
Abstract
Pluripotent embryonic stem cells (ESCs) are able to differentiate into all cell types in the organism including cortical neurons. To follow the dynamic generation of progenitors of the dorsal forebrain in vitro, we generated ESCs from D6-GFP mice in which GFP marks neocortical progenitors and neurons after embryonic day (E) 10.5. We used several cell culture protocols for differentiation of ESCs into progenitors and neurons of the dorsal forebrain. In cell culture, GFP-positive cells were induced under differentiation conditions in quickly formed embryoid bodies (qEBs) after 10–12 day incubation. Activation of Wnt signaling during ESC differentiation further stimulated generation of D6-GFP-positive cortical cells. In contrast, differentiation protocols using normal embryoid bodies (nEBs) yielded only a few D6-GFP-positive cells. Gene expression analysis revealed that multiple components of the canonical Wnt signaling pathway were expressed during the development of embryoid bodies. As shown by immunohistochemistry and quantitative qRT-PCR, D6-GFP-positive cells from qEBs expressed genes that are characteristic for the dorsal forebrain such as Pax6, Dach1, Tbr1, Tbr2, or Sox5. qEBs culture allowed the formation of a D6-GFP positive pseudo-polarized neuroepithelium with the characteristic presence of N-cadherin at the apical pole resembling the structure of the developing neocortex.
Similar content being viewed by others


Introduction
The formation of the neocortex (corticogenesis) starts at embryonic day (E) 10 from undifferentiated neural progenitors of the dorsal telencephalon. Proliferating progenitors reside in the ventricular and subventricular zone (VZ and SVZ) while newly generated neurons migrate from the germinal VZ towards the outer margin. At birth, the cortical plate consists of six layers that were generated by inside-first and outside-last succession, i.e., early-born neurons settle in the deepest layer while late-born neurons migrate along radial glial cells towards the superficial layers (Rallu et al. 2002; Wilson and Houart 2004; Fishell and Kriegstein 2005; Gotz and Huttner 2005; Rash and Grove 2006). Genetic mechanisms that control the regionalization of the telencephalon have been partly elucidated showing the involvement of the regulatory pathways Wnt, Bmp, Fgf and Shh (Shimogori et al. 2004).
Wnt signaling is important for both cell specification and cell proliferation during development of neural tissue. At early developmental stages, the expression gradient of Wnts in the neural tube increasing from anterior to posterior is capable of inducing a caudal character, i.e., the midbrain and hindbrain (Wilson and Edlund 2001; Nordstrom et al. 2002). In accordance, inhibition of Wnt signaling at early developmental stages is required for the specification of an anterior character in the forebrain (Houart et al. 2002; Lagutin et al. 2003). At later developmental stages, experiments with chicken tissue explants and conditional β-catenin knock-out mice showed that canonical Wnt signaling determines dorsal telencephalic character at the expense of the ventral telencephalon (Gunhaga et al. 2003; Backman et al. 2005). Furthermore, Wnt-3a controls cell proliferation in the hippocampal primordium (Lee et al. 2000b). At the level of transcription control, two nuclear factors, Pax6 and Emx2, that are expressed in opposing gradients along the anterior-posterior axis play a crucial role in the specification of the cortex (Muzio et al. 2002).
The developmental transition of undifferentiated cells in the early embryo into committed neurons has been partially mimicked by in vitro differentiation of embryonic stem cells (ESCs). Lee et al. (2000a) cultured ESCs to form embryoid bodies that contained multi-potent cells present in three germ layers. From this poorly defined pool of tissue-specific cells, neural stem cells (NSCs) can be expanded in the presence of EGF and FGF2 and their subsequent exposure to Shh with Fgf8b triggers differentiation into neurons of midbrain/hindbrain character including dopaminergic neurons. Differentiation of ESCs into neural precursors in adherent culture yielded a remarkably high number of neural precursors labeled with Sox1-GFP without the requirement of exogenous factors (Ying et al. 2003) suggesting that neural fate may be a default differentiation program for pluripotent stem cells (Munoz-Sanjuan and Brivanlou 2002; Levine and Brivanlou 2007). Naïve neural cells which arise from differentiating ESCs can respond to inductive signals that can further influence their fate decisions. For instance, inhibition of canonical Wnt signaling with the antagonist Dkk1 promoted telencephalic character by suppressing caudal fate in neural cells (Watanabe et al. 2005). In addition, the same authors showed that subsequent treatment with recombinant Wnt3a increased the proportion of cortex-specific neurons while Shh induced the expression of markers typical of the basal (ventral) telencephalon. In agreement with these data, inhibition of the Shh pathway with cyclopamine remarkably increased the expression of dorsal markers such as Pax6 or Vglut in cultured neuronal cells that were generated by controlled differentiation of ESCs (Gaspard et al. 2008, 2009). The role of Wnts in ESC differentiation towards neurons appears more complex because Wnt1 and Lrp6 are dispensable for generation of dopaminergic neurons and their absence rather elevates their numbers (Cajanek et al. 2009). Interestingly, in vitro differentiated neurons do not only show cell fate specific markers but also resemble the cellular structure of mature organs. Eiraku et al. (2008) showed that differentiating neural cells self-arranged in a manner that resembled the developing cortex with an apical neuroepithelium and a basal margin when they were cultured in tight contact within large cell aggregates.
We previously created transgenic reporter mice D6-GFP where GFP was specifically expressed in cortical progenitor cells from E10.5 onwards and in mature neuron descendant from these cells in the adult hippocampus and cortex (Machon et al. 2002). Thus D6-GFP serves as a highly specific marker for neural progenitors and neurons that originate in the dorsal telencephalon and that are predominantly excitatory glutamatergic. In this study, we used ES cells derived from transgenic mice containing the D6-GFP marker to directly observe differentiation of ESCs into cortical neurons in tissue culture. GFP+ cortical neuronal progenitors were efficiently generated in vitro as clusters within qEBs. In contrast normal embryoid bodies (nEBs) yielded GFP+ cortical neuronal progenitors only with an extremely low efficiency. Transcription factors that are specific for the dorsal forebrain were activated in D6-GFP+ cells, further confirming correct fate determination. After cell sorting, GFP+ cells derived from qEBs formed neurospheres in cell culture that were indistinguishable from normal neural stem cells derived from the developing telencephalon. Finally, additional stimulation of the canonical Wnt signaling pathway during the later period of ESC differentiation increased the percentage of D6-GFP+ cells but was not essential for their appearance. These results suggest that the in vitro generation of cells with a specific dorsal forebrain fate can be directed by signals that are present within qEBs while increased stimulation of canonical Wnt signaling will further enhance dorsal fate specification in qEBs.
Materials and Methods
ESC Culture
ESCs were derived from the inner cell mass of the blastocyst of transgenic mice carrying a D6-driven GFP reporter (Machon et al. 2002). ESCs were expanded on feeder cells (primary mouse embryonic fibroblasts) and were subsequently adapted to feeder-free culture conditions on 0.1% gelatin-coated plastic. The ESC medium consisted of knock-out Dulbecco’s Modified Eagle Medium (DMEM, Invitrogen) supplemented with 10% ES-grade fetal calf serum (FCS, Speciality Media), ESGRO (1000 U/ml) (Millipore), non-essential amino acids (NEAA, Invitrogen), 0.1 mM 2-mercaptoethanol (2-ME, Sigma), 2 mM l-glutamine (Invitrogen), and 100 unit/ml penicillin/streptomycin (Invitrogen). Cells were plated at the density of 0.5 × 105 per ml and were passaged before reaching confluence.
Differentiation of ESCs
All cell culture protocols are schematically illustrated in Fig. 4a.
For nEBs culture, adherent ESCs were dissociated in trypsin–EDTA and plated in uncoated 100-mm plastic dishes at a density of 500,000 ESCs per 10 ml EB media. This day was set as the differentiation day 0 (d0). For qEBs culture, dissociated ESCs were grown in 96-well plates with a round bottom at the density of 2000 cells per well in the EB medium (qEBs culture 1, see Fig. 4a) or in the Neuro-medium from d0 (qEBs culture 2). qEBs were cultured in round-bottom wells for the whole culture period without adhering to the bottom and the medium was changed every third day. The EB medium was composed of GMEM (Invitrogen) with 5% knock-out serum replacement (KSR, Invitrogen); NEAA (Invitrogen), 0.1 mM 2-mercaptoethanol (2-ME, Sigma) and 2 mM l-glutamine (Invitrogen). Neuro-medium consisted of the Neurobasal-A medium (Invitrogen) supplemented with B27 (Invitrogen), l-glutamine, penicillin/streptomycin and growth factors bFGF 10 ng/ml, EGF 20 ng/ml (R&D Systems). For additional treatment, Wnt3a was applied to the EB medium at 10 ng/ml (qEBs culture 3), Dkk1 50 ng/ml (qEBs culture 4) between d6 and d12. 0.5 μM retinoid acid (RA) was applied from d5 to d9 in the EB medium (qEBs culture 5). In growth assays, diameters of qEBs were measured from d1 to d20. The images of growing clusters were taken every day and their diameter was set as a function of the pixel size using Zeiss Axiovision software.
Immunofluorescence
To prepare samples for staining, ESCs were plated onto gelatin-coated round-shape coverslips and incubated for 2 days in the ESC medium. The whole nEBs were placed on coverslips coated with retronectin (AH Diagnostics) in the EB medium. NSCs from neurosphere cultures of FACS-sorted qEBs were attached on poly-l-lysine (Sigma)-coated coverslips and incubated for 24 h in Neuro-medium without EGF. For cryosections, qEBs were collected and fixed in suspension in 4% paraformaldehyde in PBS and dehydrated overnight in 30% sucrose in PBS. Subsequently, qEBs were embedded in OCT (Sakura) and 8 μm cryosections were prepared using a cryostat (Leica).
For immunostaining, cells were fixed in 4% paraformaldehyde for 30 min. Specimens were permeabilized in 0.1% TritonX-100 in PBS for 15 min. After saturation in 5% goat serum, 5% bovine serum albumin (BSA, Invitrogen), 0.1% TritonX-100 in PBS for 30 min, cells were incubated overnight at 4°C with a primary antibody diluted in PBS containing 0.5% goat serum, 0.5% BSA, and 0.1% TritonX-100. After washing with PBS, incubation with a secondary antibody followed for 1 h at room temperature in the same buffer. 1 μg/ml 4,6-diamidino-2-phenylindol (DAPI, Roche) in PBS was used to visualize cell nuclei.
Primary antibodies: rabbit anti-Pax6 (1:200) (Covance), rabbit anti-Tbr1 (1:200) (Chemicon), rabbit anti-Tbr2 (1:200) (Chemicon), mouse anti-N-cadherin (1:400) (BD Biosciences), rabbit anti-Oct4 (1:100) (Santa Cruz, rabbit anti-Sox1 (1:200) (Chemicon), rabbit anti-Sox2 (1:200) (Cell Signaling), mouse anti-Nestin (1:400) (Millipore), mouse anti-Tuj1 (1:400) (R&D), mouse anti-GFAP (1:400) (Sigma). Secondary antibodies: anti-mouse or anti-rabbit IgG conjugated with Alexa 488/594 (1:400) (Invitrogen). Stained sections were examined using a fluorescent microscope Axioskop2 (Zeiss) and images were taken with Axiovision software (Zeiss).
Quantitative qRT-PCR
RNA was extracted using a total RNA isolation kit (Sigma). cDNA was synthesized from 1 μg RNA using Oligo primer p(NT)6 (Roche) and reverse transcriptase kit (Stratagene) according to manufacturer’s instructions. Real-time polymerase chain reaction (qRT-PCR) was performed using the SYBR green PCR master mix kit (Stratagene). Amplification: initial denaturation for 5 min at 95°C, then 40 cycles: denaturation 95°C 30 s, annealing 55°C 40 s, elongation 72°C 30 s. Primer sequences for genes are listed in Supplementary Table 1. Statistical analysis: The values show relative values of expression that was normalized to GAPDH as the average of three independent with standard deviations.
Flow Cytometry Analysis and FACS Sorting
For SSEA-1 labeling, dissociated ESCs were suspended in PBS containing 0.2% BSA with mouse anti-SSEA-1 antibody (1:200, Millipore) and incubated on ice for 30 min. Anti-mouse IgM-Alexa594 in the same buffer was applied for another 30 min after washing. Cells were analyzed by flow cytometry after washing in PBS.
qEBs (with or without D6-GFP+ cell clusters) were dissociated with trypsin/EDTA at 37°C, and trypsinization was stopped using trypsin neutralizer solution (Gibco). Single cells thus obtained were washed and re-suspended in PBS. Cells were immediately measured using a FACS Aria flow cytometer (BD Biosciences). Both GFP− and GFP+ cells were collected and data were analyzed with FACSDiva software.
Neurosphere Culture and Differentiation
GFP− and GFP+ cells were isolated by FACS, resuspended in Neuro-medium containing bFGF and EGF, and plated at a density of 40,000 cells per ml media. Neurospheres formed within 1–2 weeks after FACS and were passaged further using trypsinization. For differentiation, dissociated neurosphere cells were plated onto poly-l-lysine-coated coverslips in Neuro-medium without EGF (seeding density 40,000 cells per cm2). After 24 h, differentiated cells were fixed in 4% paraformaldehyde and stained using immunofluorescence as described above.
Results
Neural Differentiation of ESCs in nEBs
To study differentiation of ESCs into cortex-specific neural cells, we generated ESCs from blastocysts derived from transgenic D6-GFP mice containing a GFP reporter that is driven by the forebrain-specific enhancer of the Dach1 gene D6 (Machon et al. 2002). During embryogenesis, D6 enhancer activity was detectable from embryonic day (E) 10.5 and expanded throughout the developing neocortex and the hippocampus by E12.5 and E15 (Fig. 1a–b). Immunohistochemistry with antibodies against several cortex-specific transcription factors was performed on tissue sections from E15 D6-GFP transgenic mice. An anti-Pax6 antibody was used to visualize neural progenitors along radial columns in the VZ and an anti-Tbr2 antibody was used for intermediate progenitors in the SVZ and the intermediate zone (IZ) (Englund et al. 2005). Figure 1c and d shows an overlapping expression of Pax6 or Tbr2 with D6-GFP. Pax6 in the VZ and Tbr2 in the SVZ were co-labeled with the D6-GFP marker, which is, apart from the VZ and SVZ, also expressed in cortical postmitotic neurons (the outer layer of the cortex). D6-GFP therefore marks both neocortical VZ and SVZ progenitors and mature neurons in the cortical layers at critical stages of neurogenesis. This implies that ESCs derived from D6-GFP transgenic mice provide a simple and explicit model for in vitro cell differentiation into embryonic cortical neurons.

Characterization of D6-GFP+ cells in the developing dorsal forebrain. a D6-GFP mouse embryo at E12.5 showing D6 enhancer activity exclusively in the developing dorsal forebrain. b Dorsal view of the E15 D6-GFP+ brain with a strong fluorescence in the cortical hemispheres (Cx). c–d Coronal sections of the dorsal forebrain from D6-GFP embryos at E15 showing D6-GFP+ cells throughout the neocortical layers. Immunofluorescent staining (red) of Pax6 (c′) or Tbr2 (d′) demarcates VZ and SVZ, which overlaps with D6-GFP+ cells (green) as shown in merged images (c″, d″). e Immunostaining of N-cadherin (N-cad, green) in the apical pole of neuroepithelial cells (adherens junctions, arrows) in the dorsal forebrain. A higher magnification of the cortical wall corresponding to the white rectangle in c
D6-GFP ESCs were maintained on a gelatin-coated surface in the ESC medium containing ESGRO. These ESCs showed the typical morphology and growth properties of pluripotent cells (Supplementary Fig. 1a). Immunofluorescence with antibodies recognizing the pluripotency markers Oct4 and Sox2 documented that D6-GFP ESCs strongly expressed these transcription factors (Supplementary Fig. 1c–d′). Further, FACS quantification of the SSEA-1 surface marker, which is specific for mouse ESCs, showed a high percentage of SSEA-1 positive cells in the D6-GFP derived ESCs culture (Supplementary Fig. 1b). In contrast, the early neuroectodermal marker Sox1 and the neural progenitor marker Nestin were not detected (Supplementary Fig. 1e–f′). In conclusion, ESCs derived from D6-GFP blastocysts displayed standard characteristics of pluripotency.
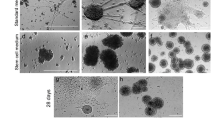
To test whether D6-GFP-derived ESCs could be differentiated into neurons of the cortex we tested several cell culture strategies. At first, we used a protocol based on generation of nEBs. At day 0 (d0), ESCs were dissociated with trypsin and seeded in uncoated plastic dishes in the EB medium (nEBs culture, see Fig. 4a). The absence of ESGRO, replacement of FCS with KSR, and the growth in suspension triggered cell differentiation. Floating cells formed middle-size aggregate nEBs (Fig. 3a–b). To examine whether a differentiation towards a neural lineage occurred, fluorescent immunohistochemistry of nEBs was performed after 12 days of incubation. As shown in Fig. 2a, many cells in D6-GFP ESCs derived nEBs were positive for the markers for neuronal precursors Nestin, the marker for differentiated neurons Tuj1 and for the astrocyte-specific protein GFAP, demonstrating a neural identity of many cells within nEBs.

Differentiation of ESCs in normal embryoid bodies (nEBs) initiates neural-specific gene expression. a Immunofluorescence of nEBs at day 12 (d12) expressing markers of neural progenitor cells (Nestin), mature neurons (Tuj1), astrocytes (GFAP), and D6-GFP (objective ×10). b Quantitative qRT-PCR of RNA from parental ESCs, nEBs at day 6 (nEB6d), nEBs at day 12 (nEB12d), and neural stem cells (NSCs) cultured as neurospheres. Pluripotency markers Oct4, Nanog, and Klf4 disappeared by day 12 while neural markers Sox1 and Nestin were strongly upregulated. The marker for the anterior neural tissue FoxG1 and cortex-specific markers Pax6, Sox5, and Dach1 were also remarkably increased during days 6–12. Markers Nkx2.1, Hoxb9, and GFAP indicated the presence of neural cells in general while Brachyury (Bra) and Vimentin indicated non-neural tissue. c Upregulation of components of the Wnt canonical pathway in nEBs. Relative values show values of expression that were normalized to GAPDH using the average of three independent experiments with standard deviations
To track the differentiation process in nEBs, qRT-PCR on mRNA obtained from days 6 and 12 of differentiation was carried out and the expression profile was compared to the parental ESCs. As a control, mRNA from E13 mouse cortex derived neurospheres was used. Our results showed that cells grown under conditions to form nEBs lost their pluripotency character between d6 and d12 as reflected by the reduced expression of Oct4, Nanog, and Klf4 (Fig. 2b). Sox2, which was highly expressed in ESCs, decreased during the first days of differentiation but its expression almost recovered at d12. As Sox2 expression is abundant both in ESCs and in neural progenitors (Zappone et al. 2000), the temporary decrease in the expression of Sox2 may reflect a spontaneous differentiation into multiple cell types at earlier stages of differentiation while the neural fate starts to dominate later. Further, the decrease of pluripotency was accompanied with an increase of markers for neuroectoderm, Nestin and Sox1 (Fig. 2b), which is in line with other reports (Pevny and Nicolis 2010). We also measured remarkably increased expression the forebrain-specific gene FoxG1 (Ahlgren et al. 2003; Hanashima et al. 2002; Wataya et al. 2008); and genes expressed in the developing the cortex such as Pax6 (Bishop et al. 2000; Englund et al. 2005) and Sox5 (Kwan et al. 2008; Lai et al. 2008; Hanashima et al. 2002) (Fig. 2b).
In addition, nEBs mRNA analysis showed an upregulation of several other neural genes such as the ventral forebrain marker Nkx2.1 (Sussel et al. 1999), the hindbrain marker Hoxb9 (Irioka et al. 2005), and the astrocyte-specific marker GFAP indicated a presence non-cortical cell types (Hanashima et al. 2002). The elevation of Brachyury (Bra) and Vimentin suggested a presence of non-neural cell identity. Altogether, qRT-PCR data reflected an expected cell heterogeneity in nEBs that includes dorsal, ventral, anterior, and posterior cell types of the neural tissue and other cell types. All the genes related to neural character that were tested in nEBs approached levels of expression close to the levels obtained from NSCs that were directly derived from E13 mouse forebrains.
Along with induction of cell-type specific genes in nEBs, genetic components of the Wnt pathway were activated and showed dynamic changes in their expression (Fig. 2c). For instance, Wnt1, Dkk1, and sFrp2 were induced upon ESC differentiation whereas Wnt3a, which was expressed in pluripotent ESCs, was downregulated in nEBs. Induction of components of the canonical Wnt signaling during differentiation led to activation of Axin2 (Fig. 2c), a well-known Wnt target gene (Fujimura et al. 2007; Jho et al. 2002), indicating activation of the canonical Wnt pathway in spontaneously differentiating cells.
During more than a 12-day culture period, cells in nEBs were monitored for the expression of GFP that would indicate differentiation of ESCs into neural cells of the neocortical fate. Although the Dach1 mRNA was induced in 12d nEBs (Fig. 2b), live D6-GFP signal was barely visible. Only a few GFP-positive cells were found in selected nEBs, and immunostaining with an anti-GFP antibody was required to visualize those cells (Fig. 2a, right). In conclusion, an ESC differentiation protocol using nEBs was prone to generate neural tissue but the telencephalic/neocortical character as marked by the D6-GFP reporter was established with low efficiency.
Forebrain-Specific D6-GFP+ Cells were Generated in qEBs
To find culture conditions that would yield neural cells of the dorsal forebrain with high efficiency, we adapted a culture protocol from Eiraku et al. (2008). In short, dissociated ESCs were allocated in round-bottom 96-well plates where they aggregated in large clusters within 1 day. These aggregates are called qEBs (Fig. 3c–d). Within qEBs as well as in nEBs, cell differentiation is thought to be a stochastic process that is influenced by a close cell–cell contact.

Differentiation of ESCs into neocortical progenitor cells in quick embryoid bodies (qEBs). EB size comparison during ESC differentiation: a Normal EBs after 2 days (nEBs 2d). b Normal EBs after 12 days (nEBs 12d). c Quick EBs after 2 days (qEBs 2d). d Quick EBs after 12 days (qEBs 12d). e–h qEBs after 12 days with corresponding D6-GFP fluorescence in right panels (f, h). Scale bar: 100 μm
In contrast to nEBs in which few D6-GFP+ cells were found during the culture period, qEBs culture yielded frequently fluorescent cells (Fig. 3e–h). Commonly, one or two clusters of D6-GFP+ cells were present within one qEB. Images of several D6-GFP+ qEBs are shown in Supplementary Fig. 2 with several clusters of fluorescent cells within a qEB.
Two different culture medias were tested for the frequency of D6-GFP+ induction in qEBs: (i) EB medium containing knock-out serum replacement (KSR) and (ii) Neuro-medium which was serum-free but contained EGF and bFGF. The size of qEB was significantly larger in Neuro-medium (up to 700 μm in diameter) at day 18 whereas those in EB media were smaller (around 400 μm in diameter) (Fig. 4b). Clusters from 8 parallel wells were measured every day during a 20-day incubation period.

Quantification of the generation of D6-GFP neuronal progenitors in different media. a A schematic illustration of the cell culture protocols that were used in this work. b A growth curve of qEB during 20 days (d1–d20). qEBs were incubated in EB medium or in Neuro-medium, and the size of qEBs was measured daily for 20 days (diameter in μm). On d18, clusters stopped growing. The average diameter of qEBs in Neuro-medium was 800 μm while only half the size was reached in the EB medium. c Incidence of GFP+ qEBs shows higher percentage of fluorescent clusters in Neuro-medium (%) compared to EB medium (40%). d Flow cytometry analysis showing a relatively lower percentage of D6-GFP+ cells in qEBs grown in Neuro-medium (19.6%) compared to qEBs grown in EB medium (35%). n number of independent experiments
Generally, the first D6-GFP+ cells in qEBs emerged around d10 to d12, in rare cases as early as at d7. After 18 days, no further clusters with D6-GFP+ cells emerged although the GFP signal persisted for at least another week. The incidence of D6-GFP-positive clusters (regarded those containing at least one fluorescent cell) was higher in Neuro-medium. On average 75% qEBs contained D6-GFP-positive cells in 2 independent experiments (n = 2). On the other hand in EB medium, on average 40% qEBs contained positive cells in six experiments (n = 6) (Fig. 4c). Nevertheless, the relative number of GFP+ cells in pooled qEBs was lower in Neuro-medium and fluorescence was relatively weaker. The peak of the GFP intensity in the FACS histograms was around 104 in Neuro-medium and 5 × 104 in EB medium (Fig. 4d). This indicates that EB medium is more suitable for generation of D6-GFP+ cells in qEBs while Neuro-medium, containing the growth factors EGF and bFGF, may support growth of noncortical neuronal types at the expense of the cortex-specific ones.
ESC-Derived D6-GFP+ Cells Grown in qEBs Expressed Typical Markers of Neural Progenitors and Neurons of the Dorsal Telencelphalon
To further characterize qEBs-derived D6-GFP+ cells, the presence of markers that are specifically activated in the developing neocortex was examined by immunofluorescence (Englund et al. 2005). Cryosections of qEBs containing clusters of D6-GFP+ cells were analyzed with antibodies against Sox2, Pax6, Tbr1, and Tbr2. As shown in Fig. 5a–d, a substantial number of D6-GFP+ cells expressed the markers Sox2, Pax6, Tbr2, and Tbr1, further confirming the telencephalic/neocortical character of the D6-GFP cells.

Characterization of D6-GFP+ cells in cryosections of qEBs by immunofluorescence. a–d Immunofluorescence of D6-GFP+ cell clusters in qEBs. a′–d′ Immunostaining of the markers Sox2, Pax6, Tbr2, Tbr1 on the same sections. a″–d″ A higher magnification of merged images shows overlapping expression of D6-GFP and differentiation markers, indicated by rectangles in a–d. Sox2+ and Pax6+ cells are scattered within the GFP+ cluster but Tbr2+ and Tbr1+ cells are organized in radial layers around a presumptive center (arrows in c′ and d′). Scale bars 100 μm in a–d and a′–d′ and 50 μm in a″–d″ panels. e–e″ Immunostaining of N-cadherin (Ncad, red) with D6-GFP showing a neuroepithelial organization of qEB with N-cadherin in the apical pole of the rosette (arrows in e′–e″). f–f″ Immunostaining of N-cadherin (Ncad, red) with Pax6 (green) showing a rosette with neuroepithelial structure with apical N-cadherin and basal Pax6
Next, we isolated mRNA from GFP+ and GFP− cells and real-time qRT-PCR analysis of other genes that are specifically expressed in the telencephalon was performed. Some qEB contained up to one third of fluorescent cells as quantified by FACS analysis (Fig. 6a). However, viable cells were not easily collected by FACS and mRNA was isolated instead from manually separated fluorescent cells under the UV-light microscope. Results in Fig. 6b confirmed that FoxG1, Dach1, Pax6, and Sox5 showed increased expression in D6-GFP+ cells.

D6-GFP+ cell quantification and gene expression analysis. a Flow cytometry analysis of qEBs without (left, negative control) and with D6-GFP+ cells (right). Selected samples from qEBs at d15–d18 show that up to one third of all cells are D6-GFP+. b Quantitative qRT-PCR of RNA from D6-GFP negative cells (gray columns) and positive cells (green columns) showing up-regulation of genes that are characteristic for the forebrain (FoxG1), or dorsal forebrain (Dach1, Pax6, Sox5)
The cortical neuroepithelium in the mouse embryo is polarized, i.e., cells at the apical pole of the ventricular zone express N-cadherin that is concentrated in adherens junctions (Fig. 1e). N-cadherin is essential for the proper function of the adherens junctions since loss-of-function mutant mice exhibit a disrupted neuroepithelial polar structure (Kadowaki et al. 2007). Labeling of qEBs with N-cadherin revealed that some clusters self-assembled in rosette-like structures. In these structures, D6-GFP+ cells surrounded a cavity with a central circle of apical N-cadherin which was similar to the organization of the neuroepithelium with adherens junctions (Fig. 5e). Clusters of Pax6+ cells around a central N-cadherin-positive rosette showed an arrangement similar to the developing neocortical wall (Fig. 5f) indicating that differentiating cells within qEBs may be able to establish a cellular architecture reminiscent of the developing neocortex.
Wnt Signaling Pathway Enhances the Efficiency of Corticogenesis In Vitro
Wnt signaling plays an important role in embryonic patterning of the telencephalon, since it promotes dorsal telencephalic identity while suppressing ventral fate (Backman et al. 2005; Guillemot 2007). We noticed that during the differentiation of ESCs in nEBs many genes of the canonical Wnt signaling pathway were induced (Fig. 2c). To test a direct impact of canonical Wnt signaling on cell differentiation, qEBs were treated with soluble Wnt3a or Dkk1 and the percentage of D6-GFP+ cells in qEBs was quantified by FACS. Representative histograms are depicted in Fig. 7. Wnt3a treatment (10 ng/ml) during d6–d12 increased the number of fluorescent cells from (35.8 ± 2.9)% (untreated samples) to (57.5 ± 3.6)% (treated samples). In contrast, treatment with the antagonist Dkk1 (50 ng/ml) decreased the number of positive cells to (18.1 ± 4.2)%. Finally, qEBs were treated with 0.5 μM retinoic acid (RA) between d5 and d9. Although RA has been shown to trigger neural induction (Bain et al. 1995; Kim et al. 2009) it completely hindered the generation of D6-GFP+ cells in our assays. This is in line with previous reports showing that RA imposes a caudal character of neural cells at the expense of anterior cell fates including the dorsal forebrain (Irioka et al. 2005).

Activation of the canonical Wnt pathway during differentiation of qEBs promotes D6-GFP+ cell fate. Flow cytometry analysis of qEBs treated with recombinant Wnt3a (10 ng/ml) or Wnt-antagonist Dkk1 (50 ng/ml) at days 6–12 shows higher number of fluorescent cells in Wnt3a-treated qEBs and lower numbers in Dkk1-treated qEBs compared to untreated controls. As a control, retinoic acid (RA, 0.5 μM) was used to impose a caudal character in cultured neural cells which strongly inhibited neocortical fate as measured by D6-GFP+ cells
ESC-Derived D6-GFP+ Cells Formed Neurospheres with Properties of NSCs
In our previous work (Machon et al. 2002, 2005), we showed that D6-GFP+ cells from E13 and neonatal forebrains could be propagated and differentiated into neurons and astrocytes. We therefore tested these properties in D6-GFP+ NSCs cells derived from qEBs. After FACS isolation of d15 qEBs, GFP+ cells formed neurospheres of various sizes when grown in standard Neurobasal A-based medium supplemented with EGF and bFGF (Fig. 8a). During expansion, the inner core of the neurospheres remained positive for GFP while the surface cells gradually lost fluorescence (rectangles in Fig. 8a, a′). NSCs in neurospheres lost fluorescence after repeated passages and thus behaved similar to NSCs isolated from the developing neocortex under the caudalizing effect of bFGF or EGF (Prajerova et al. 2010; Machon et al. 2002, 2005).
Neural stem cells meet two major criteria: the capability to proliferate and the ability to differentiate into various cell types of the nervous system. We therefore tested the differentiation potential of 2nd passage neurospheres derived from FACS-sorted D6-GFP+ cells. Dissociated neurosphere cells were plated onto polysine-coated coverslips and grown in Neuro-medium without EGF for 24 h. As shown in Fig. 8b, most of the cells were stained positive for the pan-neural progenitor marker Nestin, while some cells expressed the TujI marker of postmitotic neurons or the astrocyte-specific marker GFAP. Quantification of immunostaining documented that (87.7 ± 1.7)% cells were positive for Nestin, (9.6 ± 1.8)% for Tuj1 and (4.2 ± 1.1)% for GFAP (Fig. 8c). Thus, qEB derived D6-GFP-positive cells corresponded well to their counterparts from the mouse embryonic brain when grown under neurosphere culture conditions.

D6-GFP+ cells from qEBs retain properties of neural stem cells. a–a′ D6-GFP+ cells sorted from qEBs by FACS show similar properties as NSCs derived from the mouse embryonic forebrain. A phase contrast image (a) with a higher magnification window (below) and GFP fluorescence with a higher magnification window show that neurospheres are GFP positive. After passaging, only cells in the inner core remained positive while surface cells lost fluorescence. The rectangles depict magnified area in respective lower panels. b Immunofluorescence of differentiated cells from qEBs-derived neurospheres using the neural progenitor marker Nestin, the neuronal marker Tuj1 and the marker for astrocytes GFAP. c Quantification of Nestin, Tuj1, and GFAP positive cells after 24-h differentiation: Nestin (87.7 ± 1.7)%, Tuj1 (9.6 ± 1.8)%, and GFAP (4.2 ± 1.1)%
Discussion
In this study, we studied cell culture conditions that would yield the highest numbers of neocortex-specific neuronal cells. For this purpose, ESCs were derived from transgenic mice carrying the neocortex-specific D6-GFP reporter. This reporter turned out to be useful for tracking neocortical cells in differentiating ESCs using embryoid bodies. Interestingly, the cortical fate marked by D6-GFP was rarely reached in differentiation protocols using nEBs. In contrast, the qEBs culture, producing substantially larger cell aggregates, yielded a frequent induction of neocortex-specific cells. A related approach was used by Eiraku et al. (2008) for differentiation of ESCs into the neocortical tissue where the cell identity was monitored by immunohistochemistry with the standard cellular markers Pax6, Sox1, Bf1, or Tbr2. In addition, Eiraku employed a GFP knock-in into the Bf1 locus to directly observe cell differentiation. In comparison to Bf1-GFP, D6-GFP appears to be a more stringent marker for the neocortex as it visualizes with high specificity neocortical progenitors including radial glial cells and descendant neurons.
We think that cell communication plays a crucial role in acquirement of the neocortical cell identity. A larger mass of cells in qEBs probably resulted in proper cell signaling and self-organization that promoted cell differentiation towards the neocortical cell fate. This hypothesis is supported by the fact that GFP-positive cells were always found in large clusters rather than scattered single cells. Further, a longer time of in vitro culture is required for the induction of the neocortical cell fate compared to in vivo. The appearance of fluorescent cells peaked between 12 and 16 days whereas only 9 days are sufficient in the mouse embryo (from the blastocyst stage to E12).
Signals that directly control cell identity and organization of the neocortical tissue are not completely elucidated. Nonetheless, several reports have shown that signals including Fgf8 (Fukuchi-Shimogori and Grove 2001) and canonical Wnt signaling (Gunhaga et al. 2003; Backman et al. 2005) play an important role in this process. Our results suggest that the required inductive signals are produced within qEBs in a spatial and temporal manner that allows the induction of neocortical fate. The induction itself did not require supplementary signaling molecules but the addition of the recombinant Wnt3a in culture enhanced the process. Conversely, Dkk1, an inhibitor of the canonical Wnt signaling, reduced the number of D6-GFP-positive cells, which indicates that the Wnt pathway was activated at a physiological level in qEBs. Our experimental setup, however, was not able to unravel whether Wnt3a affected cell proliferation of cortical progenitors or an enhanced differentiation into cortical cells.
It is very intriguing that cell aggregates were organized in a way that resembled the polarized neuroepithelium in the embryonic brain (Abranches et al. 2009). It may well be that a few neuroepithelial cells with the polarized distribution of N-cadherin in the membrane induce a similar cell polarity in neighboring cells. Rosette-like structures can thus be formed via N-cadherin positive adherens junctions and this may provide a basis for the neocortical induction. Further experiments are necessary to examine the inner structure of the in vitro generated neuroepithelium. For example, genetic markers for all six layers of the cortex or various signaling molecules can be used to study its organization in more detail.
D6-GFP-positive neocortical tissue can be maintained in cell culture for more than 2 weeks. This rather broad time window can provide sufficient experimental time in tissue culture for studies on neuronal networks, discovery of new inductive signals in the neuroepithelium, or studies on small-molecule drugs that enhance or interfere with the process.
References
Abranches E, Silva M, Pradier L, Schulz H, Hummel O, Henrique D, Bekman E (2009) Neural differentiation of embryonic stem cells in vitro: a road map to neurogenesis in the embryo. PLoS One 4(7):e6286
Ahlgren S, Vogt P, Bronner-Fraser M (2003) Excess FoxG1 causes overgrowth of the neural tube. J Neurobiol 57(3):337–349
Backman M, Machon O, Mygland L, van den Bout CJ, Zhong W, Taketo MM, Krauss S (2005) Effects of canonical Wnt signaling on dorso-ventral specification of the mouse telencephalon. Dev Biol 279(1):155–168
Bain G, Kitchens D, Yao M, Huettner JE, Gottlieb DI (1995) Embryonic stem cells express neuronal properties in vitro. Dev Biol 168(2):342–357
Bishop KM, Goudreau G, O’Leary DD (2000) Regulation of area identity in the mammalian neocortex by Emx2 and Pax6. Science 288(5464):334–349
Cajanek L, Ribeiro D, Liste I, Parish CL, Bryja V, Arenas E (2009) Wnt/beta-catenin signaling blockade promotes neuronal induction and dopaminergic differentiation in embryonic stem cells. Stem Cells 27(12):2917–2927
Eiraku M, Watanabe K, Matsuo-Takasaki M, Kawada M, Yonemura S, Matsumura M, Wataya T, Nishiyama A, Muguruma K, Sasai Y (2008) Self-organized formation of polarized cortical tissues from ESCs and its active manipulation by extrinsic signals. Cell Stem Cell 3(5):519–532
Englund C, Fink A, Lau C, Pham D, Daza RA, Bulfone A, Kowalczyk T, Hevner RF (2005) Pax6, Tbr2, and Tbr1 are expressed sequentially by radial glia, intermediate progenitor cells, and postmitotic neurons in developing neocortex. J Neurosci 25(1):247–251
Fishell G, Kriegstein A (2005) Cortical development: new concepts. Neuron 46(3):361–362
Fujimura N, Vacik T, Machon O, Vlcek C, Scalabrin S, Speth M, Diep D, Krauss S, Kozmik Z (2007) Wnt-mediated down-regulation of Sp1 target genes by a transcriptional repressor Sp5. J Biol Chem 282(2):1225–1237
Fukuchi-Shimogori T, Grove EA (2001) Neocortex patterning by the secreted signaling molecule FGF8. Science 294(5544):1071–1074
Gaspard N, Bouschet T, Hourez R, Dimidschstein J, Naeije G, van den Ameele J, Espuny-Camacho I, Herpoel A, Passante L, Schiffmann SN, Gaillard A, Vanderhaeghen P (2008) An intrinsic mechanism of corticogenesis from embryonic stem cells. Nature 455(7211):351–357
Gaspard N, Bouschet T, Herpoel A, Naeije G, van den Ameele J, Vanderhaeghen P (2009) Generation of cortical neurons from mouse embryonic stem cells. Nat Protoc 4(10):1454–1463
Gotz M, Huttner WB (2005) The cell biology of neurogenesis. Nat Rev Mol Cell Biol 6(10):777–788
Guillemot F (2007) Cell fate specification in the mammalian telencephalon. Prog Neurobiol 83(1):37–52
Gunhaga L, Marklund M, Sjodal M, Hsieh JC, Jessell TM, Edlund T (2003) Specification of dorsal telencephalic character by sequential Wnt and FGF signaling. Nat Neurosci 6(7):701–707
Hanashima C, Shen L, Li SC, Lai E (2002) Brain factor-1 controls the proliferation and differentiation of neocortical progenitor cells through independent mechanisms. J Neurosci 22(15):6526–6536
Houart C, Caneparo L, Heisenberg C, Barth K, Take-Uchi M, Wilson S (2002) Establishment of the telencephalon during gastrulation by local antagonism of Wnt signaling. Neuron 35(2):255–265
Irioka T, Watanabe K, Mizusawa H, Mizuseki K, Sasai Y (2005) Distinct effects of caudalizing factors on regional specification of embryonic stem cell-derived neural precursors. Dev Brain Res 154(1):63–70
Jho EH, Zhang T, Domon C, Joo CK, Freund JN, Costantini F (2002) Wnt/beta-catenin/Tcf signaling induces the transcription of Axin2, a negative regulator of the signaling pathway. Mol Cell Biol 22(4):1172–1183
Kadowaki M, Nakamura S, Machon O, Krauss S, Radice GL, Takeichi M (2007) N-cadherin mediates cortical organization in the mouse brain. Dev Biol 304(1):22–33
Kim M, Habiba A, Doherty JM, Mills JC, Mercer RW, Huettner JE (2009) Regulation of mouse embryonic stem cell neural differentiation by retinoic acid. Dev Biol 328(2):456–471
Kwan KY, Lam MM, Krsnik Z, Kawasawa YI, Lefebvre V, Sestan N (2008) SOX5 postmitotically regulates migration, postmigratory differentiation, and projections of subplate and deep-layer neocortical neurons. Proc Natl Acad Sci USA 105(41):16021–16026
Lagutin OV, Zhu CC, Kobayashi D, Topczewski J, Shimamura K, Puelles L, Russell HR, McKinnon PJ, Solnica-Krezel L, Oliver G (2003) Six3 repression of Wnt signaling in the anterior neuroectoderm is essential for vertebrate forebrain development. Genes Dev 17(3):368–379
Lai T, Jabaudon D, Molyneaux BJ, Azim E, Arlotta P, Menezes JR, Macklis JD (2008) SOX5 controls the sequential generation of distinct corticofugal neuron subtypes. Neuron 57(2):232–247
Lee SH, Lumelsky N, Studer L, Auerbach JM, McKay RD (2000a) Efficient generation of midbrain and hindbrain neurons from mouse embryonic stem cells. Nat Biotechnol 18(6):675–679
Lee SM, Tole S, Grove E, McMahon AP (2000b) A local Wnt-3a signal is required for development of the mammalian hippocampus. Development 127(3):457–467
Levine AJ, Brivanlou AH (2007) Proposal of a model of mammalian neural induction. Dev Biol 308(2):247–256
Machon O, van den Bout CJ, Backman M, Rosok O, Caubit X, Fromm SH, Geronimo B, Krauss S (2002) Forebrain-specific promoter/enhancer D6 derived from the mouse Dach1 gene controls expression in neural stem cells. Neuroscience 112(4):951–966
Machon O, Backman M, Krauss S, Kozmik Z (2005) The cellular fate of cortical progenitors is not maintained in neurosphere cultures. Mol Cell Neurosci 30(3):388–397
Munoz-Sanjuan I, Brivanlou AH (2002) Neural induction, the default model and embryonic stem cells. Nat Rev Neurosci 3(4):271–280
Muzio L, DiBenedetto B, Stoykova A, Boncinelli E, Gruss P, Mallamaci A (2002) Conversion of cerebral cortex into basal ganglia in Emx2(−/−) Pax6(Sey/Sey) double-mutant mice. Nat Neurosci 5(8):737–745
Nordstrom U, Jessell TM, Edlund T (2002) Progressive induction of caudal neural character by graded Wnt signaling. Nat Neurosci 5(6):525–532
Pevny LH, Nicolis SK (2010) Sox2 roles in neural stem cells. Int J Biochem Cell Biol 42(3):421–424
Prajerova I, Honsa P, Chvatal A, Anderova M (2010) Neural stem/progenitor cells derived from the embryonic dorsal telencephalon of D6/GFP mice differentiate primarily into neurons after transplantation into a cortical lesion. Cell Mol Neurobiol 30(2):199–218
Rallu M, Corbin JG, Fishell G (2002) Parsing the prosencephalon. Nat Rev Neurosci 3(12):943–951
Rash BG, Grove EA (2006) Area and layer patterning in the developing cerebral cortex. Curr Opin Neurobiol 16(1):25–34
Shimogori T, Banuchi V, Ng HY, Strauss JB, Grove EA (2004) Embryonic signaling centers expressing BMP, WNT and FGF proteins interact to pattern the cerebral cortex. Development 131(22):5639–5647
Sussel L, Marin O, Kimura S, Rubenstein JL (1999) Loss of Nkx2.1 homeobox gene function results in a ventral to dorsal molecular respecification within the basal telencephalon: evidence for a transformation of the pallidum into the striatum. Development 126(15):3359–3370
Watanabe K, Kamiya D, Nishiyama A, Katayama T, Nozaki S, Kawasaki H, Watanabe Y, Mizuseki K, Sasai Y (2005) Directed differentiation of telencephalic precursors from embryonic stem cells. Nat Neurosci 8(3):288–296
Wataya T, Ando S, Muguruma K, Ikeda H, Watanabe K, Eiraku M, Kawada M, Takahashi J, Hashimoto N, Sasai Y (2008) Minimization of exogenous signals in ES cell culture induces rostral hypothalamic differentiation. Proc Natl Acad Sci USA 105(33):11796–11801
Wilson SI, Edlund T (2001) Neural induction: toward a unifying mechanism. Nat Neurosci 4(suppl):1161–1168
Wilson SW, Houart C (2004) Early steps in the development of the forebrain. Dev Cell 6(2):167–181
Ying QL, Stavridis M, Griffiths D, Li M, Smith A (2003) Conversion of embryonic stem cells into neuroectodermal precursors in adherent monoculture. Nat Biotechnol 21(2):183–186
Zappone MV, Galli R, Catena R, Meani N, De BS, Mattei E, Tiveron C, Vescovi AL, Lovell-Badge R, Ottolenghi S, Nicolis SK (2000) Sox2 regulatory sequences direct expression of a (beta)-geo transgene to telencephalic neural stem cells and precursors of the mouse embryo, revealing regionalization of gene expression in CNS stem cells. Development 127(11):2367–2382
Acknowledgments
This work was supported by the Norwegian Research Council and Grant Agency of the Czech Republic (grant # GACR 204/08/1618).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Fig. 1
Assay for pluripotency of ESCs derived from D6-GFP blastocysts. ESCs were grown on gelatin-coated surfaces and showed typical morphology of pluripotent cells (phase contrast image). b Flow cytometry analysis of pluripotency marker and membrane protein SSEA-1 documented a high percentage of pluripotent ESCs in the cultures. c–d Immunofluorescence of pluripotency markers Oct4 and Sox2. e–f Differentiation markers (Nestin in neural progenitors) and Sox1 (early neuroectoderm) were not detectable. c′–f′ Illustration of corresponding nuclei stained with DAPI. Magnification ×40. (TIFF 4326 kb)
Supplementary Fig. 2
Several examples of D6-GFP+ clusters in qEBs. qEB were daily examined for the D6-GFP fluorescence in the course of a 20-day culture. Fluorescent cells were observed in clusters within qEBs (magnification ×10). In rare cases, several distinct zones of D6-GFP+ were seen within one qEB. (TIFF 4919 kb)
Supplementary Table 1
A table showing sequences of qRT-PCR primers and their annealing temperature. (DOC 53 kb)
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Jing, Y., Machon, O., Hampl, A. et al. In Vitro Differentiation of Mouse Embryonic Stem Cells into Neurons of the Dorsal Forebrain. Cell Mol Neurobiol 31, 715–727 (2011). https://doi.org/10.1007/s10571-011-9669-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10571-011-9669-2