
Abstract
Use of low-quality (and inexpensive) feedstock such as waste oils and animal fats for biodiesel synthesis has been continuously researched as a prospective means of improving the economic efficiency of the process. Pretreatment of free fatty acids by catalytic esterification is necessary for achieving this purpose. This paper reviews some relevant studies on heterogeneous acid catalysts that have been shown to be effective for this reaction.
Similar content being viewed by others

1 Introduction
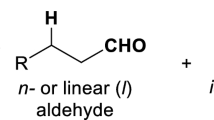
The esterification of (free) fatty acids into fatty esters is one of the “classic” organic reactions with a long history. Fatty esters are widely utilized in the production of cosmetics and detergents in the chemical industry. Recently, this reaction has gained prominence with the emergence of biodiesel as a promising renewable energy source. Transesterification of triglycerides, which are the major constituents of biological fats and oils, is the main part of biodiesel synthesis. The process has been commercialized in various ways and still continues to be upgraded to meet the emerging requirements of the industry and market. One of the urgent requirements in this upgradation is the development of a protocol for treating low-quality feedstock such as waste vegetable oils and animal fats with high content of free fatty acids (FFAs). In order to utilize low-quality feedstock, the FFAs should be pretreated or removed because they create problems in the biodiesel synthesis process. In many respects, esterification of FFAs is the most efficient and effective method for pretreating FFAs, where judicious choice of a (heterogeneous) catalyst is a prerequisite for their commercial implementation. This paper reviews recent studies on heterogeneous catalysts for FFA esterification. Initially, certain basic facts and common issues related to the development of biodiesel-production catalysts are elaborated, followed by an introduction on FFAs, and a final summary of the key findings of the studies on heterogeneous catalysts for FFA esterification.
2 Catalytic Biodiesel Synthesis
With the growing concerns about the sustainability of traditional petroleum resources, several untraditional resources such as shale gas, oils sands, extra heavy oils and biomass-derived fuels have gained attentions as alternatives [1, 2].
Biodiesel is a sustainable and environmentally benign alternative to petroleum diesel oil. Unlike petroleum diesel, carbon dioxide emitted from (the combustion of) biodiesel is exempted from greenhouse gas penalties because the carbons in biodiesel are active in the natural carbon cycle, where their combustion into carbon dioxide is offset by their return to the bioenergy resources by photosynthesis [3]. Moreover, the use of biodiesel improves the efficiency of internal combustion and reduces harmful emissions such as particulate matter, carbon monoxide, and unburned hydrocarbons [4, 5] (except for NOx, whose emission is slightly increased by 2–4 %, because the increased bulk modulus of biodiesel-mixed fuels advances the injection timing of fuel [6, 7]). Though biodiesel is used as a fuel for compression–ignition engines, it is rarely used in a neat form; instead it is used in a mixture with petroleum diesel [8]. Though pure biodiesel (B100 or BD100) can be used in the neat form, it causes some problems in handling and use because of its high freezing temperature (it starts to cloud at 275–288 K) [9], high solvency (pure biodiesel may contain impurities derived from the dissolution of sediments in the storage tank or fuel lines), and incompatibility with certain rubber compounds used in the engine parts (such as Buna-N, nitrile, and natural rubber). Thus, most of the commercially available biodiesel is used as a blend of 5–20 wt% biodiesel (B5–B20).
Biodiesel is an alkyl ester produced by the transesterification of triglycerides with alcohol (Eq. 1).

Triglycerides are usually sourced from natural lipid feedstock such as vegetable oils and animal fats. The most efficient sources of triglycerides are vegetable oils, whose top three sources are soybean, palm, and rapeseed oils. However, because of the economic and social concerns about the impact of biofuels on food and crop prices, the target raw materials have been changed to inedible feedstock such as Jatropha curcas, Milletia pinnata (karanja), Sapindus (soapnut), Ricinus communis (castor), single cell (algae), and waste cooking oils [7].
To achieve reasonable productivity of biodiesel by transesterification, certain requirements should be fulfilled. (i) An efficient catalyst should be utilized. Usually, an acid, a base, or an enzyme catalyst is used based on the process conditions and requirements. The catalyst should be sufficiently active, recyclable, environmentally benign, and economically feasible. (ii) The ideal stoichiometry of alcohol to triglyceride is 3; however, in practice, an overstoichiometric amount (usually 5–50, sometimes more than 100) of alcohol should be utilized to get reasonable productivity of the alkyl ester (i.e., to overcome the thermodynamic limitation of the reaction equilibrium). (iii) In some cases, high temperature (333–523 K) and pressure (5–80 atm) conditions are adopted to meet the requirements of productivity. (iv) By-product issues should be carefully handled because the separation and purification of biodiesel has a huge impact on the economic feasibility of the process. Further details about the catalytic transesterification for biodiesel synthesis can be found in more than a dozen review papers that have been published in the last decade [10–26].
The selection of a transesterification catalyst is strongly dependent on the feedstock conditions and other process requirements. The first consideration is the choice between an acid and a base as the catalyst. An enzyme catalyst is another possible choice [27, 28], but the transesterification rate achieved with these biological catalysts is usually slower than that achieved with acid/base catalysts [29, 30]. Acid and base catalysts are generally used as homogeneous catalysts. The most commonly utilized catalysts are H2SO4, H2SO3, KOH, NaOH, and CH3ONa, which are soluble in an oil–alcohol mixture. The acid catalysts generally afford high transesterification yield, but give rise to slower reaction rates than the base catalysts, require high temperature and pressure, and an excess of alcohol to achieve complete conversion of the feedstock within a reasonable time. When considering the transesterification rate only, base catalysts are more desirable for rapid termination of the conversion under mild temperature and pressure conditions. A comparison of the acid- and base-catalyzed processes (in terms of process conditions) is presented in Table 1. Because of the high reaction rate, base catalysts are being increasingly adopted in commercial biodiesel-production processes. However, in terms of sensitivity to feedstock quality, acid catalysts offer some advantages over base catalysts. A fatty acid is a carboxylic acid with a long unbranched aliphatic chain (saturated or unsaturated). A fatty acid that is chemically detached and transfers freely from its original chemical substrate is called a “free fatty acid.” Low-quality (and thus cheap) feedstock usually contains a large amount of FFA; waste cooking oils contain 1–8 % FFA and animal fats in excess of 5–20 %. FFAs cause undesirable saponification when a (homogeneous) base catalyst is used for transesterification. Saponification is a kind of soap (ester salt) formation (Eq. 2), which consumes the base catalyst (NaOH, KOH) and reduces the efficiency of transesterification.
More detrimentally, the resulting soaps cause difficulty in separating the biodiesel from the glycerol phase, i.e., the soap makes the water-wash process (for removing the remaining methanol) inefficient by promoting emulsion formation [3]. Consequently, base catalysts can be adopted only when the feedstock is comparatively pure and dry, and contains minor amounts of FFA (≤2 wt%, or more favorably, ≤0.5 wt%) and water (≤0.4 wt%) [31].
In contrast, acid catalysts are almost insensitive to FFA and present less danger of saponification, making them a better choice than the base catalysts when the objective is to produce biodiesel from a feedstock with a high FFA content. In principle, the acid catalysts are favored if the cost penalty because of the slow reaction rate could be compensated by the reduced cost of lower purity feedstock with highly acidic oil. However, even with an acid catalyst, the oil conversion is depressed if the FFA content in the feedstock is very high (Fig. 1). Though FFA does not directly poison the acid catalyst, the water produced from esterification retards the triglyceride conversion by promoting reverse esterification [32]. (Notably, the esterification of FFA usually proceeds at a faster rate than transesterification of triglycerides.)

Effect of FFA content on triglyceride conversion. Reaction conditions: methanol:oil (palmitic acid+soybean oil) = 6:1; catalyst: 3 wt% H2SO4; temperature = 333 K; time = 96 h; plot was made by authors using data from [32]
3 Main Issues in Development of Catalysts for Biodiesel Synthesis
3.1 Heterogeneous Catalyst for Transesterification of (Pure) Vegetable Oil
Regardless of whether an acid or a base catalyst is used, homogeneous catalysts present certain inherent disadvantages. The first is that homogeneous catalysts may corrode the reactor and flow lines (acid catalysts are more corrosive than base catalysts [33]). The second is the difficulty associated with catalyst recovery; because the catalyst is dissolved in the liquid media, it is not easily recovered, and therefore, must be neutralized and discarded by adding a counter base or acid. The third drawback is the limitation in establishing a continuous process [28]. Because of these disadvantages, the replacement of homogeneous catalysts with heterogeneous solid catalysts is on the increase [28].
The well-known heterogeneous acid catalysts for transesterification include ion-exchange resins (Amberlyst-15 [34–36], Nafion [37]), sulfated inorganic oxides (SO4 2−/ZrO2, SO4 2−/TiO2, SO4 2−/SnO2) [38, 39], and inorganic superacids such as WO3/ZrO2 and WO3/ZrO2–Al2O3. Similar to the case of homogeneous catalysts, the transesterification rate achieved with heterogeneous base catalysts is superior to that achieved with (heterogeneous) acid catalysts. Among the recognized heterogeneous base catalysts, alkaline earth oxides (CaO, MgO) [40], alkali-supported catalysts (KF-, K2CO3-, or KNO3-supported Al2O3), Na/NaOH/Al2O3 [41], zeolite-X [42–44], titanosilicates (ETS-4 and 10) [43, 45], SBA-15 [46, 47], hydrotalcites [48–50], and guanidine-supported catalysts [51–53] have been extensively studied.
Despite the unresolved issues such as low-reaction rate (compared to homogeneous catalysts), high temperature/pressure requirements, and leaching of active species into the liquid phase, the attempts to utilize heterogeneous catalysis and continuous fixed-bed operation have been continuing in establishing a commercial process. The Esterfip-H process introduced by the French company, Axens, is a known example with a production capacity of 160,000 ton/year [54, 55]. Most of the implemented processes adopt solid-base catalysts for the transesterification of vegetable oil.
A review on heterogeneous base catalysts for biodiesel synthesis was published by the authors in 2009 [26].
3.2 Development of Transesterification Processes for Waste Edible Oils
Because more than 70 % of the biodiesel price is allocated to the feedstock cost, the profitability of the biodiesel production process is critically contingent on the cost of feedstock [21, 56]. Consequently, the price of biodiesel is heavily dependent on the market price of agricultural sources such as soybean, rapeseed, and palm, some of which are connected to the food supply chain, thus making the forecast of the profitability of biodiesel production ambiguous. The bright prospect of biodiesel is based in part on the prediction that the price of petroleum oil will keep on increasing, which would make biodiesel competitive with petroleum diesel at some point, allowing it to become profitable without tax exemption. However, this simple prospect is overly optimistic because the feedstock price is largely influenced by the petroleum price [56].
From an economic standpoint, waste cooking oil is the most desirable alternative as a feedstock for biodiesel. Since the use of waste cooking oils in livestock feed was banned in the EU in 2002 [21], a new route for treating (or utilizing) waste oils has been highly demanded. Disposal of highly acidic waste oils by neutralization with aqueous base (NaOH, KOH) is not an adequate solution, because the metallic soaps that are generated as by-products require additional treatment and disposal. An alternative and attractive means of utilizing waste oils is the esterification of FFAs into fatty esters using alcohol in the presence of an acid catalyst (Eq. 3).
Other methods such as steam injection [57], chromatography [58], and film vacuum evaporation [59] are suggested for removing FFA from waste oils. However, the esterification method offers definite advantages over these other methods because it produces fatty esters (R–COO–R′ in Eq. 3) that directly constitute biodiesel.
If the FFA in waste oil is properly esterified or removed, a subsequent transesterification can be devised using a base catalyst. In addition to the FFAs, other impurities such as water and polymers should also be removed prior to this process [21, 32, 59, 60]. Base-catalyzed transesterification coupled with acid-catalyzed FFA esterification is a feasible process for the synthesis of biodiesel from waste cooking oils. However, the entire process becomes complicated as the number of equipment increases. The direct transesterification of waste cooking oil using an acid catalyst is also feasible, whereby the esterification of FFAs and transesterification of triglycerides can be accomplished using a single acid catalyst [16, 61]. This process is relatively simple and has some advantages in terms of process integration. However, the transesterification rate is slow, and thus, the process requires a large amount of methanol with a consequent increase in the number of required reactors (or an increase in the reactor volume) and the need for a larger methanol-recovery column [61]. Because each process has its own advantages and disadvantages, the choice of a process is governed by both the technological and economical aspects under the given conditions [61, 62].
4 FFAs in Waste Vegetable Oils
When food materials are fried or cooked with vegetable oils, three major types of chemical reactions occur: (i) pyrolytic degradation, (ii) oxidative degradation, and (iii) hydrolysis [63, 64].
Most food materials have a large amount of moisture; thus, when they are cooked with vegetable oils at elevated temperatures, triglycerides are hydrolyzed to form a diglyceride and a carboxylic acid [64] (Fig. 2). The diglyceride is then hydrolyzed to obtain another acid molecule and a monoglyceride; however, it is known that the hydrolysis of diglycerides is not as active as that of triglyceride. Hence, the content of monoglyceride in the used oil is very minor [65]. The generated carboxylic acids are the FFAs that are to be removed in the biodiesel production process. Some unsaturated FFAs such as linoleic and linolenic acids are converted to monomeric “cyclic” fatty acids via intramolecular cyclization involving the double bonds in the unsaturated fatty chain [66]. Cyclic fatty acids represent a minor (below 50 ppm) contribution, but there is debate about their biological toxicity.

Formation of FFAs by hydrolysis of fatty acids
It is known that about 20 wt% of the triglycerides is hydrolyzed into FFAs during frying [64]. Because the fatty acids are generally more volatile than tri- or diglycerides, most of the fatty acids are vaporized during frying and only 0.5–2 wt% remains in the liquid oil [64, 67]. Moreover, because of the volatility of FFAs, the hydrolysis of triglycerides is almost free from thermodynamic limitations and is kinetically driven in the forward direction, resulting in a rapid reaction rate.
The hydrolysis rate of short and unsaturated fatty glycerides is faster than that of long and saturated fatty glycerides because of the higher solvency of the unsaturated fatty acids in water [64]. Hence, it is supposed that unsaturated acids such as oleic and linoleic acid are more adequate as model molecules for FFA than the saturated acids (capric, lauric, palmitic, and stearic acid). In fact, it is reported that unsaturated acids (60 % C18:1, 26 % C18:2 and C18:3) constitute 86 % of FFAs in a waste cooking oil. Among these, oleic acid (59.7 %) and linoleic/linolenic acid (26.1 %) are present in the highest concentration [68].
If the FFA content in waste oil is over 15 %, the oil is classified as yellow grease; otherwise, it is termed brown grease [21, 69]. FFAs are generally quantified by a titrimetric method employing potassium hydroxide. The acid value (or acid number) of oils and fats is defined as the amount of KOH (mg) required to neutralize the acid components dissolved in the oils or fats (per gram). The standard methods for determining the acid value of biodiesel include ASTM D664 (in the US) and EN14104 (in Europe), both of which are based on the KOH titration principle. The acid value of commercial biodiesel is generally below 0.5 mg KOH/g. Base-catalyzed transesterification is appropriate for biodiesel synthesis when the acid value of the waste oil is below 1.0 mg KOH/g [68] (the acid value of 1.0 mg KOH/g is equivalent to 0.5 wt% FFA). Most of waste cooking oils have acid values exceeding 2.5 mg KOH/g on average. Chromatographic, colorimetric, and enzymatic methods are commonly used for determining the FFA concentration [70].
5 Heterogeneous Solid Acid Catalysts for FFA Conversion
The FFA esterification mechanism consists of several reaction steps (Fig. 3). Protonation of a carboxylic oxygen in the fatty acid initiates the reaction, generating an electro-deficient carbon. This carbon is attacked by the (nucleophilic) oxygen atom of an alcohol molecule, followed by an intra-molecule proton transfer. The intermediate undergoes rearrangement with the evolution of water; the subsequent de-protonation furnishes the alkyl ester.

The reaction mechanism of Fischer esterification
The FFA esterification reaction is largely influenced by the steric bulk of the reactants, favoring less crowded primary acids and alcohols [71]. The acid strength of the carboxylic acid (FFA) exerts a relatively minor impact on the esterification rate [71]. Because esterification is a reversible reaction, it is generally performed in excess methanol to promote the forward reaction [72]. Water, which is coproduced with the alkyl ester, inhibits the forward reaction, hydrolyzing the ester to regenerate the acid.
Even though the initial report of esterification was presented more than a century ago, intensive efforts are still expended in the search for a better catalyst [10], particularly in the pharmaceutical and cosmetic industries [73]. For example, the synthesis of a monoglyceride-type emulsifier by the esterification of fatty acids with glycerol, where the key issue is finding an apposite catalyst for promoting the reaction under near equimolar acid/alcohol ratio and mild temperature conditions [74].
For biodiesel synthesis, homogeneous acid catalysts such as sulfuric, phosphoric, hydrofluoric, alkyl benzene sulfonic, and p-toluene sulfonic acids are employed for FFA esterification [32, 68, 75–77]. Upon completion of the reaction, the remaining acids are generally neutralized using a large amount of alkaline water [61, 75, 76]. The alkali often corrodes the metallic parts of the reactor to produce metallic sludge, which contaminates the ester phase [76]. Consequently, heterogeneous solid catalysts are favored because they are easily separated from the liquid phase and do not exert a direct caustic effect on the metallic equipment.
Lotero et al. [16] suggested three vital elements in the design of heterogeneous acid catalysts for biodiesel synthesis: (i) an interconnected system of large pores, (ii) a moderate-to-high density (concentration) of strong acid sites, and (iii) hydrophobic character. These elements are likewise applicable in the design of acid catalysts for transesterification of triglycerides and esterification of FFAs because the two reactions are chemically similar and involve relatively large-sized organic reactants. A large pore structure is required in the catalysts to alleviate mass transfer resistance of molecules having long alkyl chains, and strong acid sites are a requisite for promoting the reaction rate. The last design element, i.e. a hydrophobic surface, is essential for promoting the preferential adsorption of fatty materials on the catalyst surface while preventing the adsorption of the products such as glycerol and water.
Based on our investigation, we found that the most frequently studied or applied solid acid catalysts for FFA esterification are acidic organic resins, zeolites, mesoporous silicates, sulfated zirconia, heteropolyacids, and binary metal oxides. The catalysts in each category exhibit individually characteristic pore structure, surface acidity, and hydrophobicity. The following section summarizes the major findings in the studies on each category of catalysts, and discusses the relationship between the characteristics of the catalysts and the esterification activity.
5.1 Acidic Organic Resins
The term acidic organic resin, in most cases, refers to Nafion and Amberlyst-15, both of which are commercial cation-exchange resins used as solid acid catalysts in various industrial applications such as MTBE synthesis, dehydration of tert-butanol into isobutylene, bisphenol-A synthesis, and hydration of propane to 2-propanol [78].
Nafion is a perfluorinated resin sulfonic acid, which is a copolymer of tetrafluoroethylene and perfluoro-2-(fluorosulfonylethoxy)propylvinyl ether. Amberlyst-15 is a sulfonated resin based on the styrene–divinylbenzene copolymer. Both resins have terminal sulfonic acid (SO −3 H+) groups (Fig. 4), which are the source of the acidity, and release acidic protons when the resins are sufficiently wet. However, the two catalysts differ in terms of acid properties: it is generally accepted that the acidic strength of Nafion is higher than that of Amberlyst-15 (the Hammett acidity value of Nafion is between −11 and −13, whereas that of Amberlyst-15 is ca. −2.2 [78]). On the contrary, the number of acid sites in Amberlyst-15 is about five times more than that of Nafion [78]. The two catalysts also differ in terms of thermal stability: Nafion is functional to a maximum allowable temperature of about 553 K whereas that of Amberlyst-15 is restricted to 393–413 K [78, 79].

Structural unit of Amberlyst-15 and Nafion
These differences in acid strength and thermal durability lead to a differentiation in the type of reaction to which each catalyst is applied: Nafion exhibits high activity for alkylation, which requires high acid strength and temperature, whereas Amberlyst-15 is adequate for esterification, which does not strictly require a strong acid catalyst and utilizes relatively mild (330–520 K) reaction temperatures [78].
The esterification activities of the organic resin catalysts have been estimated for the esterification of acrylic acid with 1-butanol [71, 80, 81], in which the catalysts were ranked in the activity order: Nb2O5 < ZSM-5 < Amberlyst-15 < Cs2.5H0.5PW12O40 ≒ SO4 2−/ZrO2 ≪ Nafion. Certain studies claim that Nafion and SO4 2−/ZrO2 are as active as sulfuric acid (on the basis of a single active site) for biodiesel-related esterification and transesterification [37, 45].
On the other hand, the low porosities and specific areas of Nafion and Amberlyst-15 (less than 5 cm3/g and 50 m2/g, respectively) are seemingly inadequate for industrial catalysts. However, these materials swell and become macroporous when immersed in polar liquids. The extent of this swelling is dependent on the cross-linking degree of polymer backbone: a lower degree of cross-linking leads to greater swelling of the resin [82, 83]. An excess of alcohol is applied in the esterification or transesterification for biodiesel synthesis in order to promote the forward reaction. The alcohol-rich environment prompts swelling of the acidic resins, which permits access of large-sized fatty molecules to the internal acid sites of the resin [16]. In addition to the acidity and stability, the swelling property is a decisive factor in the choice of ion-exchange resins as catalysts for esterification of fatty molecules.
The initial studies on the esterification of fatty molecules using acidic organic resins can be dated back to the mid-1980s [84, 85]. These studies adequately demonstrate the contradictory nature of the effects of water on the acid-resin catalysis. A certain degree of wetness is indispensable for the (acid) catalytic activity of the sulfonated resins, but the presence of water promotes reverse esterification, which lowers the reaction rate and the yield of the product (ester). When the objective is to use sulfonated resin catalysts in esterification, the wetness level should be determined, considering the balance between the catalytic activity and the reaction equilibrium.
The hydrophobicity of a catalyst is critical for sulfonated-resin catalysis. Goto et al. [86] prepared pellets of Amberlyst-15 using polyethylene as a binder. Evaluation of these catalysts in the esterification of palmitic acid demonstrated that the catalytic activity of the pellets was greater than that of the particulates, even though diffusion resistance was more severe in the pellet-catalyzed reaction. The enhanced activity of the pellets was attributed to the increased hydrophobicity derived from mixing with the polyethylene binder. Park et al. [87] evaluated the hydrophobicity effect in more detail in the Amberlyst-15-catalyzed esterification of oleic acid. The adverse effect of water on the acid-catalyzed esterification is common for all types of acid catalysts. However, the water tolerance of resin sulfonic-acid catalysts such as Amberlyst-15 is a particular challenge because the hydrophilicity of the sulfonated surface causes the pores to fill up with water, thereby hindering the access of fatty acids to the acid sites. It was found that a water content of even 1 % decreased the ester yield with Amberlyst-15. As a solution, the authors suggested two-step esterification, which includes the exchange of methanol and the resin catalyst with fresh batches in the middle of the reaction. By using the two-step method, the time required to reach 85 % conversion could be reduced from 6 to 1.8 h.
Over the last decade, ion-exchange organic resins have been improved to meet the customized demands of catalyst-based industries. In particular, styrene-based resins have been diversified to afford a wide spectrum of acidity, porosity, and swelling ability. It is anticipated that the number of studies will further increase, paving the way for the development of new resin catalysts for esterification of fatty matters. Recently, a resin sulfonic-acid catalyst that can reportedly be applied to the transesterification/esterification of a feedstock of any FFA content (0.5–100 %) without the drawbacks of suppressed yield and by-products was developed specifically for biodiesel synthesis and patented under the name “Amberlyst BD20 [88].” At the end of 2009, Bayer Technology Services GmbH and Dow Water & Process Solutions announced the cooperative development of the “BayFAME” process that produces biodiesel from the transesterification of acidic oils (waste cooking oils) using the Amberlyst BD20 catalyst [89]. The recent work by Park et al. [90] demonstrated that Amberlyst BD20 was superior to Amberlyst-15 in terms of water tolerance. Comparison of the activity of these catalysts over five cycles for the conversion of oleic acid (3-h batch reaction per cycle) demonstrated a 3 % decrease (from 93 to 90 %) in the conversion when Amberlyst BD20 was used, whereas with Amberlyst-15, the conversion decreased by 30 % (from 96 to 90 %; note: the conversion values were evaluated by the current authors using the data suggested in the literature). BET analysis shows that Amberlyst BD20 is almost nonporous (<0.1 m2/g) compared to Amberlyst-15 (40–55 m2/g). The authors claimed that the porosity of the catalyst had a minor impact on the water tolerance of the Amberlyst catalysts because the surface of the catalysts is so hydrophilic that the pores become filled with water, which hinders the access of fatty molecules to the acid sites located inside the pores. However, a contradictory report on the influence of porosity was presented by Özbay et al. [83], in which the higher average pore diameter of the Amberlyst catalyst led to a higher FFA conversion: Amberlyst-15 (30 nm, 53 m2/g) >Amberlyst-35 (30 nm, 50 m2/g) >Amberlyst-16 (25 nm, 30 m2/g). We propose that this contradictory result may have been derived from the comparatively small amount of water produced inside the catalyst. In the study by Özbay et al., the FFA conversions were comparatively low (less 45 %) because the tests were performed using an acidic oil containing a low concentration of FFA (0.41–0.47 wt%) and a small amount of methanol (20 vol% based on acidic oil). Thus, the reaction would produce a comparatively small amount of water, which would present a lesser obstruction to the access of FFA to the inside of the pore. However, the effect of porosity on the esterification performance of Amberlyst BD20 is not yet clearly understood, and is one of the topics under debate. Furthermore, structural and chemical data on Amberlyst BD20 are not currently available, and thus, further studies geared at elucidating the relationships between the structure, properties, and activity of Amberlyst BD20 are anticipated.
The drawbacks of using resin sulfonic-acid catalysts in biodiesel synthesis include: (i) the inferior thermal stability, which limits the reaction temperature to below 553 K (at this temperature, the sulfonic acid chains start to decompose) [37], and (ii) the deactivation caused by the entanglement of long-chain organic reactants with the nano-domain polymeric chains on the catalyst surface [91]. However, this class of catalysts is still considered as one of the promising catalysts for biodiesel synthesis based on the high performance and reasonable price.
5.2 Zeolites
The microporous structure of a zeolite is seemingly not adequate for reactions such as FFA esterification where a large-sized molecule is involved. However, many studies use zeolites as a benchmark for evaluating catalysts designed for FFA esterification. In spite of their small pore size, zeolites exhibit high activity for esterification. Chung et al. [92, 93] reported that 80 % conversion of oleic acid could be achieved at 333 K using 1 g of H-ZSM5 (H-MFI, Si/Al = 25) or H-MOR (Si/Al = 10) catalyst in a batch reactor (ca. 600 mL, oleic acid:methanol = 1:16 (molar ratio) within 90 min of initiating the reaction.
Kirumakki et al. [94] proved by kinetic studies that esterification (of acetic acid) over zeolites (H-β, H-Y, H-ZSM5) proceeds by the Eley–Rideal mechanism in which acid molecules are adsorbed on the acid sites to react with alcohol in the gaseous phase (Fig. 5).

Eley–Rideal mechanism for esterification of acetic acid over zeolites [94]
The esterification activity of a zeolite is strongly related to its acid properties and hydrophobicity, both of which are dependent on the Si/Al ratio of the zeolite framework [16, 92].
For most zeolite materials, the number of acid sites increases as the Si/Al ratio decreases because both the Brönsted and Lewis acid sites originate from the Si–O–Al sites. The dependence of the acid strength of zeolites on the Si/Al ratio follows various trends depending on the type (structure) of zeolite, range of Si/Al ratio, and measurement method. Evaluation of the acid strength of H-MFI and H-MOR zeolites via NH3 temperature programmed desorption (NH3-TPD) demonstrated that the acid strength of the strong acid sites decreased as the Si/Al ratio increased (from 10 to 350), indicated by a decrease in the maximum peak temperature (Tmax) for NH3 desorportion [92]. However, certain researchers asserted that there is some ambiguity in determining the acid strength of zeolites by the NH3-TPD method because the re-adsorption of NH3 may take place during a TPR run under high w/f conditions [95, 96]. Okuhara [96] suggested that more precise evaluation of the acid strength of zeolite materials could be achieved via microcalorimetry because the heat of NH3 adsorption can be determined close to the theoretical value by this technique [97]. Based on the microcalorimetric analysis, it was concluded that an increase in the Si/Al ratio of H-ZSM5 (from 2 to 10) increased the acid strength [96]. In terms of the esterification activity, increasing the Si/Al ratio generally decreases the FFA conversion because of the decline in the surface acid sites (Fig. 6a). In certain cases, as presented in Fig. 6b, the FFA conversion does not decline significantly with an increase in the Si/Al ratio. In such a case, the acid strength of the zeolite (in this case, H-MOR) is strong enough to maintain a constant conversion, even with the reduced number of acid sites.

Conversion of oleic acid over zeolite catalysts: a H-MFI and b H-MOR. Reaction conditions: oleic acid:methanol = 1:16; temperature = 333 K; reaction time = 3 h; plot was made by authors using data from [92]
The hydrophobic character of a zeolite is very important in determining its esterification activity. The hydrophobicity originates largely from the nonpolar Si–O–Si linkages in the zeolite framework [96]. The ideal zeolite pores surrounded by SiO4 tetrahedrons do not possess silanol (–SiOH) groups on the surface and should thus be highly hydrophobic. Therefore, zeolites are classified as water-tolerant catalysts in which the acid sites are protected from the adsorption of water molecules and the thermodynamic limitation on the product yield is alleviated (for the reversible reactions where water is produced and impedes the forward reaction) [96]. The hydrophobicity of a zeolite can be reduced by partial substitution of Si4+ by Al3+. A negative charge develops on an oxygen atom of AlO4, to which a counter cation (IA/IIA metal cation or proton) becomes attached and induces affinity to water. Hence, the Si/Al ratio governs the esterification activity by altering the acidity and hydrophobicity of zeolite catalysts. In a similar manner, zeolites exhibit good performance in other water-related reversible reactions such as alcohol dehydration.
In addition to the influences of the Si/Al ratio, the characteristic pore structure of certain zeolite catalysts influences the FFA esterification activity. Chung et al. [92] reported that despite the weaker acid strength and fewer acid sites of H-MFI (Si/Al = 25) relative to H-MOR (Si/Al = 10), the two zeolites exhibited similar FFA esterification activity (80 % conversion of oleic acid at 333 K). It was proposed that the narrow pore entrance and bent (i.e., zigzag) pore structure of MFI extended the residence time of fatty acid molecules inside the pore, thereby promoting C–C bond dissociation and esterification of the captured molecules, which culminated in the FFA conversion being comparable to that of the more acidic H-MOR catalyst [92, 93].
The acid strength of un-doped zeolites may span a wide distribution. Impregnation of zeolites with metal oxides narrows this distribution and enhances the stability of the zeolites. In the evaluation of a WO3-supported USY (FAU) zeolite catalyst for FFA esterification by Costa et al. [98], it was claimed that although WO3 itself is weaker in acidity than USY, the former moderates the surface acidity to a level that is more favorable for esterification and improves the hydrophobicity of the catalyst, thereby enhancing the catalytic activity for oleic acid esterification (with ethanol). They reported that the promotion effect was optimal at a WO3 loading of 11.4 wt%, where 74 % oleic acid conversion was achieved within 2 h in a batch reactor at 473 K. Attempts by Vieira et al. [99] to enhance the esterification activity of H-ZSM5 by incorporating sulfated lanthanum oxide (SO4 2−/La2O3) on the surface showed that the oleic acid conversion of SO4 2−/La2O3/H-ZSM5 (100 %) was higher than that achieved with SO4 2−/La2O3 (96 %), H-ZSM5 (80 %), and La2O3 (67 %). However, in both of the aforementioned studies, the activity of the impregnated zeolites declined with repetition of the reaction, which was attributed to the leaching of the impregnated species [98] and accumulation of products [99], respectively.
5.3 Mesoporous Silicates
Catalyzed biodiesel synthesis is hampered by the mass transfer limitation during catalysis, which is unavoidable because of the large size of fatty molecules. The number of carbon in the triglycerides in conventional feedstock generally range from 12 (lauric) to 18 (linoleic) [26]. Even though some FFAs are formed by the fragmentation of the fatty chains of triglycerides, the molecular size of FFAs is not significantly different from that of triglycerides, which is large enough to cause diffusion resistance in most heterogeneous esterification reactions. As discussed in Sect. 4.2, zeolites are generally good catalysts for esterification; however, the esterification of large acid molecules takes place primarily on the external surface of zeolites because of their microporosity [100]. Thus, alleviation of diffusion resistance by employing a mesoporous structure is a fundamental strategy in the development of FFA esterification catalysts.
Mesoporous silicates with amorphous pore walls, such as the MCM and SBA silicate series, are generally not acidic enough to catalyze esterification reactions. Similar to the case of microporous zeolites, interlinking silicon oxides with other transition metal oxides such as aluminum, titanium, and zirconium oxides generates an appreciable amount of acidic sites on the mesoporous silicates. When a transition metal or an acidic functional group is incorporated into the silicates, the acid functional groups are dispersed on the mesoporous silicates in a denser, more homogeneous manner than on microporous silicates [101]. The development of (metal-incorporated) mesoporous silicates with a homogeneous distribution of strong acid sites on the inner surface of the pores while maintaining the mesoporous dimensions is necessary for further enhancement of the esterification activity of the catalysts [16, 102].
However, the incorporation of transition metals into amorphous silicates does not generate acid sites that are effective for FFA esterification. The aluminum-incorporated species Al-MCM41 is accepted as an active catalyst for a variety of acid-catalyzed reactions; however, it was reported that the activity of Al-MCM41 for the esterification of oleic acid with glycerol was significantly lower than that of zeolite USY and β, both of which had similar Si/Al ratios (Si/Al ~ 15) to Al-MCM41 [102].
Encapsulation of an acidic complex within the pores of mesoporous silicates is another method for enhancing the surface acidity. Mesoporous silicates have a high density of surface silanol (Si–OH) groups. In the case of MCM-41, when a surface area of 800 m2/g is assumed and one Si–OH is present per three Si atoms, the surface silanol density is approximately 4 Si–OH/nm2 [102, 103]. These silanol groups are utilized as the anchoring sites for a large variety of functional groups, among which organosulfonic groups are mostly favored for acidic functionalization. The grafting of organosulfonic groups with the surface silanol groups is a rudimentary method for the preparation of sulfonic acid-functionalized mesoporous silicates. However, the co-condensation method is more effective for achieving homogeneous distribution of the sulfonic acid groups than the grafting method [104–107]. The co-condensation method proceeds via simultaneous precipitation of a mixture of precursors such as tetraethoxysilane and mercaptoalkyl-alkoxy silane. This method facilitates the production of a periodically ordered, uniform organosulfonic-functionalized mesosilicate with a narrow pore size distribution and high acid-exchange capacity [104, 107]. Furthermore, the co-condensation method induces a higher surface acid concentration than the grafting methods [102]. The acid strength of the co-condensed mesoporous silicate can be roughly manipulated by the judicious selection of the organosulfonic precursor [105]. For example, co-condensed SBA-15 functionalized with an arenesulfonic acid group exhibited higher acid strength than congeners functionalized with a mercapto-alkyl group because the electron-withdrawing ability of the phenyl group increases the acid strength of sulfonic groups. Another advantage of the co-condensation method is that hydrophobicity may effectively be induced by the homogeneous coverage of the catalyst surface with (hydrophobic) organosulfonic groups [107].
Mbaraka et al. systematically studied the application of sulfonic-acid-functionalized mesoporous silicates to FFA esterification using 15 wt% palmitic acid dispersed in soybean oil and excess methanol (FFA:methanol = 1:20) at 358 K [106, 108]. Evaluation of the various sulfonic-acid-functionalized catalysts demonstrated that the mesoporous silicate species with a high sulfonic acid density and large median pore was typically active for FFA esterification [106]. The sulfonic-acid density was found to be largely dependent on the surfactant used in the synthesis; the choice of surfactant also influenced the pore size characteristics. The use of a non-ionic polymer surfactant (Pluronic P123) offered the advantage of simultaneous accomplishment of a high sulfonic acid density and large pore diameter compared to the ionic surfactant (n-dodecylamine). An increase in the median pore size of sulfonated mesoporous silicates (from 22 to 35 Å) decreased the activation energy (from 75 to 40 kJ/mol), indicating the impact of activated diffusion (i.e., alleviation of diffusion resistance) on the FFA esterification activity of mesoporous silicates. The acid strength also has a pivotal effect on the activity of sulfonated mesoporous silicates, as exemplified by SBA-15 functionalized with arene-sulfonic acid, which in spite of having a smaller number of acid sites was significantly more active than SBA-15 functionalized with alkyl-sulfonic acid. The arene-sulfonated SBA-15 showed an activity close to that of the homogenous acid H2SO4 and superior activity to that of p-toluenesulfonic acid. Kinetic evaluation confirmed that the enhanced acidic strength, large median pore diameter, and surface hydrophobicity were the main sources of the high activity of arene-sulfonated SBA-15 [106]. The hydrophobicity of sulfonated SBA-15 was demonstrated to be further enhanced by the introduction of additional organic functional groups (methyl, ethyl, or phenyl) by applying the co-condensation or post-grafting method [107]. The co-condensation method was effective for dispersal of the functional groups over the inner surface of the pores, whereas the post-grafting method distributed the functional groups preferentially over the external surface. Both methods enhanced the hydrophobicity of sulfonated SBA-15 without a significant change in the acid site density. However, the additional introduction of a phenyl group decreases the activation energy, attributed to mass transfer resistance derived from the electronic interaction between the phenyl group and the reactants.
5.4 Heteropolyacids
Heteropolyacids are structured multi-oxide clusters that act as strong Brönsted acids in both the homogeneous and heterogeneous phases [108, 109]. Heteropolyacids are composed of a proton (or metal cation) and a heteropolyanion that comprises oxides of an addendum atom (M: V, Mo, W, and so on) and heteroatom (X: P, Si, and so on). The structures of the heteropolyanions can be classified into several groups based on the composition and structure: Keggin-type, (XM12O n−40 ), Silverton-type (XM12O n−42 ), Dawson-type (X2M18O n−62 ), Strandberg-type (X2M5O n−23 ), and Anderson-type (XM6O n−24 ) [108]. Among these structures, Keggin-type anions and their salts are the most widely applied acid catalysts because of their stability and facile synthesis [109]. Numerous examples of the application of heteropolyacids to acid-catalyzed reactions have been documented [108–110]. The hydration of propylene is the first commercialized reaction employing heteropolyacids [110]. The catalytic activity of Keggin-type heteropolyacids (proton form) is essentially dependent on the acid strength, which roughly follows the order: H3PW12O40 > H3SiW12O40 ≥ H3PMo12O40 > H3SiMo12O40 [110]. A selectivity of 91 % (90 % conversion) was achieved with the most acidic of these, H3PW12O40, in the esterification of acetic acid with ethanol at 423 K [111]. When this catalyst was applied to the esterification of fatty acids (myritic, palmitic, stearic, oleic, and linoleic acids) with ethanol, its activity was remarkable, which was close to that achieved with sulfuric acid and p-toluene sulfonic acid [112, 113].
However, the proton form of heteropolyacids is inherently disadvantageous for use as a heterogeneous catalyst because of its small specific surface area (1–10 m2/g) and is also inadequate for use as a heterogeneous catalyst for FFA esterification because it dissolves well in polar solvents such as alcohol.
Both these disadvantages could be resolved by the formation of (NH4–, Cs–, etc.) salts of the heteropolyacids. For instance, the partial substitution of protons in the Keggin-type heteropolyacids with Cs (such as CsxH3-xPW12O40 and CsxH3-xPMo12O40) makes the catalysts insoluble in polar solvents and enhances the specific surface area [114]. The incorporation of Cs in Cs2.5H0.5PW12O40 increased the specific surface area to 157 m2/g [115]. (The specific area of the parent acid H3PW12O40 was merely 6 m2/g.) The most distinctive benefit of making Cs salts is the increase in the surface acidity that is only obtained at a specific degree of Cs substitution: In the case of CsxH4-xPMO11VO40, the amount of surface proton (H+) was ~160 μmol/g at 2 ≤ x ≤ 3, whereas the corresponding value for the mother acid (H4PMO11VO40) was ~10 μmol/g at most [116]. As the Cs substitution (x) increases, the amount of available protons reduces. However, at an appropriate degree of substitution (2 ≤ x ≤ 3, for CsxH4−xPMO11VO40), the increase in the specific surface area outweighs the decrease in the available protons with a consequent increase in the surface acidity (Fig. 7). It follows that such Cs salts are more active in the acid-catalyzed reaction than their proton forms [108, 109, 114, 116]. Moreover, the Cs salts are hydrophobic, which is beneficial for their application to FFA esterification [116].

Surface acidity (amount of surface H+) of CsnH4−nPMo11VO40: opened circle, BET area: closed circle, amount of H+ per anion (PMo11VO40 4−); amount of surface H+ per gram of catalyst: triangle [114]
Wilson and coworkers studied the application of Cs-doped heteropolyacids to the esterification of palmitic acid [117, 118]. CsxH3−xPW12O40 was active toward the esterification of palmitic acid and the maximum activity was achieved at Cs substitutions (x) between 2.0 and 2.3, when the density of accessible acid sites also reached a maximum [117]. Cs incorporation also led to the formation of mesoporous voids ranging 3–4 nm in size, which facilitated the access of palmitic acid to the acid sites. It was claimed that Cs2.3H0.7PW12O40 exhibited superior activity relative to commercial acid catalysts such as SO4/ZrO2, Nafion, and HZSM-5. The authors extended the evaluation of the catalyst to the transesterification of tributyrin in the presence of fatty acid (palmitic acid), with the objective to devise a one-step protocol for biodiesel synthesis from waste oils. As a result, 50.2 % tributyrin conversion, 100 % palmitic acid conversion, and 90 % selectivity of the fatty ester were achieved after 6 h of reaction using a reactant mixture comprising oil (FFA+triglyceride):alcohol = 1:30. The Cs salts are composed of a core–shell structure, wherein the Cs-salt cores are capped with the shell of a proton-form layer. Considering that the proton form is soluble in alcohol, leaching of the acid species may be expected during the reaction. However, it was reported that the catalysts were resistant to leaching, and could be recycled without major loss of activity when x > 1 for CsxH3−xPW12O40 [117] and x > 0.8 for CsxH4−xSiW12O40 [118].
The proton-form heteropolyacids have been immobilized on supports such as activated carbon [119], neutral alumina [120], MCM-41 [121], zirconia [122, 123], niobia [124], and silica [125] for applying the catalysts to esterification. In most cases, the heteropolyacids were well dispersed over the supports, and the catalysts exhibited an acceptable level of surface acidity. However, the supported acids may undergo undesired leaching during the reaction. To preclude leaching of the acid, caution must be exercised in the selection of the support material and immobilization method. Among the common support materials, γ-alumina is unsuitable owing to its surface basicity, which decomposes the heteropolyacids [119]. Adequate calcination of neutral alumina may be used to circumvent this drawback, allowing the heteropolyacids to remain intact after immobilization. Minimal loss of esterification activity was observed for 20–70 % H4PW12O40-supported neutral alumina even after several repetitions of hot-water washing [120]. However, when MCM-41 was used as the support, the catalyst underwent deactivation in a different manner [121]. The initial activity of H3PW12O40/MCM-41 was quite high because of the high dispersion of the heteropolyacid over the MCM-41 internal surface; as the reaction continued, the heteropolyacids migrated toward the external surface of the support and agglomerated into large clusters (~10 nm), accompanied by a decrease in the esterification activity. This clustering phenomenon was expected to be caused by the water produced from esterification, which dissolved heteropolyacids and leached them out to the external surface of the support. Hydrous zirconia (ZrO2·nH2O, or zirconium hydroxide, Zr(OH)4·nH2O) is a good support for heteropolyacids [122]. The strong interaction between H3PW12O40 and the surface hydroxyl groups of hydrous zirconia not only resulted in satisfactory leaching resistance, but also generated Lewis acid sites that were quite effective for the transesterification of canola oil containing 20 wt% FFAs. The esterification/transesterification activity of H3PW12O40/ZrO2·nH2O was better than that of the analogous supported catalysts (H3PW12O40-supported SiO2, Al2O3, or activated carbon). Silica is generally not an adequate support for heteropolyacids because the acids are readily leached in a polar liquid environment. Caetano et al. [125] reported that the immobilization strength could be improved when heteropolyacids (H3PW12O40, H4SiW12O40, H3PMo12O40) were impregnated on silica via the sol–gel method. Prolonged contact of the catalyst with pure methanol over 72 h at 333 K resulted in a loss of only 3 % of the supported heteropolyacids. The FFA esterification activity did not decrease after four consecutive reaction cycles interspersed with methanol washings.
5.5 Sulfated Metal Oxides
Sulfated zirconia (SO4 2−/ZrO2) is the most used catalyst among the sulfated metal oxides. Sulfation imparts several characteristics to zirconia; first, sulfation generates both Brönsted and Lewis acid sites on the zirconia surface (Fig. 8), with an increase in the surface acidity to the super-acidity level (H0 ≤ −16.4) [126, 127]. SO4 2−/ZrO2 is highly active in the isomerization of n-butane where a high level of acidity is required for the catalyst for achieving reasonable activity [128]. Some researchers argued that SO4 2−/ZrO2 is in the league of H-ZSM5 in terms of catalytic activity because the super-acid sites of SO4 2−/ZrO2 do not participate in the acid catalysis [129, 130]. SO4 2−/ZrO2 catalysts are widely used as catalysts or support materials in petroleum refinery, especially for acid-catalyzed reactions such as isomerization, alkylation, and cracking [131]. The surface acidity of SO4 2−/ZrO2 is strongly dependent on the crystallographic phase of zirconia. In the sulfated state, tetragonal zirconia exhibits higher surface acidity and catalytic activity than the more stable, monoclinic zirconia [130, 132]. Secondly, sulfation increases the BET area and improves the thermal stability of zirconia, preventing the reduction of the BET surface area during high-temperature applications. The well-accepted explanation is that the attached SO4 2− ions increase the interatomic Zr–Zr distance, which hinders the agglomeration of ZrO2 crystallites [133]. The enhancement of the BET area and the thermal stability become more pronounced as the fraction of the tetragonal phase in zirconia increases, because the non-bridging hydroxyl groups are prominent on the surface of tetragonal ZrO2, which facilitates the bridging of the surface Zr atoms by the sulfate ions [134]. The activity of sulfated zirconia is strongly affected by the calcination temperature because of the possible dissociation or detachment of the sulfate species from the surface at high temperatures. Loss of the sulfate species results in the loss of the catalytic activity because of a decrease in the acid site density and surface area [135]. In general, the optimal temperature for calcination of sulfated zirconia is 723–773 K.

Generation of Brönsted and Lewis acids on sulfated zirconia; drawing was made by authors based on figure in [126]
López et al. [45] reported that the activity of SO4 2−/ZrO2 was comparable to that of the homogeneous H2SO4 catalyst on a rate-per-site basis (i.e., turnover frequency) in the transesterification of triacetin with ethanol. It was observed that SO4 2−/ZrO2 lost its initial activity accompanied by the leaching of 20 % of the original sulfur after five reaction cycles. Although the authors claimed that the blockage of the active sites was the more probable cause for the loss of activity, leaching of sulfate ions is detrimental for SO4 2−/ZrO2 catalysts, especially when water is present during the reaction. Sulfur leaching becomes particularly substantial at high temperature. Jitputti et al. [136] reported that sulfated zirconia lost its activity after single-cycle transtesterification of palm oil with methanol at 473 K. Goodwin and coworkers [137] later reported that SO4 2−/ZrO2 was more active than other modified zirconia (TiO2–ZrO2 and WO3–ZrO2) species, both in transesterification (of tricaprylin with ethanol) and esterification (of caprylic acid with ethanol), but suffered decline in activity because of sulfur leaching. Omota et al. [138] reported that the leaching of sulfur from SO4 2−/ZrO2 is largely caused by the hydrolysis of the sulfates by water; thus, leaching is minimized when the organic phase contains a small amount of water. Sulfur leaching is a potentially serious problem when SO4 2−/ZrO2 is used in FFA esterification because water is produced in proportion to FFA conversion. López et al. [137] further speculated that alcohol can react with the sulfate ions with the consequent leaching of the ions as H2SO4 (Eq. 4). The authors proposed that the esterification activity of SO4 2−/ZrO2 is not strictly heterogeneous because of mixed catalysis by the sulfate ions attached to the solid catalyst and those dissolved in the liquid phase.
The SO4 2−/ZrO2 system offers the notable advantage of high selectivity for FFA esterification even in the presence of excess alcohol. The use of excess alcohol (alcohol/acid ~100×) in FFA esterification promotes side reactions such as alcohol dehydration (to form dimeric ether). It is known that SO4 2−/ZrO2 exhibits relatively low activity for alcohol dehydration and high selectivity for esterification in the presence of excess alcohol, and is thus suitable as a catalyst for reactive-distillation-assisted FFA esterification, in which the alcohol/acid ratio varies over several orders of magnitude [19].
A common approach for improving the activity and durability of SO4 2−/ZrO2 is forming mixed oxides with other metal oxide components. Jiang et al. [139] reported significant enhancement of the esterification activity and sulfur-leaching resistance of SO4 2−/ZrO2 by forming a mixed oxide with MoO3–Nd2O3. MoO3–Nd2O3 species are homogeneously mixed with ZrO2 and stabilize the tetragonal ZrO2 phase, with preventing the transformation into the more stable monoclinic ZrO2. They claimed that the sulfur species combines with ZrO2 very strongly when the tetragonal ZrO2 phase is dominant. Its mixing with rare-earth oxides confers certain beneficial properties and activity to SO4 2−/ZrO2. First of all, because of the relatively large ionic radius (ca. 10 Å) of the rare-earth cations, incorporation of rare-earth oxides (such as La2O3, Ce2O3, and Yb2O3) increases the surface area by approximately 10–20 % on average compared to that of sulfated zirconia [140]. The surface acidity decreases if a single rare-earth oxide is used to form a mixed oxide with SO4 2−/ZrO2, but the amount of moderate-to-superacid sites increases if a rare-earth oxide and Al2O3 are combined with sulfated zirconia (e.g., SO4 2−/ZrO2–Yb2O3–Al2O3 [140]). The (sulfated) triple oxides such as SO4 2−/ZrO2–Yb2O3–Al2O3 [140] and SO4 2−/ZrO2–TiO2–La2O3 [141] showed improved activity and stability (reusability or sulfur-leaching resistance) during esterification compared to SO4 2−/ZrO2.
Sulfated titania (SO4 2−/TiO2) is another sulfated metal oxide catalyst that is increasingly being studied as an esterification catalyst. Its acidity falls within the level of superacids (H0 < −11.93, [142]). The sulfation procedure is comparatively simple, involving the contact of the titania support with a sulfuric acid solution, followed by drying and calcination of the collected catalyst. Ropero-Vega et al. [143] affirmed that both Brönsted and Lewis sites are developed when the titania is sulfated using ammonium sulfate, whereas only Lewis sites develop when sulfation is performed using sulfuric acid. The distribution of acid strength was wider in the ammonium sulfate catalyst (H0 < +1.2) than in the sulfuric acid catalyst (H0 < +0.8). The authors also emphasized that the sulfated titania was comparable to other acid catalysts in terms of its activity for FFA esterification (oleic acid) because high FFA conversion (58.7 %) and ester selectivity (100 %) could be achieved at low temperature (353 K) using a low catalyst loading (2 wt%).
Sulfated tin oxide (SO4 2−/SnO2) is another superacid catalyst with high potential. The acid strength of most of the acid sites on sulfated tin oxide fall within a narrow distribution, whereas the acid sites on sulfated zirconia cover a relatively wide range of acid strengths [144]. Hence, the average acidity of sulfated tin oxide is higher than that of sulfated zirconia [144, 145]. Furuta et al. [144] reported that sulfated tin oxide was more active than sulfated zirconia for the esterification of n-octanoic acid, especially at low temperatures (below 423 K). A major drawback of sulfated tin oxide is its difficult synthesis because of the tendency of tiny SnO2 particles obtained from the tin-oxide gel, which cannot be collected by conventional filtration. An electrostatic coagulant such as ammonium acetate is required in the washing stage, which neutralizes the surface charge of the SnO2 precipitate and thereby condenses the particles into aggregates large enough to be collected using conventional filtration techniques [146].
5.6 Binary Metal Oxides
Tungstate-zirconia (WO3–ZrO2) is a superacid catalyst (H0 ≈ −14.5) that is especially active for low-alkane isomerization and Friedel–Craft acylation [147]. Tungstate-zirconia exhibits activity comparable to that of sulfuric acid, SO4 2−/ZrO2, and Nafion in the transesterification reaction on the basis of per-site activity [45]. WO3–ZrO2 possesses both Brönsted and Lewis acid sites; however, López et al. [148] employed a selective poisoning technique (using 2,6-di-tert-butylpyridine, which is selectively adsorbed on the Brönsted acid sites) to prove that esterification is catalyzed only by the Brönsted acid sites. It was reported that WO3–ZrO2 was inferior to sulfated zirconia (SO4 2−/ZrO2) in terms of the initial rate of esterification (of caprylic acid with ethanol), whereas WO3–ZrO2 is advantageous over SO4 2−/ZrO2 when long-time reaction or repeated use of the catalyst is required because the activity and textural properties of the former can be easily regenerated by a simple procedure: washing, drying, and re-calcination [137].
In many cases, the surface acidity and acid catalytic activity of WO3–ZrO2 is governed primarily by the surface density of tungstate (W atoms/nm2) [148–150]. It is known that the number of acid sites reaches the maximum when the surface W density is about 5–7 W atoms/nm2, which is roughly consistent with the saturation of tungstate coverage over the zirconia surface (i.e., the formation of a tungstate monolayer) [148, 151, 152]. When this surface W density was exceeded, the tungstate phase was transformed from the monomeric to the polymeric phase with a concomitant decrease in the specific surface area and acid-site density of tungstate–zirconia. López et al. [148] demonstrated the direct correlation of the surface W density with the initial rate of esterification (of acetic acid with methanol) for a tungstate–zirconia catalyst. They asserted that the calcination temperature and tungsten loading are crucial for determining the surface W density and esterification activity of the catalyst. Ramu et al. [153] presented a similar argument about the importance of calcination temperature, but emphasized that the composition of the tetragonal phase in zirconia ultimately determines the surface acidity and esterification activity of the catalyst (Fig. 9). A plethora of evidence has been presented in support of the fact that the monomeric phase of tungstate stabilizes tetragonal zirconia by preventing its transformation into the monoclinic phase [153, 154]. This stabilizing ability of tungstate declines when the temperature exceeds 1,073 K because of the agglomeration of monomeric tungstate into the polymeric species [153, 154]. Hence, the surface W density and tetragonal ZrO2 concentration are like two sides of the same coin in determining the acid catalytic activity of WO3–ZrO2.

Dependence of tetragonal ZrO2 fraction (in entire ZrO2) and esterification activity (palmitic acid conversion) of 5.0 wt% WO3/ZrO2 (impregnated) on calcination temperature; plot was made by authors using data from [153]
Molybdenia-mixed oxide catalysts (MoO3–SiO2, MoO3–ZrO2, MoO3–Al2O3) have also been applied to a diverse acid-catalyzed reactions, including esterification. Compared to typical solid acids, Mo(VI) oxide catalysts are mildly acidic. NH3-TPD analysis of a MoO3/Al2O3 catalyst (impregnated) demonstrated that the acid strength of MoO3 is relatively weaker than that of Al2O3 [155]. It was also reported that the (average) acid strength of MoO3–ZrO2 (9 and 23 atm.% of Mo) was lower than that of SiO2–Al2O3 (Al 13.8 %), H-ZSM5 (Si/Al = 40), and a heteropolyacid salt (Cs2.5H0.5PW12O40) [156]. Moreover, MoO3–ZrO2 (0.07 and 0.12 mmol/g) contained fewer acid sites than SiO2–Al2O3 (0.35 mmol/g) and H-ZSM5 (0.40 mmol/g) [156]. In spite of their mild acidity, the catalytic activity of the molybdenia catalysts was comparable to that of other solid acids, especially for hydrolysis, dehydration, and esterification reactions where water is involved as a reactant or a product [156, 157]. Water exerts a detrimental effect on acid catalysis by temporarily poisoning the acid sites of the catalyst or (in the case of esterification and dehydration) promoting a reverse reaction. Okuhara and coworkers [156] argued that the acid catalytic activity of MoO3–ZrO2 should be explained not only in terms of the acidity but also hydrophobicity. They evaluated the hydrophobicity of several oxide catalysts using the reciprocal density of adsorbed water (1/D) as a parameter, and demonstrated that MoO3–ZrO2 was more hydrophobic than H-ZSM5 (Si/Al = 40) and Cs2.5H0.5PW12O40 [156]. They emphasized that calcination at a temperature exceeding 773 K was a requisite for achieving a high level of catalytic activity with MoO3–ZrO2, which was not related to the generation of new acid sites but to the induction of hydrophobicity, thereby imparting water-tolerance character to the catalyst (Fig. 10).

Dependence of esterification rate (of acetic acid with ethanol) of 23 at.% MoO3–ZrO2 (impregnated) on hydrophobicity; plot was made by authors using data from [156]
It is also notable that when water was present in excess (over 20 % in reaction fluid), the water molecules temporarily generated additional acid sites on MoO3–ZrO2, which accelerated the esterification rate [157]. The accelerating effect was curtailed when water was removed, indicating that the generation of the acid sites was temporary and reversible. It was proposed that Lewis acid sites were generated by the dehydration between Mo–OH and Zr–OH on the catalyst, and the sites were converted into Brönsted acid sites in the presence of water.
The activity of molybdenia catalysts is sensitive to the size of the reactant molecules, culminating in mass transfer limitation during catalysis. A high loading of MoO3 is generally required for the catalysts to retain sufficient acidity, but this high loading blocks the pores of the matrix oxide (SiO2 [158], ZrO2 [159]) impeding the access of large-size reactant molecules. The mass transfer limitation was observed even in the esterification of acetic acid with ethanol (over MoO3/SiO3, impregnated with more than 20 wt% MoO3), where the smallest carboxylic acid was used as a reactant [158]. When fatty acids such as lauric and oleic acid were esterified, the mass transfer limitation became more serious [159]. Thus, for using a molybdenia-based catalyst in FFA esterification, a cautious balance between the acid properties (plus hydrophobicity) and textural properties must be considered in devising the synthesis protocol for the catalyst.
6 Summary and Discussions
Table 2 presents a comparison of the FFA esterification activity of representative catalysts from various categories. A wide spectrum of reaction conditions was adopted in the various studies of FFA esterification; thus, certain standards had to be applied in selecting the studies for the list. Studies in which palmitic or oleic acid was used as the FFA reactant and the FFA conversion was carried out for at 2 or 3 h (in a batch reactor) were selected. As presented in Table 2, all of the catalysts exhibited good activity, the lowest FFA conversion being 70 %. It is thus believed that complete conversion may be achieved in a reasonable reaction time using any of the listed catalysts, if the FFA/Alcohol ratio, temperature, and catalyst loading are optimized to exploit the full activity of the catalysts. In a practical sense, it seems less meaningful to determine which catalyst offers the best FFA conversion, but instead, to select or modify the catalyst in line with the reaction conditions or requirements under consideration. However, if we must choose the most useful catalysts based on the literature data, organic resins and heteropolyacid salts appear to be most effective. Organic resins have been mostly applied in commercial applications because of their high activity and low material cost. In terms of laboratory-scale performance, heteropolyacid salts rank foremost among the catalysts, as they offer the highest initial activity and are quite durable even with repeated use.
The activity of a heterogeneous catalyst for FFA esterification is generally dependent on the surface acidity (acid strength and density), pore dimension, water tolerance (hydrophobicity), and durability (leaching resistance). The major issues are summarized on the basis of the catalyst properties as outlined below.
6.1 Surface Acidity
Surface acidity is the determining factor for the initial activity of heterogeneous FFA esterification catalysts. Heterogeneous acid catalysts such as Nafion, SO4 2−/ZrO2, and WO3-ZrO2 are super-acidic and comparable to homogeneous catalysts (H2SO4) in terms of their per-site esterification activity. In many studies, the acid density is regarded as more important than the acid strength when explaining the activity of a catalyst in terms of its surface acidity. The most common approach for developing FFA esterification catalysts is to choose a support material with a high BET surface area and apply methods to homogenously distribute an acid species over the surface (e.g., supported heteropolyacids, arene-sulfonated SBA-15, and WO3/USY). A number of solid superacids have been utilized as catalysts for FFA esterification, but super or strong acid sites are not considered essential for the catalyst to be active. Catalysts such as Amberlyst-15 and H-MFI are satisfactorily active even though they possess moderate acid sites. More importantly, most of these catalysts feature a high level of surface acid density. Conversely, even when the acid density is comparatively low, the catalyst can be active if its acid sites are sufficiently strong. A good example is H-MOR (Si/Al = 100), which is easily de-aluminated, thereby becoming typically low in surface acid density, but is as active as the H-MOR catalysts with lower Si/Al values (10 ≤ Si/Al ≤ 64).
For the influence of Brönsted and Lewis acid sites, both are active for esterification of FFAs, but several authors claimed that Brönsted site is kinetically better than Lewis site [160, 161] (i.e., Lewis acid sites usually require higher pressure and temperature than Brönsted acid sites to get a comparable esterification activity.). However, it needs more specific researches about this topic for each category of catalyst. Because many esterification catalysts contain both Brönsted and Lewis acid sites, the selective formation of a specific acid site could be a key to improve the activity of each catalyst.
6.2 Pore Dimension
The issue of pore dimension is raised particularly for zeolite and molybdenia-embedded catalysts. Molybdenia-embedded catalysts suffer from mass transfer resistance because the molybdenia blocks the intergranular spaces of the matrix. The microporosity of zeolite catalysts is a handicap in terms of the mass transfer (pore diffusion) rate, but in the case of certain pore geometries (e.g., the zigzag pore tunnels of H-MFI), the microporosity may enhance FFA conversion by prolonging the residence time of FFA molecules inside the micropores. From another perspective, it is supposed that such an effect may be possible for the zeolites because the acid sites are uniformly distributed over the pore surface. In contrast, the mesoporous multioxides have an inherent advantage in terms of the pore dimension and the diffusion resistance of fatty reactants. Organic resin catalysts are also advantageous because they swell and become macroporous when exposed to an alcohol-excess environment. However, the mass transfer limitation may originate from other sources such as entanglement of FFA with the polymeric nano-domain on the resin surface. In the case of mesoporous acid oxides such as arene-sulfonated SBA-15, the dipole–dipole interaction between the surface phenyl groups and the reactants increases the mass transfer resistance.
6.3 Water Tolerance
Water tolerance is essential for FFA esterification catalysts. The hydrophobicity of the catalyst assumes increasing importance when the reaction is conducted at low temperature. At temperatures below 373 K, progressive buildup of water in the pores impedes the forward reaction by hindering the access of the reactant molecules and promoting the hydrolysis of the fatty esters (i.e., reverse esterification). The majority of FFA esterification catalysts are hydrophobic. However, the sulfonated catalysts containing resin sulfonic acids are largely hydrophilic. Their hydrophilicity can be reduced by incorporating hydrophobic components into the catalysts, such as achieved by surface modification of sulfonated silicates with alkyl functional groups, and using organic polymer binders in pelletizing resin sulfonic acids. In the synthesis of sulfonic-acid-functionalized catalysts, the choice of the precursor for the sulfonic acid group and the technique for synthesizing the catalyst (cocondensation or postgrafting) influence the hydrophobicity of the final catalysts. For zeolites and mesoporous aluminosilicates, the Si/Al ratio should be determined based on a balance of the surface acidity and hydrophobicity. The hydrophobicity increases as the Si/Al ratio increases, whereas the acid density follows the opposite trend. The Cs salts of heteropolyacids are hydrophobic. The acidity can be modified by changing the degree of Cs substitution, with little influence on the hydrophobicity. Among the catalysts discussed in this paper, the molybdenia–zirconia (MoO3–ZrO2) catalyst may be the most hydrophobic. The esterification activity of MoO3–ZrO2 is more strongly dependent on its hydrophobicity than on the surface acidity.
6.4 Durability (Leaching Resistance)
The durability of FFA esterification catalysts is measured in terms of the extent of the change in FFA conversion after 3–5 reaction cycles. Leaching of active species is the major reason for the decrease in conversion, which is substantial for the sulfonic-acid-functionalized catalysts. The sulfonic acid species in SO4 2−/ZrO2 are not only leached out by water but also by the alcohol used as a reactant. The leaching resistance of the catalysts can be improved by the judicious formation of mixed oxides (e.g., SO4 2−/ZrO2–MoO3–Nd2O3, SO4 2−/ZrO2–Yb2O3–Al2O3, and SO4 2−/ZrO2–TiO2–La2O3). Most organic resins suffer leaching of sulfonic acid during the reaction, with Amberlyst BD-20 (and its analogues) being an exception; the latter exhibit outstanding leaching resistance among the resin sulfonic acid catalysts. Cs salts of the heteropolyacids with a salt-acid core–shell structure show resistance to leaching within a specific range of Cs substitution, depending on the heteropolyanion. Immobilization of heteropolyacids using neutral alumina or hydrous zirconia as the support material provides satisfactory leaching resistance of the heteropolyacid.
References
Rana MS, Sámano V, Ancheyta J, Diaz JAI (2007) Fuel 86:1216
Ko CH, Park SH, Jeon J-K, Suh DJ, Jeong K-E, Park Y-K (2012) Korean J Chem Eng 29(12):1657
Van Gerpen J (2005) Fuel Process Technol 86:1097
McCormick RL, Graboski MS, Alleman TL, Herring AM (2001) Environ Sci Technol 35:1742
Wang WG, Lyons DW, Clark NN, Gautam M (2000) Environ Sci Technol 34:933
Tat ME, Van Gerpen JH, Soylu S, Canakci M, Monyem A, Wormley S (2000) J Am Oil Chem Soc 77:285
Ban-Weiss GA, Chen JY, Buchholz BA, Dibble RW (2007) Fuel Process Technol 88:659
Biodiesel Handling and Use Guide. 4th edn. National Renewable Energy Laboratory, NREL/TP-540-43672, 2009)
Timilsin GR, Shresth A (2011) Energy 36(4):2055
Narasimharao K, Lee A, Wilson K (2007) J Biobased Mater Bioenergy 1:19
Ma F, Hanna MA (1999) Bioresour Technol 70:1
Schuchardta U, Serchelia R, Vargas RM (1989) J Braz Chem Soc 9:199
Barrault J, Pouilloux Y, Clacens JM, Vanhove C, Bancquart S (2002) Catal Today 75:177
Bondioli P (2004) Top Catal 27:77
Pinto AC, Guarieiro LLN, Rezende MJC, Ribeiro NM, Torres EA, Lopes WA, Pereira PA, Andrade JB (2005) J Braz Chem Soc 16:1313
Lotero E, Liu Y, Lopez DE, Suwannakarn K, Bruce DA, Goodwin JG (2005) Ind Eng Chem Res 44:5353
Huber GW, Iborra S, Corma A (2006) Chem Rev 106:4044
Meher LC, Sagar DV, Naik SN SN (2006) Renew Sustain Energy Rev 10:248
Kiss AA, Dimian AC, Rothenberg G (2005) Adv Synth Catal 348:75
Lotero E, Goodwin JG, Bruce DA, Suwannakarn K, Liu Y, Lopez DE (2006) The catalysis of biodiesel synthesis. In: Spivey J (ed) Catalysis, vol 19. The Royal Society of Chemistry, London, p 41
Kulkarni MG, Dalai AK (2006) Ind Eng Chem Res 45:2901
Saraf S, Thomas B (2007) Process Saf Environ Prot 85:360
Demirbas A (2008) Energy Conserv Manag 49:125
Balat M, Balat H (2008) Energy Conserv Manag 49:2727
Di Serio M, Tesser R, Pengmei L, Santacesaria E (2008) Energy Fuels 22:207
Lee D-W, Park Y-M, Lee K-Y (2009) Catal Surv Asia 13:63
Malilas W, Kang SW, Kim SB, Yoo HY, Chulalaksananukul W, Kim SW (2013) Korean J Chem Eng 30(2):405
Lee JH, Kim SB, Yoo HY, Lee JH, Park C, Han SO, Kim SW (2013) Korean J Chem Eng 30(6):1272
Nelson LA, Foglia TA, Marmer WN (1996) J Am Oil Chem Soc 73(8):1191
Watanabe Y, Shimada Y, Sugihara A, Tominaga Y (2001) J Am Oil Chem Soc 78(2):703
Lee J (2007) News Inform Chem Eng 25(6):613
Canakci M, Van Gerpen J (1999) Trans ASAE 42:1203
Freedman B, Pryde EH, Mounts TL (1986) J Am Oil Chem Soc 63(10):1375
Abro S, Pouilloux Y, Barrault J (1997) Stud Surf Sci Catal 108:539
Choi J, Kim D, Park J, Rhee Y, Lee J, Kor J (2008) Chem Eng Res 46(1):194
Talukder MMR, Wu JC, Lau SK, Cui LC, Shimin G, Lim A (2009) Energy Fuels 23(1):1
López DE, Goodwin JG Jr, Bruce DA (2007) J Catal 245(2):381
Kiss AA, Omota FO, Dimian AC, Rothenberg G (2006) Top Catal 40(1–4):141
Furuta S, Matsuhashi H, Arata K (2004) Catal Commun 5:721
Buasri A, Ksapabutr B, Panapoy M, Chaiyut N (2012) Korean J Chem Eng 29(12):1708
Kim H-K, Kang B-S, Kim M-J, Kim D-K, Lee J-S, Lee K-Y (2004) Stud Surf Sci Catal 150:201
Leclercq E, Finiels A, Moreau C (2001) J Am Oil Chem Soc 78:1161
Suppes GJ, Dasari MA, Doskocil EJ, Mankidy PJ, Goff MJ (2004) Appl Catal A 257:213
Xie W, Huang X, Li H (2007) Bioresour Technol 98:936
López DE, Goodwin JG Jr, Bruce DA, Lotero E (2005) Appl Catal A 295:97
Albuquerque MCG, Jiménez-Urbistondo I, Santamaría-González J, Mérida-Robles JM, Moreno-Tost R, Rodríguez-Castellón E, Jiménez-López A, Azevedo DCS, Cavalcante CL Jr, Maireles-Torres P (2008) Appl Catal A 334:35
Saravanamurugan S, Sujandi D-S, Han J-BK, Park S-E (2008) Catal Commun 9:158
Cantrell DG, Gillie LJ, Lee AF, Wilson K (2005) Appl Catal A 287:183
Xie W, Peng H, Chen LJ (2006) J Mol Catal A 246:24
Liu Y, Lotero E, Goodwin JG Jr, Mo X (2007) Appl Catal A 331:138
Sercheli R, Ferreira ALB, Guerreiro MC, Vargas RM, Sheldon RA, Gelbard G (1997) Tetrahedron Lett 38:1325
Sercheli R, Vargas RM, Schuchardt U (1999) J Am Oil Chem Soc 76:1207
Peter SKF, Ganswindt R, Neuner H-P, Weidner E (2002) Eur J Lipid Sci Technol 104:324
Bournay L, Casanave D, Delfort B, Hillion G, Chodorge J (2005) Catal Today 106:190
Di Serio M, Tesser R, Pengmei L, Santacesaria E (2008) Energy Fuels 22:207
Cha W (2007) News Inform Chem Eng 25(6):609
Supple B, Holward-Hildige R, Gonzalez-Gomez E, Leahy JJ (2002) J Am Oil Chem Soc 79(2):175
Cvengros J, Cvengrosova Z (2004) Biomass Bioenergy 27:173
Ki-Teak L, Foglia TA, Chang K (2002) J Am Oil Chem Soc 79(2):191
Zappi M, Hernandez R, Sparks D, Horne J, Brough M, Arora SM, Motsenbocker WD (2003) A review of the engineering aspects of the biodiesel industry. Mississippi Biomass Council, Jackson, p 71
Zhang Y, Dubé MA, McLean DD, Kates M (2003) Bioresour Technol 89(1):1
Zhang Y, Dubé MA, McLean DD, Kates M (2003) Bioresour Technol 90(2):229
Ledahudec J, Pokomý J (1991) Die Nahrung 35(10):1071
Choe E, Min DB (2007) J Food Sci 72(5):R77
Dobarganes MC (2009) Formation of new compounds during frying—general observation. http://lipidlibrary.aocs.org/frying/c-newcpds/index.htm. Accessed 2 Feb 2009
Christie WW, Dobson G (2011) Formation of cyclic fatty acids during frying. http://lipidlibrary.aocs.org/frying/c-cyclic/index.htm. Accessed 15 June 2011
Orthoefer FT, Gurkin S, Liu L (1996) In: Perkins EG, Erickson MD (eds) Deep frying: chemistry, nutrition, and practical applications. AOCS Press, Champaign, p 223
Chhetri AB, Watts KC, Islam MR (2008) Energies 1:3
Canakci M, Gerpen JV (2001) Trans ASAE 44:1429
Mulder C, Shouten JA, Popp-Snijders C (1983) J Clim Chem Clin Biochem 21:823
Hoydoncks HE, De Vos DE, Chavan SA, Jacob PA (2004) Top Catal 27:83
Freedman B, Pryde EH, Mounts TL (1984) J Am Chem Soc 61:1638
Sonntag N (1982) J Am Oil Chem Soc 59(10):735A
Otera J (2001) Angew Chem 113:2099
Lepper H, Friesenhagen L (1986) US patent 4,608,202
Koono S, Moriya O, Noguchi T, Okamura H (1993) EP patent 0566047
Hayyan A, Hashim MA, Mirghani MES, Hayyan M, AlNashef IM (2013) Korean J Chem Eng 30(6):1229
Harmer MA, Sun Q (2001) Appl Catal A 221:45
Samms SR, Wasmus S, Savinell RF (1996) J Electrochem Soc 143:1498
Hino M, Arata K (1981) Chem Lett 1671
Okuhara T, Kimura M, Kawai T, Xu Z, Nakato T (1998) Catal Today 45:73
Honkela ML, Root A, Lindblad M, Krause AOI (2005) Appl Catal A 245:289
Özbay N, Oktar N, Tapan NA (2008) Fuel 87:1789
Marchal P, Masson C, Moulongui Z, Delmas M, Gaset A (1985) Rev Fr Corps Gras 32:429
Asdih M, Marchal P, Moulongui Z, Gharbi RL, Delmas M, Gaset A (1990) Rev Fr Corps Gras 37:241
Goto S, Takeuchi M, Matouq MH (1992) Int J Chem Kinet 24:587
Park J, Wang Z, Kim D, Lee J (2010) Ren. Eng. 35:614
http://www.energytype.com/2009/12/biodiesel-process-improvement-by.html
Park J, Kim D, Lee J (2010) Bioresour Technol 101:S62
Liu Y, Loteo E, Goodwin JG Jr (2006) J Catal 243:221
Chung K-H, Chang D-R, Park B-G (2008) Bioresour Technol 99:7438
Chung K-H, Park B-G (2009) J Ind Eng Chem 15:388
Kirumakki S, Nagaraju N, Chary KVR (2006) Appl Catal A 299:185
Niwa M, Katada N (1997) Catal Surv Jpn 1:215
Okuhara T (2002) Chem Rev 102:3641
Tsutsumi K, Nishiyama K (1989) Thermochim Acta 143:299
Costa AA, Braga PRS, de Macedo JL, Dias JA, Dias SCL (2012) Microporous Mesoporous Mater 147:142
Vieira SS, Magriotis ZM, Santos NAV, Saczk AA, Hori CE, Arroyo PA (2013) Bioresour Technol 133:248
Corma A, Rodriguez M, Sanchez N, Aracil J (1994) WO Patent, WO9413617
Viswanathan B, Jacob B (2005) Catal Rev Sci Eng 47:1
Pérez-Pariente J, Díaz I, Mohino F, Sastre E (2003) Appl Catal A 254:173
Kolodziejski W, Corma A, Navarro M, Pérez-Pariente J (1993) Solid State Nucl Mag Res 2:253
Margolese D, Melero JA, Christiansen SC, Chmelka BF, Stucky GD (2000) Chem Mater 12:2448
Melero JA, Stucky GD, van Griekena R, Morales G (2002) J Mater Chem 12:1664
Mbaraka IK, Radu DR, Lin VS, Shanks BH (2003) J Catal 219:329
Mbaraka IK, Shanks BH (2005) J Catal 229:365
Okuhara T, Mizuno N, Misono M (1996) Adv Catal 41:113
Lee K-Y, Misono M (2007) Heteropoly compounds. In: Ertl G, Knözinger H, Weitkamp J (eds) Handbook of heterogeneous catalysis, vol 1. VCH, Weinheim, p 118
Kozhevnikov IV (2002) Catalysts for fine chemical synthesis: catalysis by polyoxometalates, vol 2. Wiley, Chichester, pp 67–68
Izumi Y, Hasebe R, Urabe K (1983) J Catal 84:402
Cardoso AL, Augusti R, Da Silva MJ (2008) J Am Oil Chem Soc 85:555
Cao F, Chen Y, Zhai F, Li J, Wang J, Wang X, Wang S, Zhu W (2008) Biotechnol Bioeng 101:93
Lee K, Oishi S, Igarashi H, Misono M (1997) Catal Today 33:183
Nishimura T, Okuhara T, Misono M (1991) Appl Catal 73:L7
Yang J-I, Lee D-W, Lee J-H, Hyun J-C, Lee K-Y (2000) Appl Catal A 194–195:123
Narasimharao K, Brown DR, Lee AF, Newman AD, Siril PF, Tavener SJ, Wilson K (2007) J Catal 248:226
Pesaresi L, Brown DR, Lee AF, Montero JM, Williams H, Wilson K (2009) K. Appl Catal A 360:50
Izumi Y, Urabe K (1981) Chem Lett 663
Sharma P, Vyas S, Patel A (2004) J Mol Catal A 214:281
Verhoef M, Kooyman PJ, Peters JA, van Bekkum H (1999) Microporous Mesoporous Mater 27:365
Kulkani MG, Gopinath R, Meher LC, Dalai AK (2006) Green Chem 8:1056
Rao KN, Sridhar A, Lee AF, Tavener SJ, Young NA, Wilson K (2006) Green Chem 8:790
Srilatha K, Lingaiah N, Prabhavathi Devi BLA, Prasad RBN, Venkateswar S, Sai Prasad PS (2009) Appl Catal A 365:28
Caetano CS, Fonseca IM, Ramos AM, Vital J, Castanheiro JE (2008) Catal Commun 9:1966
Lunsford JH, Sang H, Campbell SM, Liang C-H, Anthony RG (1994) Catal Lett 27:305
Corma A (1997) Curr Opin Solid State Mater Sci 2(1):63
Hino M, Kobayashi S, Arata K (1979) J Am Chem Soc 101:6439
Morterra C, Cerrato G, Emanuel C, Bolls V (1993) J Catal 142:349
Spielbauer D, Mekhemer GAH, Knözinger H (1996) Catal Lett 40:71
Song X, Sayari A (1996) Catal Rev Sci Eng 38:329
Huang BC, Huang ZT, Ma ZF (1999) J Mol Catal (China) 13:383
Norman CJ, Goulding PA, McAlpine I (1994) Catal Today 20:313
Ward DA, Ko EI (1995) J Catal 157:321
Li X, Nagaoka K, Simon LJ, Olindo R, Lercher JA (2007) Catal Lett 113:34
Jitputti J, Kitiyanan B, Rangsunvigit P, Bunyakiat K, Attanatho L, Jenvanitpanjakul P (2006) Chem Eng J 116:61
López DE, Goodwin JG Jr, Bruce DA, Furuta S (2008) Appl Catal A 339:76
Omota F, Dimian AC, Bliek A (2003) Chem Eng Sci 58:3175
Jiang K, Tong D, Tang J, Song R, Hu C (2010) Appl Catal A 389:46
Yu GX, Zhou XL, Li CL, Chen LF, Wang JA (2009) Catal Today 148:169
Li Y, Zhang X-D, Sun L, Zhang J, Xu H-P (2010) Appl Energy 87:156
Moghaddam MK, Gholami MR (2006) Mater Lett 60:715
Ropero-Vega JL, Aldana-Pérez A, Gómez R, Niño-Gómez ME (2010) Appl Catal A 379:24
Furuta S, Matsuhashi H, Arata K (2004) Appl Catal A 269:187
Wang GW, Hattori H, Tanabe K (1983) Chem Lett 277
Matsuhashi H, Miyazaki H, Kawamura Y, Nakamura H, Arata K (2001) Chem Mater 13:3038
Hino M, Arata K (1988) J Chem Soc Chem Commun 1259
López DE, Suwannakarn K, Bruce DA, Goodwin JG Jr (2007) J Catal 247:43
Wilson RD, Barton DG, Baertsch CD, Iglesia E (2000) J Catal 194:175
Baertsch CD, Komala KT, Chua Y-H, Iglesia E (2002) J Catal 44:44
Wachs IE, Kim T, Ross EI (2006) Catal Today 116:162
Barton DG, Soled SL, Iglesia E (1998) Top Catal 6:87
Ramu S, Lingaiah N, Prabhavathi BLA, Prasad RBN, Suryananrayana I, Sai Prasad PS (2004) Appl Catal A 276:163
Park Y-M, Chung S-H, Eom HJ, Lee J-S, Lee K-Y (2010) Bioresour Technol 101:6589
Mitran G, Makó É, Rédey Á, Marcu I-C (2010) Catal Lett 140:32
Li L, Yoshinaga Y, Okuhara T (1999) Phys Chem Chem Phys 1:4913
Li L, Yoshinaga Y, Okuhara T (2002) Catal Lett 83(3–4):231
E-A A, Said A, El-Wahab MMA (2006) J Chem Technol Biotechnol 81:329
Bail A, Santos VC, Freitas MR, Ramos LP, Schreiner WH, Ricci GP, Ciuffi KJ, Nakagaki S (2013) Appl Catal B 130–131:314
Cardoso AL, Neves SCG, da Silva MJ (2008) Energies 1:79
Suwannakarn K (2008) Biodiesel Production from High Free Acid Content Feedstocks. PhD dissertation, Clemson University, South Carolina
Acknowledgments
This work was supported by the MSIP (Ministry of Science, ICT & Future Planning) of Korea Grant funded by the Korean Government (NRF-2012R1A2A1A03009667).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Lee, DW., Lee, KY. Heterogeneous Solid Acid Catalysts for Esterification of Free Fatty Acids. Catal Surv Asia 18, 55–74 (2014). https://doi.org/10.1007/s10563-014-9166-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10563-014-9166-y