherry a 568-nm krypton laser was used for excitation. Emitted fluorescence was detected with a 585-nm-long pass filter. For imaging DAPI-stained DNA, a 364-nm UV laser was used. Emitted fluorescence was detected with a 385–470-nm band pass filter.
In vivo microscopic observations
In vivo observations were carried out on a Zeiss 200 M (Zeiss) inverted fluorescence microscope with a 20X Plan-N (0.75 NA) air objective (Zeiss). For imaging green fluorescence (eGFP) a 490-nm short-pass excitation filter was used. The emitted fluorescence was detected by a 525–565-nm band pass filter. Images were recorded with a cooled CCD camera (Coolsnap HQ, Roper Scientific, Tucson, AZ, USA).
Image analysis
Image analysis on confocal three-dimensional z-stacks was carried out using Argos software (http://homepages.cwi.nl/~wimc/argos). The Mann–Whitney U test for two non-parametric, independent groups was carried out using GraphPad Prism software (GraphPad Software, San Diego, CA, USA). The integrated density of individual mCherry-tagged episomal spots was measured on confocal 3D image stacks. For each spot the intensity was measured in all relevant optical sections (usually just one section), using ImageJ freeware (http://rsb.info.nih.gov/ij/). Regions of interest (ROIs) were defined containing the spot and values were corrected for the intensity in ROIs of the same size in the same section, not containing an episomal spot.
FACS analysis
Cells were trypsinized and resuspended in PBS-B (1 × BS; 0.5% (w/v) BSA). For fluorescence analysis, eGFP and mCherry fluorescence were excited by lasers with 488- and 543-nm wavelengths, respectively. FACS distributions were analyzed by segmentation of the distribution of mCherry fluorescence intensity versus eGFP fluorescence intensity. For quantification of eGFP fluorescence, 2D distributions were segmented as shown in Figs. 4b and 7b. Points in the lower right section represented cells with above background eGFP expression.
Trichostatin A and 5-aza-2′-deoxycytidine treatments
CHO-K1, IMEFs, and Suv39h1 −/− cells were grown in medium supplemented with trichostatin A (TSA; 25, 50, and 100 ng/ml; Sigma, Zwijndrecht, The Netherlands), 5-aza-2′-deoxycytidine (5, 25, and 50 μM; Fluka), or both drugs simultaneously. eGFP fluorescence was evaluated microscopically or by FACS analysis after 48, 72, and 96 h growth in the presence of the drugs. For the two-tailed Student’s t test (MS Excel, Microsoft, Redmond, WA, USA) was used to identify statistical significance differences (Fig. 6).
Chromatin immunoprecipitation
Chromatin immunoprecipitation was performed as described previously (Hatzis et al. 2008). In brief, CHO-K1/pEPI-eGFP cells were cross-linked with 1% formaldehyde for 20 min. at room temperature. The reaction was quenched with glycine at a final concentration of 125 mM. The cells were subsequently washed with phosphate-buffered saline, buffer B (0.25% Triton-X 100, 10 mM ethylenediaminetetraacetic acid (EDTA), 0.5 mM ethylene glycol tetraacetic acid (EGTA), 20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES; pH 7.6)), and buffer C (0.15 M NaCl, 1 mM EDTA, 0.5 mM EGTA, and 20 mM HEPES (pH 7.6)) at 4°C for 10 min each. The cells were then resuspended in ChIP incubation buffer (0.3% sodium dodecyl sulfate (SDS), 1% Triton-X 100, 0.15 M NaCl, 1 mM EDTA, 0.5 mM EGTA, and 20 mM HEPES (pH 7.6)) and sheared using a Covaris S2 (Covaris, Woburn, MA, USA) sonicator for 10 min with following settings: duty cycle, max; intensity, max; cycles/burst, max. The final average size of chromatin fragments was 500–1,000 bp. Sonicated chromatin was incubated for 12 h at 4°C with a rabbit-raised polyclonal anti-H3 antibody (ab 1791; Abcam plc, Cambridge, UK)—this antibody recognizes both the non-acetylated state as the acetylated form of histone H3—or a rabbit-raised polyclonal anti-pan-acetyl-H3 (06-599, Upstate-Millipore, Billerica, MA, USA) at a concentration of 2 μg of antibody per 106 cells with protein G beads (Upstate, Lake Placid, NY, USA). The beads were successively washed two times with buffer 1 (0.1% SDS, 0.1% deoxycholate, 1% Triton-X 100, 0.15 M NaCl, 1 mM EDTA, 0.5 mM EGTA, and 20 mM HEPES (pH 7.6)), one time with buffer 2 (0.1% SDS, 0.1% sodium deoxycholate, 1% Triton-X 100, 0.5 M NaCl, 1 mM EDTA, 0.5 mM EGTA, 20 mM HEPES (pH 7.6)), one time with buffer 3 (0.25 M LiCl, 0.5% sodium deoxycholate, 0.5% NP-40, 1 mM EDTA, 0.5 mM EGTA, 20 mM HEPES (pH 7.6)), and two times with buffer 4 (1 mM EDTA, 0.5 mM EGTA, 20 mM HEPES (pH 7.6)) for 5 min each at 4°C. Precipitated chromatin was eluted by incubation of the beads with elution buffer (1% SDS, 0.1 M NaHCO3) at room temperature for 20 min. After washing and elution, immune-precipitated chromatin was de-cross-linked by incubation at 65°C for 5 h in the presence of 200 mM NaCl, extracted with phenol-chloroform, and ethanol precipitated.
Quantitative PCR
Quantitative PCR (qPCR) in an iCycler iQ Real-Time PCR Detection System (BIORAD, Hercules, CA, USA), using iQ SYBR Green Supermix (BIORAD), was carried out for analysis of ChiP experiments. The manufacturer’s recommendations were followed. The reaction volume of each qPCR reaction was 25 μl with a final concentration of 0.8 μM for each primer. All primers used in this work have been published previously in Stehle et al. (2007) and Rupprecht and Lipps (2009) with the exception of primers for the Neo/Kan sequence on pEPI-eGFP (5′-GATCTGGACGAAGAGCATCAG-3′ and 5′- GCCAACGCTATGTCCTGATAG-3′) and the primers on intron 2 of the C. griseus Rhodopsin gene (GenBank accession number: X61084.1; 5′-TAGGGACAGTGGTTTGGT-3′ and 5′-GGTGCGGCTTTTAGAAACA-3′) used as endogenous control. ChIP values are expressed as % of input. The results were collected over a minimum of three technical replicates.
Results
Experimental system
To allow in vivo visualization and manipulation of pEPI-eGFP we constructed the modified episome pELO64 (Fig. 1a), based on pEPI-eGFP (Baiker et al. 2000; Piechaczek et al. 1999). pEPI-eGFP contains the following functional elements: (1) a human S/MAR sequence (isolated from the human INF-beta gene), which is essential for episomal persistence, (2) a selection marker conveying G418 resistance to transfected cells, and (3) the reporter gene eGFP that reads through into the S/MAR sequence. pELO64 (Fig. 1b) in addition carries a tandem array of 64 copies of the lac operator (lacO) sequence (2.5 kb) to visualize the episome in vivo and to target chromatin modifiers, using the lacO–lacR technology (Belmont 2001). Cell lines that persistently carry pELO64 or pEPI-eGFP were made as described previously (Baiker et al. 2000; Jenke et al. 2004b; Rupprecht and Lipps 2009; Stehle et al. 2007). Three weeks after transfection with the episome and culturing in selection medium (500 μg/ml G418 for CHO-K1 cells), Southern analysis showed that pELO64 is present as a single band (Fig. 1d; 9.2 kb), which is the expected result for the episomal state (Piechaczek et al. 1999; Stehle et al. 2003). Integration in the host genome would result in multiple bands of different sizes, as shown by others (Piechaczek et al. 1999; Stehle et al. 2003; Jenke et al. 2004b). Removing the selection pressure does not affect the persistence of the episome in the host cells (Piechaczek et al. 1999; data not shown). However, to ensure the presence of pELO64 in all cells, the concentration of the selecting agent G418 was kept at 250 μg/ml throughout all experiments. Transient expression of the mCherry-lac repressor fusion protein (mCherry-lacR; Fig. 1c; Shaner et al. 2004) allows visualization of the pELO64 episome in vivo and in fixed cells. The mCherry-lacR gene is under control of a weak polyoma promoter to avoid high background fluorescence of unbound protein.
Visualization of the pELO64 episome in the interphase nucleus
To visualize pELO64 episomes in vivo, cells were transiently transfected with a vector carrying the mCherry-lacR fusion gene (Fig. 1c). Small mCherry-labeled dots could be seen in nuclei of transfected cells (Fig. 2a). Control cells that do not contain pELO64 displayed no mCherry-labeled dots (not shown), indicating that the dots represent episomes. To underscore this conclusion, we carried out FISH using the pELO64 sequence as a FISH probe. Figure 2b shows that the mCherry-labeled dots observed in vivo coincide with FISH-labeled spots, further confirming that the mCherry-lacR protein specifically labels pELO64. The average number of dots per cell that contained at least one detectable pELO64 was 1.8 ± 1.3 (mean ± SD; n = 95). Less than 15% of the cells contained more than two pELO64 spots per nucleus. In about 25% of the cells, no mCherry-tagged pELO64 was detectable (Fig. 2c), probably due to the background fluorescence of unbound mCherry-lacR protein in the nucleus. Using FISH, 2.5 ± 0.9 (mean ± SD; n = 31) pELO64 spots were detected in cells that contained at least one recognizable FISH signal, very similar to what is detected by in vivo labeling of pEPI with mCherry-lacR (Fig. 2b). No FISH signal was detected in about 9% of the observed cells. This number of spots per cell is similar to what has been found for pEPI-eGFP in previous studies (Nehlsen et al. 2006; Piechaczek et al. 1999). FISH probably results in overestimation of the number of episomal spots per cell due to background noise whereas the number of in vivo observed fluorescent pELO64 spots per cell probably is an underestimate due to the background of unbound mCherry. Figure 2d (open bars) shows the distribution of the integrated density per nucleus of mCherry-labeled spots in CHO-K1 cells. The histogram shows a maximum at low intensity values and over 60% of the integrated intensities measured per cell are contained in the 3 lower bins of the histogram. The distribution is skewed towards higher integrated intensities.

In vivo visualization of the episome and quantification. a Projection of z-stack of a confocal image of a nucleus of a living CHO-K1 cell that persistently contains pELO64 and that had been transiently transfected with a vector expressing mCherry-lacR to visualize the episomes. Three mCherry-labeled dots containing episomes are observed. b Visualization of episomes by FISH and mCherry-lacR. The FISH signal for pELO64 (green, top left) and mCherry-lacR signal (red, top right) co-localize. The nucleus was counterstained with DAPI (blue, bottom left). The bottom-right image is a merge of the three other panels. To underscore the colocalization of the two signals a rare cell containing 5 episomal spots was selected. c Number of mCherry-labeled pELO64 dots per nucleus in CHO-K1/pELO64 cells, measured 48 h after transient transfection with mCherry-lacR (open bars) and with mCherry-lacR-VP16-acidic domain (gray bars). d Distribution of the fluorescent intensity per mCherry-tagged episomal spot in CHO-K1/pELO64 nuclei, measured 48 h after transient transfection with mCherry-lacR (open bars) and with mCherry-lacR-VP16-acidic domain (gray bars). Scale bars = 5 μm
Our results on in vivo mCherry labeling and FISH analysis in fixed cells show that the CHO-K1 cells contain on average about two pELO64 episomes per nucleus.
Expression of the episomal eGFP reporter gene
Figure 3 shows CHO-K1 cells that persistently carry pELO64 episomes. Only 5% of the cells expresses the episomal reporter gene eGFP at a detectable level, showing a wide distribution of expression levels (FACS data in Fig. 4b; control). Results in Fig. 1d (Southern blot) make a loss of the eGFP gene due to recombination events unlikely. More so, many studies have shown that the pEPI episome replicated synchronously with the host genome and is faithfully transferred to both daughter cells (Stehle et al. 2007; Jenke et al. 2004b; Schaarschmidt et al. 2004; Baiker et al. 2000). Evidently, many cells carry episomes that are in a state that allows only weak gene expression, sufficient to obtain G418 resistance, but not sufficient for expression of eGFP at a level that is detectable by fluorescence microscopy or FACS analysis. Similar results have been reported for MEL cells (Papapetrou et al. 2006). The expression of the episomal Neo/Kan resistance gene may suppress eGFP expression via promoter, a process called interference, i.e., by inducing downstream changes in episomal chromatin structure that hamper transcription of the neighboring eGFP (Emerman and Temin 1984). However, removal of the selection pressure did not result in an increase in eGFP expression (data not shown), making such interaction between the two neighboring expression units unlikely.

Expression of the episomal reporter gene eGFP. eGFP-expressing cells in colonies of CHO-K1/pELO64 cells. Upper panels, phase contrast; lower panels, eGFP. Scale bar in upper left panel, 10 μm

Chromatin manipulation in CHO-K1 cells. a Histogram of FACS analyses of eGFP expression. Effect of TSA and 5-aza-dC incubation (48 h treatment) on the expression of eGFP in CHO-K1/pELO64 cells. The percentage of eGFP-expressing cells (open bars) and the mean fluorescence intensity per cell (gray bars, arbitrary units) are presented. b FACS data on which histogram in (a) is based. Dots in the lower right region, representing eGFP-expressing cells, are quantified in (a). For each measurement at least 104 cells were analyzed. Control, CHO-K1/pELO64 cells in standard culture medium
Chromatin-modifying drugs affect episomal expression levels
We have manipulated the chromatin state of the episome and analyzed its effect on gene eGFP expression. This was done in four different ways: (1) increasing histone acetylation by inhibiting histone deacetylase (HDAC) activity by culturing in the presence of the HDAC inhibitor TSA, (2) decreasing the DNA methylation level by inhibiting DNA methyl transferase (DMT) activity by culturing cells in the presence of 5-aza-2′-deoxycytidine, (3) decreasing histone H3 lysine 9 tri-methylation (H3K9Me3) by establishing episomes in IMEFs deficient for the heterochromatic histone methyltransferase (HMT) Su(Var)3–9, using the parent wild-type strain as a control (Peters et al. 2001), and (4) targeting the acidic activation domain of the viral transcriptional activator VP16 to the lacO array on the episome (Chuang et al. 2006; Tumbar and Belmont 2001). The first three approaches affect episomal and host chromatin alike, the fourth specifically targets the episome.
CHO-K1/pELO64 cells were treated with the DMT inhibitor 5-aza-2′-deoxycytidine (5-aza-dC; 48 h, 5 and 50 μM); (Jones 1985), with the HDAC inhibitor TSA (48 h, 25 and 100 ng/ml); (Yoshida et al. 1990), and with a combination of the two drugs. In these experiments, we measured two aspects of eGFP expression by FACS analysis: (1) the fraction of cells that express eGFP (open bars), and (2) the average expression per cell (filled bars, Fig. 4a). The percentage of CHO-K1 cells that contain detectable levels of eGFP is 5% in non-treated cultures (Fig. 4a; control). Treatment with TSA or 5-aza-dC (25 and 100 ng/ml TSA; 5 and 50 μM 5-aza-dC, 48 h) resulted in an up to 3.6- and a 3-fold increase in the percentage of cells with detectable eGFP expression, respectively (Fig. 4a; 100 ng/ml TSA; 50 μM 5-aza-dC, open bars). Simultaneous treatment with TSA and 5-aza-dC did not reveal synergy between the two drugs (not shown). Figure 4b shows the FACS diagrams on which the histogram is based. Clearly, there is a gradual increase in expression, rather than an on-off type of switching of the eGFP gene. In all cases there is no detectable expression in a considerable fraction of cells. These results indicate that the activity of genes on the episome is directly or indirectly affected by a reduction of histone deacetylase activity and of DNA methylation. In this sense the episomal gene behaves the same as host genes.
Increased eGFP expression is accompanied by an increased histone acetylation level on the episome
To get insight into the changes in chromatin state of the episome, we analyzed the histone H3 acetylation level of the pEPI-eGFP episome in CHO-K1 cells by ChIP before and after exposure to TSA (100 ng/ml, 48 h), followed by qPCR of co-precipitated episomal DNA fragments. We analyzed various functional elements of the episome (Rupprecht and Lipps 2009; Schaarschmidt et al. 2004; Stehle et al. 2007) by immunoprecipitation with an antibody against the core of histone H3 (Fig. 5a). The enrichment in acetylation marks was analyzed with an antibody that recognizes a variety of acetylated states of histone H3 (Fig. 5b). All functional elements of pEPI-eGFP that were analyzed produced a clear histone H3-core signal, confirming the chromatinized status of pEPI-eGFP (Fig. 5a). Treatment with TSA resulted in a decrease of the qPCR signal for H3-core throughout the episome, suggesting a loss in nucleosomal occupation of the episome, ranging from a decrease of about 13.5% of the qPCR signal for the HSV polyA sequence to close to 98% for the pCMV promoter driving the eGFP gene. An alternative explanation is that histone H3 molecules become less accessible for the antibody after TSA treatment. This result shows that the increased transcriptional activity of the episomal genes is correlated with a loss or decrease in accessibility of nucleosomes, similar to what is found for host genes (Li et al. 2007; Schones et al. 2008). Nonetheless, consistent with the observed episomal transcriptional activation, the qPCR signals for H3Ac of most episomal sequences increased significantly after TSA treatment, marking transcriptional activation (Fig. 5b; Eberharter and Becker 2002). PEPI-eGFP is thus responsive to modulation of histone acetylation, in the same way as endogenous sequences are.

ChIP analysis of pEPI-eGFP in CHO-K1 cells. a ChIP with an antibody against histone H3-core carried out on CHO-K1/ pEPI-eGFP cells cultured under standard conditions (open bars) and after TSA treatment (100 ng/ml, 48 h, gray bars). b ChIP with an antibody that recognizes various types of acetylated histone H3 on CHOK1/pEPI-eGFP cells cultured under standard conditions (open bars) and after TSA treatment (100 ng/ml, 48 h, gray bars). Results were corrected for histone H3-occupancy as described in (Haring et al. 2007). Error bars correspond to the standard error of three replicates
Episomal expression levels are controlled by various chromatin-mediated mechanisms
In addition to CHO-K1 cells, we also analyzed episomal expression in IMEFs that persistently carry pEPI-eGFP. We used the fraction of cells that express eGFP at a level that is detectable by light microscopy as a measure of episomal activity. Basal expression levels of pEPI-eGFP in IMEFs (Fig. 6a, control, all bars) were about 3-fold higher than observed for the CHO-K1 cells (Fig. 4a, control, open bar). The mouse cells were treated with the same chromatin-affecting drugs as the CHO-K1 cells in Fig. 4. In untreated IMEF cultures 18% of the cells showed eGFP fluorescence (Fig. 6a, control). After 96 h of treatment (Fig. 6a, black bars) this percentage increased to 99% in cells treated with 50 ng/ml TSA (Fig. 6a; TSA (50 ng/ml)) and to 49% after culturing in the presence of 25 μM 5-aza-dC (Fig. 6a; 5-aza-dC (25 μM)). Lower concentrations drugs and lower incubation times resulted in smaller effects on eGFP expression (Fig. 6). These results suggest that histone de-acetylation and DNA methylation contribute significantly to silencing of the episomal reporter gene. In the IMEF cells TSA-mediated blocking of histone acetylation is sufficient to induce eGFP expression in virtually all cells (Fig. 6a). Under the same conditions only 20% of the CHO-K1 cells become GFP-positive (Fig. 4a; TSA (100 ng/ml), open bar). Evidently, one or more unidentified inhibitory processes are (more) active in CHO-K1 cells, compared with IMEFs.

Chromatin manipulation in mouse embryonic fibroblasts. Expression of the episomal reporter gene eGFP in wild-type IMEFs (a) and Suv39h1 −/− mouse embryonic fibroblasts (b) in the presence of drugs that affect chromatin. Bars represent the percentage of eGFP-expressing cells 48 h (open bars), 72 h (light gray) and 96 h (dark gray) after beginning of drug treatment. Error bars, average ± standard error of the mean. *p < 0.05; **p < 0.01; ***p < 0.001 significance value in Suv39h1 deficient cells compared with wild-type IMEFs (two-tailed Student’s t test)
To analyze the role of inhibition of episomal gene expression by histone H3K9 methylation, the pEPI-eGFP episome was established in mutant mouse embryonic fibroblasts that do not express the enzyme HMT Suv39h1 (Peters et al. 2001). Our choice was motivated by the fact that, although Suv39h1 is prominently known for its role in methylating H3K9 in constitutive heterochromatin (Lehnertz et al. 2003), it has been shown that in IMEFs the activity of other HMTs depend on the presence of Suv39h1 (Fritsch et al. 2010). This includes the HMTs G9a and GLP, which are thought to be responsible for methylation of H3K9 in euchromatin (Tachibana et al. 2005). Choosing Suv39h1 −/− cells, which display a broad spectrum of H3K9 methylation defects (Fritsch et al. 2010), was thus the most likely to affect the episomal chromatin state. Indeed, these cells show an about 2-fold higher basal eGFP expression (Fig. 6b) than their parental wild-type cell line (Fig. 6a), suggesting that H3K9 methylation plays a role in repressing episomal gene expression in IMEFs. Increasing histone acetylation by inhibiting HDAC activity with TSA results in eGFP expression in close to 100% of the Suv39h1 −/− cells after 96 h (Fig. 6b; TSA (25 ng/ml) and TSA (50 ng/ml), dark gray bars), as was also observed for wild-type cells (Fig. 6a, TSA (25 ng/ml) and TSA (50 ng/ml), dark gray bars). Interestingly, the transcriptional stimulation by inhibition of DNA methyltransferase in the Suv39h1 −/− cells (Fig. 6b; 5-aza-dC (5 μM) and 5-aza-dC (25 μM)) is considerably stronger than in wild-type cells (Fig. 6a; 5-aza-dC (5 μM) and 5-aza-dC (25 μM)), irrespective of the treatment duration (48-, 72-, or 96-h treatments) resulting in eGFP expression in up to about 90% of the cells after 96 h (Fig. 6b; 5-aza-dC (25 μM), dark gray bar). This underscores that, in addition to histone H3K9 methylation and histone de-acetylation, DNA methylation also contributes to silencing of the episome and indicates that the episomal state of pEPI-eGFP and its derivatives does not exempt them from control by various cellular silencing processes.
Targeting of VP16 results in enhanced episomal gene expression
To specifically manipulate the chromatin state of the episome, we targeted the acidic activation domain of the transcriptional activator protein VP16 to the pELO64 episome in CHO-K1 cells, as a mCherry-lacR-VP16-acidic activation domain fusion protein (Fig. 1c). VP16 has been shown to enhance gene activity among others via increasing histone acetylation (Tumbar et al. 1999). Indeed, 48 h after transfection the percentage of eGFP expressing cells had increased 9.2-fold (Fig. 7a), without a change in the number of episomal spots per cell (Fig. 2c, gray bars). The somewhat increased eGFP fluorescence per spot (Fig. 2d, gray bars) compared with mCherry-lacR-targeted episomes (Fig. 2d, open bars) may be the result of enhanced accessibility of the lacO array due to the chromatin-modifying properties of VP16. The mean eGFP expression per cell increased 1.9-fold (Fig. 7a). Figure 7b shows the FACS diagrams on which the data in Fig. 7a are based. The expression of mCherry-lacR-VP16 always was somewhat lower than that of mCherry-lacR. Like the effect of TSA (Fig. 4b), targeting VP16 does not simply switch on the eGFP gene. Rather, cells with a wide range of expression levels are observed.

Manipulation of episomal chromatin in CHO-K1 cells. a Episome-specific chromatin modulation by targeting of mCherry-lacR-VP16-acidic domain to pELO64 (48 h after VP16 transfection). The percentage of eGFP-expressing cells measured by FACS analysis (open bars) and the mean fluorescence intensity per cell (gray bars, arbitrary units) are presented. b FACS data. Dots in the lower right region, representing eGFP-expressing cells, are quantified in (a). For all measurements at least 104 cells were analyzed. Control, CHO-K1/pELO64 cells transfected with mCherry-lacR in standard culture medium. mClacR refers to the mCherry-lacR plasmid, lacking VP16
Spatial distribution and mobility of the episome inside the nucleus
Transcriptionally highly active and less active chromosomes and sub-chromosomal domains have a different radial distribution in interphase nuclei, the former being on average closer to the nuclear center than the latter (Cremer and Cremer 2001; Foster and Bridger 2005; Goetze et al. 2007). To investigate whether this also holds for pELO64 episomes, we measured the 3D-radial nuclear position p n (p n = r 0/r n) of episomes, in which r 0 is the distance between a mCherry-labeled pELO64 episome spot and the geometrical center of the nucleus, and r n the distance between the center of the nucleus and the nuclear envelope measured through the episomal spot (Goetze et al. 2007). Figure 8 (open bars) shows that in a cell population in which only about 5% expresses eGFP, the radial distribution does not deviate significantly from what is expected from a random radial distribution in a scalene ellipsoid with radii a, b, and c (a and b being the equatorial radii and c the polar radius). The ratio between the radii a = 2b = 3c approximates the shape of CHO-K1 nuclei best. The distribution of the episomes significantly changed after they were transcriptionally activated by targeting mCherry-VP16-lacR, relocating the episomes on average towards the center of the nucleus (Fig. 8, gray bars; p < 0.01, non-parametric Mann–Whitney U test). Except for the radial distribution, no specific position of episomes inside the nucleus was observed. Others have shown that these episomes preferentially locate near early replication sites in the nucleus (Stehle et al. 2007), which are found in early S-phase throughout the nucleus.

Transcriptional activation induces relocation of episomes towards the nuclear center. The three-dimensional radial distribution of pELO64 in the interphase nucleus of CHO-K1 nuclei was measured by confocal microscopy. The nucleus was subdivided in ten concentric shells. Bars show the percentage of pELO64 dots in each shell in cells 48 h after transfection with mCherry-lacR (open bars, n = 95) and 48 h after transfection with mCherry-lacR-VP16-acidic domain (gray bars; n = 41). The line shows calculated values assuming a random radial distribution in a scalene ellipsoid with radii a, b, and c (a and b being the equatorial radii and c the polar radius). The ratio between the radii is: a = 2b = 3c.The distribution of the mCherry-lacR and mCherry-lacR-VP16 samples are significantly different (non-parametric Mann–Whitney U test; p < 0.01)
We have analyzed the mobility of the pELO64 episome in the G1 interphase nucleus of CHO cells to establish whether these distributions are related to rapid movement of the episomes or not. Figure 9 shows a time series of images (z-stack projections) over a period of 20 min, visualizing mCherry-tagged pELO64 episomes inside the interphase nucleus of a living CHO cell (full movie available as Electronic supplementary material (Movie 1)). Any movement that was observed was due to movement of (part of) the nucleus, rather than to the episome inside the nucleus. For instance, this is illustrated in Fig. 9 by the episome that moves due to an invagination of the nuclear envelope (arrowhead). Four independent experiments were done with the same result. Evidently, pELO64 episomes show little or no movement inside the interphase nucleus on a time scale of tens of minutes, which agrees with observations on plasmids microinjected in mammalian cells (Mearini et al. 2004).

Episomes are immobile in the nucleus at a time scale of tens of minutes. Z-stack projections of nuclei of a living CHO-K1/pELO64 cell acquired over a period of 20 min. The arrowhead indicates one of the three mCherry-labeled pELO64 episomes, which follows the movement of its nuclear environment. Scale bar = 5 μm
Discussion
Artificial S/MAR-containing episomal vectors, such as the pEPI-eGFP, have properties that make them promising for stable and safe gene transfer, including gene therapy, without integration into the host genome (Baiker et al. 2000; Jackson et al. 2006; Jenke et al. 2004b; Lufino et al. 2007; Manzini et al. 2006; Stehle et al. 2007). These episomes avoid the risk of insertional mutagenesis and the uncertainty of transgene expression due to position effects. Here we focus on the regulation of expression of episomal genes and chromatin state, using the pEPI-eGFP derived pELO64 vector (carrying a 64 tandem array of LacO sites) in two different mammalian cell lines as a model system. Our main conclusion is that the regulatory mechanisms that act on episomal genes seem to be similar to those that control host genes. In particular, trans-acting mechanisms that affect chromatin by changing histone modifications and DNA methylation levels play a role in episomal gene regulation.
In cultures of CHO-K1 cells and IMEFs only a fraction of the cells (5% to 24%, respectively) express detectable levels of the episomal eGFP reporter gene (Figs. 2, 3, 4, 5, and 6). Similar and higher values (22% up to 60%) have been reported for other mammalian host cell lines with pEPI episomes that do not contain LacO repeats (Nehlsen et al. 2006; Papapetrou et al. 2006), underscoring that pEPI expression levels are cell-type dependent. This indicates that the LacO array of the pELO episome has little or no effect on the expression of the reporter gene. Results discussed below suggest that cell-type-dependent effects may in part depend on the balance between different types of chromatin-affecting signals in the cell. It has been suggested that the CMV promoter sequence, which drives eGFP expression in pEPI-eGFP, is prone to epigenetic silencing in rodent cells (Brooks et al. 2004). However, when we established pEPI-eGFP persistence in the human colorectal carcinoma cell line HCT116, only 7% to 12% of the cells expressed eGFP to detectable levels (data not shown), indicating that the low reporter gene expression observed in CHO-K1 and IMEF cells is not rodent-specific.
The low number of eGFP-expressing cells is remarkable, since under G418 selection pressure each cell must contain one or more episomes that express the neomycin resistance gene. It may be argued that non-expressing cells lose the episome or the eGFP marker gene for instance by recombination or during mitosis. However, pEPI episomes have been shown to be persistent in mammalian cells for many cell cycles and during development (Stehle et al. 2007; Jenke et al. 2004b; Schaarschmidt et al. 2004; Baiker et al. 2000; Manzini et al. 2006). Moreover, we grow the cells in the presence of G418, which requires permanent presence of the drug resistance gene. Finally, the observation that after treatment with for instance TSA the fraction of expressing cells increases from about 20% to 99% shows that the marker gene is not lost, which is consistent with the histone deacetylation-induced silencing mechanism of pEPI-eGFP genes proposed by (Papapetrou et al. 2006). Altogether, this makes it very likely that the cells that do not express eGFP do carry the episome in a silenced state.
Expression of the pEPI-eGFP reporter gene is dependent on the chromatin state of the episome. It has been shown that pEPI chromatin undergoes the same modifications as host chromatin (Rupprecht and Lipps 2009; Stehle et al. 2007). Our results show that inhibition of HDAC activity affects the episomal chromatin by increasing the level of histone H3 acetylation similar to the host genome, i.e. it increases histone acetylation and activates gene expression (Figs. 4 and 5). FACS analyses show that the global induction of histone acetylation does not just switch on the eGFP reporter gene. Rather, it increases the average expression level, resulting in a wide range of expression levels (Fig. 4b). Interestingly, different elements of the episome are acetylated to a different extent after TSA treatment. Particularly the pCMV promoter and the polyA site become highly acetylated, probably directly related to increased transcriptional activation of the episome. In contrast to TSA and 5-aza-dC treatment, targeting of the lacO-mCherry-VP16 acidic domain chimeric protein to the lacO array is an episome-specific way of gene activation (Fig. 7). The acidic domain of VP16 is known to recruit histone acetyl-transferase activity (Tumbar et al. 1999). FACS analysis shows that, like the effect of TSA, VP16 targeting increases the average expression level of the eGFP gene, rather than just switching on the gene. The observed activation of eGFP expression by VP16 is another example showing that episomal genes behave similarly to host genes.
Global changes in histone H3K9 methylation also affect pEPI eGFP expression. Episomes in IMEF cells that are deficient in H3K9 methyl transferase are transcriptionally more active compared with the wild-type cell line, implying that H3K9 methylation has an inhibitory effect on episomal transcription (Fig. 6). Similarly, experiments using the DNA methyltransferase inhibitor 5-aza-dC show that DNA methylation suppresses episomal gene expression (Figs. 4 and 6). These effects may be direct by changing episomal chromatin or indirect via changing host properties. Although likely, it is not certain that pEPI episomes undergo DNA methylation and H3K9 methylation as literature on the subject is either contradictory (Argyros et al. 2008; Papapetrou et al. 2006; Jenke et al. 2004a) or lacks detail (Stehle et al. 2007). Interestingly, if mutant mouse cells that lack H3K9 methyltransferase were exposed to 5-aza-dC, their eGFP expression levels increased significantly more than after a similar treatment of wild-type IMEFs (Fig. 6), indicating synergy between the H3K9 methylation and DNA methylation systems (Fuks 2005).
We present microscopic evidence that cells stably contain between 1 and 3 spots that contain one or more episome molecules per interphase nucleus (Fig. 2). Earlier studies found similar numbers based on FISH labeling (Baiker et al. 2000; Nehlsen et al. 2006; Stehle et al. 2007). Our results confirm those studies using FISH on fixed cells and in vivo mCherry labeling, using the lacO/lacR method (Fig. 2; Belmont 2001). Remarkably, episomes are not motile at a time scale of tens of minutes and do not show a corralled type of local low amplitude movement, suggesting that they are firmly bound to host chromatin or other nuclear structures (Fig. 9 and Electronic supplementary material (Movie 1)). Evidence has been presented that episomes preferentially interact with early replication sites (Stehle et al. 2007), which are distributed throughout the nucleoplasm. This may be related to the finding that pEPI episomes are often attached to metaphase chromosomes in metaphase spreads (Baiker et al. 2000; Jenke et al. 2004b; Nehlsen et al. 2006; Stehle et al. 2007). Alternatively, immobilization could be due to binding to nucleoskeletal structures via the S/MAR sequence (Jenke et al. 2002). Despite this immobility, episome seem to reallocate to positions closer to the nuclear center if their gene expression is up-regulated by the targeting of mCherry-lacR-VP16 acidic domain (Fig. 8). This is similar to what is observed for genomic loci, i.e., transcriptionally highly active chromosomes and sub-chromosomal domains are located closer to the nuclear center than genomic areas that are less active (Cremer and Cremer 2001; Goetze et al. 2007; Lanctot et al. 2007).
In summary, our results seem to indicate that regulatory mechanisms affect episomal genes in a similar manner as host genes. This indicates that for application in gene therapy-specific epigenetic control elements must be incorporated in pEPI-like episomal vectors to assure reliable gene expression in a wide range of cell types and physiological conditions.
Abbreviations
- 5-aza-dC:
-
5-aza-2′-deoxycytidine
- CCD:
-
Charge-coupled device
- ChIP:
-
Chromatin immunoprecipitation
- CHO-K1:
-
Chinese hamster ovary K1
- CMV:
-
Cytomegalovirus
- DAPI:
-
4′, 6′-diamino-2-phenylindole
- DMT:
-
DNA methlytransferase
- eGFP:
-
Enhanced green fluorescence protein
- FBS:
-
Fetal bovine serum
- FISH:
-
Fluorescence in situ hybridization
- HCT116:
-
Human colorectal tumor 16
- HDAC:
-
Histone deacetylase
- HMT:
-
Histone methyltransferase
- HSV:
-
Herpes simplex virus
- IMEF:
-
Immortalized mouse embryonic fibroblast
- INF:
-
Interferon
- LacO/LacR:
-
Lac operator / Lac repressor
- MEL:
-
Mouse erythroleukemia
- NLS:
-
Nuclear localization signal
- S/MAR:
-
Scaffold/matrix attachment region
- Suv39h1:
-
Suppressor of variegation 3-9 h1
- TSA:
-
Trichostatin A
References
Argyros O, Wong SP, Niceta M, Waddington SN, Howe SJ, Coutelle C, Miller AD, Harbottle RP (2008) Persistent episomal transgene expression in liver following delivery of a scaffold/matrix attachment region containing non-viral vector. Gene Ther 15:1593–1605
Baiker A, Maercker C, Piechaczek C, Schmidt SB, Bode J, Benham C, Lipps HJ (2000) Mitotic stability of an episomal vector containing a human scaffold/matrix-attached region is provided by association with nuclear matrix. Nat Cell Biol 2:182–184
Belmont AS (2001) Visualizing chromosome dynamics with GFP. Trends Cell Biol 11:250–257
Brooks AR, Harkins RN, Wang P, Qian HS, Liu P, Rubanyi GM (2004) Transcriptional silencing is associated with extensive methylation of the CMV promoter following adenoviral gene delivery to muscle. J Gene Med 6:395–404
Chuang CH, Carpenter AE, Fuchsova B, Johnson T, De Lanerolle P, Belmont AS (2006) Long-range directional movement of an interphase chromosome site. Curr Biol 16:825–831
Conese M, Auriche C, Ascenzioni F (2004) Gene therapy progress and prospects: episomally maintained self-replicating systems. Gene Ther 11:1735–1741
Cremer T, Cremer C (2001) Chromosome territories, nuclear architecture and gene regulation in mammalian cells. Nat Rev Genet 2:292–301
Eberharter A, Becker PB (2002) Histone acetylation: a switch between repressive and permissive chromatin. Second in review series on chromatin dynamics. EMBO Rep 3:224–229
Emerman M, Temin HM (1984) Genes with promoters in retrovirus vectors can be independently suppressed by an epigenetic mechanism. Cell 39:449–467
Finlan LE, Sproul D, Thomson I, Boyle S, Kerr E, Perry P, Ylstra B, Chubb JR, Bickmore WA (2008) Recruitment to the nuclear periphery can alter expression of genes in human cells. PLoS Genet 4:e1000039
Foster HA, Bridger JM (2005) The genome and the nucleus: a marriage made by evolution. Genome organisation and nuclear architecture. Chromosoma 114:212–229
Fraser P, Bickmore W (2007) Nuclear organization of the genome and the potential for gene regulation. Nature 447:413–417
Fritsch L, Robin P, Mathieu JR, Souidi M, Hinaux H, Rougeulle C, Harel-Bellan A, Ameyar-Zazoua M, Ait-Si-Ali S (2010) A subset of the histone H3 lysine 9 methyltransferases Suv39h1, G9a, GLP, and SETDB1 participate in a multimeric complex. Mol Cell 37:46–56
Fuks F (2005) DNA methylation and histone modifications: teaming up to silence genes. Curr Opin Genet Dev 15:490–495
Glover DJ, Lipps HJ, Jans DA (2005) Towards safe, non-viral therapeutic gene expression in humans. Nat Rev Genet 6:299–311
Goetze S, Mateos-Langerak J, Gierman HJ, De Leeuw W, Giromus O, Indemans MH, Koster J, Ondrej V, Versteeg R, Van Driel R (2007) The three-dimensional structure of human interphase chromosomes is related to the transcriptome map. Mol Cell Biol 27:4475–4487
Hacein-Bey-Abina S, Von Kalle C, Schmidt M, Le Deist F, Wulffraat N, Mcintyre E, Radford I, Villeval JL, Fraser CC, Cavazzana-Calvo M, Fischer A (2003a) A serious adverse event after successful gene therapy for X-linked severe combined immunodeficiency. N Engl J Med 348:255–256
Hacein-Bey-Abina S, Von Kalle C, Schmidt M, Mccormack MP, Wulffraat N, Leboulch P, Lim A, Osborne CS, Pawliuk R, Morillon E, Sorensen R, Forster A, Fraser P, Cohen JI, De Saint Basile G, Alexander I, Wintergerst U, Frebourg T, Aurias A, Stoppa-Lyonnet D, Romana S, Radford-Weiss I, Gross F, Valensi F, Delabesse E, Macintyre E, Sigaux F, Soulier J, Leiva LE, Wissler M, Prinz C, Rabbitts TH, Le Deist F, Fischer A, Cavazzana-Calvo M (2003b) LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science 302:415–419
Haring M, Offermann S, Danker T, Horst I, Peterhansel C, Stam M (2007) Chromatin immunoprecipitation: optimization, quantitative analysis and data normalization. Plant Meth 3:11
Hatzis P, Van Der Flier LG, Van Driel MA, Guryev V, Nielsen F, Denissov S, Nijman IJ, Koster J, Santo EE, Welboren W, Versteeg R, Cuppen E, Van De Wetering M, Clevers H, Stunnenberg HG (2008) Genome-wide pattern of TCF7L2/TCF4 chromatin occupancy in colorectal cancer cells. Mol Cell Biol 28:2732–2744
Jackson DA, Juranek S, Lipps HJ (2006) Designing nonviral vectors for efficient gene transfer and long-term gene expression. Mol Ther 14:613–626
Jenke BH, Fetzer CP, Stehle IM, Jonsson F, Fackelmayer FO, Conradt H, Bode J, Lipps HJ (2002) An episomally replicating vector binds to the nuclear matrix protein SAF-A in vivo. EMBO Rep 3:349–354
Jenke AC, Scinteie MF, Stehle IM, Lipps HJ (2004a) Expression of a transgene encoded on a non-viral episomal vector is not subject to epigenetic silencing by cytosine methylation. Mol Biol Rep 31:85–90
Jenke AC, Stehle IM, Herrmann F, Eisenberger T, Baiker A, Bode J, Fackelmayer FO, Lipps HJ (2004b) Nuclear scaffold/matrix attached region modules linked to a transcription unit are sufficient for replication and maintenance of a mammalian episome. Proc Natl Acad Sci USA 101:11322–11327
Jones PA (1985) Altering gene expression with 5-azacytidine. Cell 40:485–486
Kumaran RI, Thakar R, Spector DL (2008) Chromatin dynamics and gene positioning. Cell 132:929–934
Lanctot C, Cheutin T, Cremer M, Cavalli G, Cremer T (2007) Dynamic genome architecture in the nuclear space: regulation of gene expression in three dimensions. Nat Rev Genet 8:104–115
Lehnertz B, Ueda Y, Derijck AA, Braunschweig U, Perez-Burgos L, Kubicek S, Chen T, Li E, Jenuwein T, Peters AH (2003) Suv39h-mediated histone H3 lysine 9 methylation directs DNA methylation to major satellite repeats at pericentric heterochromatin. Curr Biol 13:1192–1200
Li B, Carey M, Workman JL (2007) The role of chromatin during transcription. Cell 128:707–719
Lipps HJ, Jenke AC, Nehlsen K, Scinteie MF, Stehle IM, Bode J (2003) Chromosome-based vectors for gene therapy. Gene 304:23–33
Lufino MM, Manservigi R, Wade-Martins R (2007) An S/MAR-based infectious episomal genomic DNA expression vector provides long-term regulated functional complementation of LDLR deficiency. Nucleic Acids Res 35:e98
Manzini S, Vargiolu A, Stehle IM, Bacci ML, Cerrito MG, Giovannoni R, Zannoni A, Bianco MR, Forni M, Donini P, Papa M, Lipps HJ, Lavitrano M (2006) Genetically modified pigs produced with a nonviral episomal vector. Proc Natl Acad Sci USA 103:17672–17677
Mearini G, Nielsen PE, Fackelmayer FO (2004) Localization and dynamics of small circular DNA in live mammalian nuclei. Nucleic Acids Res 32:2642–2651
Nehlsen K, Broll S, Bode J (2006) Replicating minicircles: generation of nonviral episomes for the efficient modification of dividing cells—research article. Gene Ther Mol Biol 10B:233–243
Papapetrou EP, Ziros PG, Micheva ID, Zoumbos NC, Athanassiadou A (2006) Gene transfer into human hematopoietic progenitor cells with an episomal vector carrying an S/MAR element. Gene Ther 13:40–51
Peters AH, O’carroll D, Scherthan H, Mechtler K, Sauer S, Schofer C, Weipoltshammer K, Pagani M, Lachner M, Kohlmaier A, Opravil S, Doyle M, Sibilia M, Jenuwein T (2001) Loss of the Suv39h histone methyltransferases impairs mammalian heterochromatin and genome stability. Cell 107:323–337
Piechaczek C, Fetzer C, Baiker A, Bode J, Lipps HJ (1999) A vector based on the SV40 origin of replication and chromosomal S/MARs replicates episomally in CHO cells. Nucleic Acids Res 27:426–428
Robinett CC, Straight A, Li G, Willhelm C, Sudlow G, Murray A, Belmont AS (1996) In vivo localization of DNA sequences and visualization of large-scale chromatin organization using lac operator/repressor recognition. J Cell Biol 135:1685–1700
Rupprecht S, Lipps HJ (2009) Cell cycle dependent histone dynamics of an episomal non-viral vector. Gene 439:95–101
Schaarschmidt D, Baltin J, Stehle IM, Lipps HJ, Knippers R (2004) An episomal mammalian replicon: sequence-independent binding of the origin recognition complex. EMBO J 23:191–201
Schones DE, Cui K, Cuddapah S, Roh TY, Barski A, Wang Z, Wei G, Zhao K (2008) Dynamic regulation of nucleosome positioning in the human genome. Cell 132:887–898
Shaner NC, Campbell RE, Steinbach PA, Giepmans BN, Palmer AE, Tsien RY (2004) Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat Biotechnol 22:1567–1572
Stehle IM, Scinteie MF, Baiker A, Jenke AC, Lipps HJ (2003) Exploiting a minimal system to study the epigenetic control of DNA replication: the interplay between transcription and replication. Chromosome Res 11:413–421
Stehle IM, Postberg J, Rupprecht S, Cremer T, Jackson DA, Lipps HJ (2007) Establishment and mitotic stability of an extra-chromosomal mammalian replicon. BMC Cell Biol 8:33
Tachibana M, Ueda J, Fukuda M, Takeda N, Ohta T, Iwanari H, Sakihama T, Kodama T, Hamakubo T, Shinkai Y (2005) Histone methyltransferases G9a and GLP form heteromeric complexes and are both crucial for methylation of euchromatin at H3-K9. Genes Dev 19:815–826
Tumbar T, Belmont AS (2001) Interphase movements of a DNA chromosome region modulated by VP16 transcriptional activator. Nat Cell Biol 3:134–139
Tumbar T, Sudlow G, Belmont AS (1999) Large-scale chromatin unfolding and remodeling induced by VP16 acidic activation domain. J Cell Biol 145:1341–1354
Van Craenenbroeck K, Vanhoenacker P, Haegeman G (2000) Episomal vectors for gene expression in mammalian cells. Eur J Biochem 267:5665–5678
Verschure PJ, Van Der Kraan I, De Leeuw W, Van Der Vlag J, Carpenter AE, Belmont AS, Van Driel R (2005) In vivo HP1 targeting causes large-scale chromatin condensation and enhanced histone lysine methylation. Mol Cell Biol 25:4552–4564
Yoshida M, Kijima M, Akita M, Beppu T (1990) Potent and specific inhibition of mammalian histone deacetylase both in vivo and in vitro by trichostatin A. J Biol Chem 265:17174–17179
Zhao R, Bodnar MS, Spector DL (2009) Nuclear neighborhoods and gene expression. Curr Opin Genet Dev 19:172–179
Acknowledgments
The authors wish to thank Wim de Leeuw (University of Amsterdam, The Netherlands) for his help with image analysis and use of the Argos software, Pantelis Hatzis and Jurian Schuijers (Hubrecht Institute, Utrecht, The Netherlands) for help and advice with the ChIP and qPCR protocols, Berend Hooibrink (FACS facility, AMC, Amsterdam, The Netherlands) for the FACS analysis, Roger Tsien (UC San Diego, CA, USA) for the kind gift of the mCherry gene, Thomas Jenuwein (Research Institute of Molecular Pathology, Vienna, Austria) for the Suv39h1 −/− mouse embryonic fibroblasts and Hanil Quirindongo and Diewertje Piebes (University of Amsterdam) for their assistance in designing and making the lacR fusion proteins. This work was supported by the European Commission as part of the EPI-Vector program, contract LSH-2003-1.2.4-7.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible Editor: Wendy Bickmore.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Movie 1
(AVI 3202 kb)
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Tessadori, F., Zeng, K., Manders, E. et al. Stable S/MAR-based episomal vectors are regulated at the chromatin level. Chromosome Res 18, 757–775 (2010). https://doi.org/10.1007/s10577-010-9165-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10577-010-9165-4