
Abstract
Purpose
Dronedarone is a novel multichannel blocker with antiadrenergic and vasodilatory properties. The aim of this study was to investigate the effects of dronedarone on functional capacity in patients with severe left ventricular (LV) dysfunction and compensated stable heart failure (HF).
Methods
This was a multicentre, double-blind, randomized, placebo-controlled, dose-escalating study. Patients in sinus rhythm with impaired LV function (LV ejection fraction [LVEF] ≤ 30%) and compensated HF (New York Heart Association [NYHA] class I–II), who would continue to receive cardiovascular treatment (excluding antiarrhythmic agents), were eligible. A total of 124 patients were randomized to receive dronedarone (400 mg or 800 mg once daily or 600 mg twice daily) or placebo for 30 days. The primary objective was assessment of the effects of dronedarone on functional capacity, using the 6 min walk test. Secondary objectives included the effects of dronedarone on LVEF, cardiothoracic ratio, NYHA status, and Holter parameters.
Results
A total of 111 patients completed the study. There were no significant differences between dronedarone and placebo with respect to walking distance and LVEF. The cardiothoracic ratio was similar in all treatment groups throughout the study, and the NYHA status did not change in the majority of patients. Dronedarone was well tolerated and, as expected, decreased heart rate. No new arrhythmic events or torsades de pointes were reported.
Conclusions
Short-term treatment with dronedarone did not affect exercise capacity and did not decrease LVEF in patients with severe LV dysfunction and compensated HF.
Similar content being viewed by others


Introduction
Atrial fibrillation (AF) is the most common arrhythmia in clinical practice [1] and a major cause of morbidity and mortality [2–5]. Dronedarone is a novel multichannel blocker with antiadrenergic and vasodilatory properties that was developed for the treatment of patients with AF [6–8].
Dronedarone is effective in maintaining sinus rhythm and reducing the ventricular rate in patients with paroxysmal and persistent AF [9, 10]. A 25% to 55% reduction in the risk of recurrence of AF or atrial flutter was observed with dronedarone compared with placebo in several large controlled studies [9, 10]. Dronedarone also reduced ventricular rate during AF in these trials. In the ERATO (Efficacy and safety of dRonedArone for The cOntrol of ventricular rate during atrial fibrillation) trial, dronedarone significantly reduced the mean heart rate during submaximal and maximal exercise without decreasing exercise duration in patients with permanent AF receiving standard heart-rate lowering therapies [8, 11]. In ERATO, as well as in other studies, dronedarone was associated with a low risk of proarrhythmia, including torsades de pointes [8–10].
The ANDROMEDA (ANtiarrhythmic trial with DROnedarone in Moderate to severe congestive heart failure Evaluating morbidity DecreAse) study, conducted in patients with recent hospitalization due to severe heart failure (HF), was prematurely stopped because of a higher number of deaths in the dronedarone group at the time of an intermediate safety analysis [12]. The ATHENA (A placebo-controlled, double-blind, parallel arm Trial to assess the efficacy of dronedarone 400 mg bid for the prevention of cardiovascular Hospitalization or death from any cause in patiENts with Atrial fibrillation/atrial flutter) study, which excluded patients with New York Heart Association (NYHA) class IV or recently decompensated HF, showed a significant reduction in the combined endpoint of cardiovascular hospitalizations or death with dronedarone in patients with AF [14]. Dronedarone was approved by the US Food and Drug Administration in July 2009 to reduce the risk of cardiovascular hospitalization in patients with paroxysmal or persistent AF or atrial flutter, with a recent episode of AF/atrial flutter and associated cardiovascular risk factors, who are in sinus rhythm or who will be cardioverted [15]. It is contraindicated in patients with NYHA class IV HF, or NHYA class II–III HF with a recent decompensation requiring hospitalization or referral to a specialized HF clinic.
During the early course of development of dronedarone, an ascending dose study was specifically designed to look at tolerability in patients with compensated HF, with severe left ventricular (LV) dysfunction. This paper reports the findings from this study.
Methods
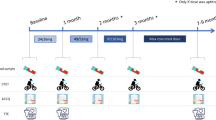
This double-blind, randomized, placebo-controlled, dose-escalating study in three sequential groups was conducted in 12 US centers between June 1996 and March 1998. All patients gave written informed consent to participate in the study, which was approved by local institutional review boards and conducted in accordance with the Helsinki principles [16] and Guidelines for Good Clinical Practice.
Patients
Patients aged 18–75 y who met the following criteria were eligible for inclusion in the study: in sinus rhythm; impaired LV systolic function (LV ejection fraction [LVEF] ≤ 30%), documented with radionuclide ventriculography (multiple uptake gated acquisition [MUGA] scan), echocardiography, or ventricular angiogram during the screening phase of the study and confirmed with a radionuclide ventriculography at baseline; known dilated cardiomyopathy of any cause; and HF (NYHA class I–II) stable during the previous 2 months. Patients were selected 1 month to 2 weeks before study entry.
The main exclusion criteria were as follows: evidence of clinically relevant hematologic, hepatic, gastrointestinal, renal, pulmonary, endocrinologic, or psychiatric disease; clinical evidence of acute HF under optimal treatment with angiotensin-converting enzyme (ACE) inhibitors and diuretics; unstable angina, recent myocardial infarction (≤3 months), atrioventricular block (second degree or more); history of ventricular tachycardia or ventricular fibrillation necessitating chronic treatment with antiarrhythmic agents; uncontrolled hypertension; abnormal laboratory tests; females of childbearing potential; concomitant therapy with antiarrhythmic agents, beta-blockers, verapamil, diltiazem, tricyclic antidepressants, anticonvulsants, or phenothiazines. Patients were allowed to receive cardiac glycosides, diuretics, ACE inhibitors, and nitrates, provided the doses were kept constant during the 14 days preceding the study and throughout the study.
Treatment
Patients were randomized in a 2:1 ratio in three sequential groups to receive dronedarone (400 mg once daily, 800 mg once daily, or 600 mg twice daily) or placebo for 30 days. Before progression to the next-dose step a safety committee blinded to treatment allocation assessed the safety endpoints (walking distance, LVEF, and NYHA status).
Evaluation criteria
The primary objective was to assess the effects of dronedarone on functional capacity, measured during a standardized 6 min walk test of submaximal exercise capacity [17] at Days 8 and 30.
Secondary objectives were to assess the effects of dronedarone on LVEF and cardiothoracic ratio, as assessed by radionuclide ventriculography and chest x-ray, respectively, at baseline and on Day 30.
The effects of dronedarone on NYHA status, hepatic and renal function, and Holter parameters (premature ventricular contraction counts, arrhythmic events, heart rate, and QT variability) were assessed. Twelve-lead electrocardiogram (ECG) measurements (heart rate; QT, corrected QT [QTc], and PR intervals) were recorded on Days 1, 8, and 30 pre-dose and 4 h post-dose; and on Days 15 and 22 post-dose at time of study visit and at the post-study visit (Day 38). Holter measurements were recorded over 24 h on Day 0 and over 48 h on Days 1/2, 8/9, and 29/30.
Patients underwent clinical examination at scheduled visits on Days 1, 2, 8, 9, 15, 22, 29, 30, and 38. Clinical laboratory parameters (hematology, biochemistry, and urinalysis) were measured at screening, baseline, and on Days 8, 30, and 38, and vital signs were monitored at scheduled visits. Adverse events were assessed at each scheduled visit and coded according to the World Health Organization Adverse Reaction Terminology.
Statistical methods
The statistical analyses included data for all patients who were randomized to treatment. Safety analyses included all available data for all patients. The main safety and efficacy parameters for the treatment groups were compared at baseline, using analysis of variance (ANOVA) or Kruskal-Wallis test according to the variable distribution for quantitative parameters, and the Fisher’s exact test when relevant. The frequency and percentage of patients with at least one adverse event were recorded by treatment. Statistical analyses of clinical data were performed using SAS software (Version 6–09; SAS Institute Inc, Cary, NC, USA).
Walking distance, LVEF, and cardiothoracic ratio were measured at baseline, and changes from baseline were calculated at each subsequent time point (walking distance, Days 8 and 30; LVEF and cardiothoracic ratio, Day 30). Changes from baseline were analyzed by ANOVA. Equivalence of dronedarone doses and placebo was concluded if the 90% confidence intervals (CIs) were entirely within the reference interval (walking distance, 0.85–1.15; LVEF and cardiothoracic ratio, 0.80–1.20).
The mean difference of the measurement of walking distance on Day 8 and Day 30 vs. placebo was calculated (see Table 2, fourth column) as well as the 90% confidence interval (see Table 2, fifth column). The difference was calculated with respect to placebo in order to take into account a potential placebo effect during the study (for example a “training effect” due to the walking test itself). A potential decrease in walking distance was considered significant if the lower limit of the 90% confidence interval was >15% the baseline value (i.e. >60.7 m). In terms of safety analyses, individual values that were outside the normal laboratory range and predefined marked increases and decreases from baseline were noted. In addition, patients with high QTc intervals (QTc >450 ms for men, >470 ms for women) were identified.
The authors had full access to the data and take responsibility for its integrity. All authors have read and agree to the manuscript as written.
Results
Of the 247 patients who were screened before the start of the study, 124 were randomized to treatment (29 dronedarone 400 mg, 30 dronedarone 800 mg, 25 dronedarone 1,200 mg, 40 placebo; Fig. 1). The main reasons 123 patients were not randomized to treatment included no current impairment of LV function (95 patients) and abnormal laboratory tests (54 patients). A total of 111 patients completed the study (27 dronedarone 400 mg, 27 dronedarone 800 mg, 19 dronedarone 1,200 mg, 38 placebo); 13 patients withdrew from the study, and the reasons for withdrawal are detailed in Fig. 1.

Patient disposition
Baseline characteristics
At baseline, demographic characteristics, walking distance, LVEF, and cardiothoracic ratio were comparable among the treatment groups (Table 1). Concomitant medication usage was equally distributed across the treatment groups and was in line with recommendations at the time the study was conducted.
Additional baseline analyses demonstrated that the treatment groups were similar in terms of duration and physical symptoms of HF (results not shown).
Exercise capacity
No significant difference was reported among treatment groups with respect to the primary endpoint of walking distance during the 6 min test, indicating no decrease in exercise capacity on Days 8 and 30 (Table 2). The mean walking distances in the dronedarone 400 mg and 800 mg groups were equivalent to placebo on Days 8 and 30 (Table 2). The results of the analysis of the mean values for walking distance at different timepoints are shown in Fig. 2.

Walking distance (meters) at baseline and during study treatment (mean ± SEM)
LVEF and cardiothoracic ratio
The difference in mean LVEF for each dronedarone treatment group compared with placebo on Day 30, along with the respective 90% confidence intervals, are shown in Fig. 3.

Difference vs. placebo for left ventricular ejection fraction (%) after 30 days’ treatment with dronedarone 400 mg, 800 mg, or 1,200 mg daily. Error bars represent 90% confidence interval of the difference vs. placebo
No significant differences for LVEF on Day 30 were noted among treatment groups (Fig. 3; dose effect from ANOVA on observed values = 0.37). The cardiothoracic ratio was similar in all treatment groups at baseline and throughout treatment.
NYHA status
The majority of patients did not experience any change in NYHA status during treatment compared with baseline. A total of 7/84 patients receiving dronedarone and 4/40 receiving placebo had changes in NYHA status while on treatment; however, no trends between the changes and treatment were detected. One patient in the dronedarone 400 mg group, treated until Day 30, had an increase in NYHA status from class I at baseline to class IV (on Day 38) in the context of angina pectoris and acute myocardial infarction leading to death on Day 42.
Electrophysiological activity
The heart rate–lowering properties of dronedarone were confirmed by the ECG and Holter measurements. A moderate decrease was observed in mean heart rate with dronedarone 800 mg and 1,200 mg, together with a moderate increase in mean PR interval, and no significant change in mean QRS and QTc intervals compared with baseline on Day 30 at 4 h post-dosing. Mean changes from baseline in heart rate on Day 30 (4 h post-dosing) were −6.5 (standard error of the mean [SEM] 2.0) beats/min (p = 0.0037) and −8.5 (SEM 3.3) beats/min (p = 0.019) in the dronedarone 800 mg and 1,200 mg groups, respectively. Heart rate returned to baseline values on Day 38, 1 week after dronedarone discontinuation, with no evidence of a rebound phenomenon.
Mean changes from baseline in PR interval on Day 30 (4 h post-dosing) were 9.6 (SEM 3.8) ms (p = 0.0193) and 12.9 (SEM 4.2) ms (p = 0.0076) in the dronedarone 800 mg and 1,200 mg groups, respectively. For the placebo and dronedarone 400 mg groups, none of the changes from baseline were significant.
A significant dose-related decrease in average heart rate was observed with the Holter measurements (Fig. 4). A significant decrease in the number of supraventricular extrasystoles was observed during daytime at the end of the treatment period (Days 29–30) in the dronedarone 1,200 mg group (p = 0.005). A pronounced decrease in ventricular extrasystoles was observed in two patients in the 1,200 mg group during the daytime at the end of the treatment period (Days 29–30).

Average heart rate during 30 days’ treatment with dronedarone 400 mg, 800 mg, 1,200 mg daily, or placebo according to Holter measurements. Bpm, beats per min
Adverse events
The global incidence of treatment-emergent adverse events (defined as all adverse events after the first dronedarone dose) was 61.9% with dronedarone and 72.5% with placebo. Cardiovascular adverse events were reported in 13/84 (15.5%) dronedarone patients overall, but in 4/30 (13.3%) of those taking 800 mg once daily and in 4/40 (10.0%) placebo patients. These events included cardiac failure, hypotension, hypertension, ECG abnormalities, and edema. With respect to cardiac failure no evidence of a dose effect was observed as these events occurred in 3/29, 1/30, and 2/25 patients in the dronedarone 400, 800, and 1,200 mg groups, respectively, and also in 2/40 patients in the placebo group. However, due to the small patient numbers in each group the interpretation of this finding is limited. A prolonged QT/QTc was observed in two patients who received dronedarone (2.4%; QT/QTc=540/561 and 560/566 ms) both in the 1,200 mg group. Importantly, no torsades de pointes was reported with dronedarone despite the intense continuous Holter monitoring. Thirteen patients (10 dronedarone, three placebo) experienced 22 serious adverse events (Table 3). Eighteen of the 22 events were considered unrelated or unlikely to be related to treatment. A total of 12 adverse events led to discontinuation of treatment in seven patients (one placebo, one dronedarone 400 mg, two dronedarone 800 mg, three dronedarone 1,200 mg). Three deaths occurred during or after completion of the study (Table 3). One death occurred during the study in a patient receiving dronedarone 1,200 mg; this patient with severe ischemic cardiomyopathy, HF, and diabetes mellitus died suddenly on Day 17. No ventricular arrhythmia or any QTc prolongation was documented during treatment with the study drug. In addition, two deaths occurred after completion of the study in patients receiving dronedarone 400 mg and 800 mg; they occurred 12 and 26 days after the end of treatment with the study drug. The first death occurred in a patient with triple vessel coronary artery disease. The second death occurred in a patient with dilated cardiomyopathy and a history of HF. The relationships with the study drug were reported as unlikely by the investigators.
No significant differences were observed between treatment groups for hematologic or biochemistry parameters. No thyroid disorders were reported during the study, and no clinically relevant changes were noted in hepatic or renal parameters.
Discussion
Arrhythmias are common in patients with severe systolic dysfunction, and it is important to determine if treatments are well tolerated in this population. The main objective of this randomized study was to investigate the effects of dronedarone on functional capacity, as assessed by exercise capacity, in patients with severe LV dysfunction and compensated HF. Dronedarone (400 mg, 800 mg, or 1,200 mg daily) did not decrease functional capacity in this patient population. This finding was consistent with the results from the ERATO study, in which dronedarone 400 mg twice daily decreased heart rate during submaximal and maximal exercise, without a reduction in exercise tolerance, in patients with permanent AF receiving standard treatments [8]. In the present study, LVEF was measured at baseline and on therapy, and no significant differences in LVEF were observed. Likewise there was no significant effect of therapy on cardiothoracic ratio. Dronedarone did not adversely impact functional capacity as measured by NYHA classification.
Another interesting aspect of this study is the significant, dose-dependent reduction in heart rate with dronedarone. The reduction in heart rate could be related to the calcium antagonist, the antiadrenergic properties of dronedarone, or possibly the inhibiting effect on the I f current which has been reported in animal models [18]. Whether the beneficial effects on outcomes reported with dronedarone in the ATHENA study could have been partially related to heart rate reduction is speculative at present [14]. However, interventions associated with heart rate reduction have been reported to improve outcomes [19].
The safety profile of new AF treatments is an important consideration given the proarrhythmia or end-organ toxicity of currently available treatments [20]. In the present study dronedarone was well tolerated, with 11/84 patients discontinuing treatment with dronedarone compared with 2/40 patients in the placebo group. The incidence of adverse events was low in patients receiving dronedarone, with no dose effect, and often similar incidence in the placebo group, indicating that the events observed were part of the natural history of the disease rather than a side effect of treatment. The safety data were consistent with the known profile of dronedarone [8–10, 14]. In terms of cardiovascular adverse events, there were no reports of new arrhythmic events, which may be supportive of an absence of a proarrhythmic effect or of torsades de pointes. This finding concurs with only one report of torsades de pointes with dronedarone in the development program overall, which included nearly 3,500 patients who received dronedarone. That event occurred in a patient in the ATHENA study who had additional risk factors for torsades de pointes (QTc at baseline of 522 ms and episodes of severe bradycardia). No thyroid disorders or liver abnormalities were reported in the present study.
Spontaneous sudden death is generally high in this patient population, and two of the three deaths in this study occurred 12 and 26 days after discontinuation of dronedarone, the elimination half-life of which is 24 h [6]. Drug-related exacerbation of HF was not evident in this study. The ANDROMEDA study investigated the long-term effects of dronedarone in hospitalized patients with symptomatic decompensated HF and severe LV dysfunction, specifically to demonstrate that dronedarone does not increase mortality in these patients [12]. Patients were to be randomly assigned to dronedarone 400 mg twice daily or placebo [12]. During a median follow-up of 2 months, more deaths were observed in the dronedarone group than in the placebo group (25 patients [8.1%] vs. 12 patients [3.8%], respectively). The excess mortality in the dronedarone group was largely due to worsening HF (10 dronedarone, two placebo). This imbalance in mortality rates led the data and safety monitoring board to recommend early trial discontinuation after inclusion of 627 patients (310 dronedarone, 317 placebo) [12]. The unstable nature of these patients has been identified as a potential factor in these findings, although the possibility that this was a chance finding cannot be ruled out [13].
In the present study there was no evidence that dronedarone had a deleterious effect on clinical parameters and LVEF, assessed by a MUGA scan, which is a precise and reliable measure.
Treatments to improve both the short- and long-term outcomes of AF are needed. The present study has several limitations. Firstly, the four groups were small and treatment duration was relatively short, making it difficult to draw safety conclusions. Secondly, the study was conducted at a time when beta-blockers were not recommended in patients with HF and it could therefore be considered that the patients were not optimally treated. Thirdly, a 6 min walk test was the only crude parameter used to evaluate the effect of the drug on exercise capacity in the study. Finally, as this study was conducted more than 10 years ago it could be considered of little interest today. However, this study provides important information with particular regard to potential changes in functional capacity and LVEF measurements after initiation of dronedarone treatment in patients with severe LV dysfunction. This study also examined dronedarone treatment at several different dosage levels (400–1,200 mg/day), thereby providing data on levels within and outside of the current recommended dosages. On entry to this study patients were asymptomatic and fully functional following aggressive ACE inhibitor therapy. It is important to point out that although beta-blockers are now indicated for patients with stable HF, they were not considered the standard of care in the mid- to late-1990s when this study was conducted.
To summarize, the results from this study show that dronedarone (400 mg, 800 mg, or 1,200 mg daily) did not affect exercise capacity and did not decrease LVEF in patients with severe LV dysfunction and stabilized compensated HF receiving other cardiovascular treatments. Dronedarone decreased the heart rate and was well tolerated with no proarrhythmic effects in this small cohort of patients.
References
Fuster V, Rydén LE, Cannom DS, et al. American College of Cardiology; American Heart Association Task Force; European Society of Cardiology Committee for Practice Guidelines; European Heart Rhythm Association; Heart Rhythm Society. ACC/AHA/ESC 2006 guidelines for the management of patients with atrial fibrillation: full text: a report of the American College of Cardiology/American Heart Association Task Force on practice guidelines and the European Society of Cardiology Committee for Practice Guidelines (Writing Committee to Revise the 2001 guidelines for the management of patients with atrial fibrillation) developed in collaboration with the European Heart Rhythm Association and the Heart Rhythm Society. Europace. 2006;8:651–745.
Wolf PA, Abbott RD, Kannel WB. Atrial fibrillation as an independent risk factor for stroke: the Framingham Study. Stroke. 1991;22:983–8.
Krahn AD, Manfreda J, Tate RB, Mathewson FA, Cuddy TE. The natural history of atrial fibrillation: incidence, risk factors, and prognosis in the Manitoba follow-up study. Am J Med. 1995;98:476–84.
Benjamin EJ, Wolf PA, D’Agostino RB, Silbershatz H, Kannel WB, Levy D. Impact of atrial fibrillation on the risk of death: the Framingham heart study. Circulation. 1998;98:946–52.
Stewart S, Hart CL, Hole DJ, McMurray JJ. A population-based study of the long-term risks associated with atrial fibrillation: 20-year follow-up of the Renfrew/Paisley study. Am J Med. 2002;113:359–64.
Zareba KM. Dronedarone: a new antiarrhythmic agent. Drugs Today (Barc). 2006;42:75–86.
Dale KM, White CM. Dronedarone: an amiodarone analog for the treatment of atrial fibrillation and atrial flutter. Ann Pharmacother. 2007;41:599–605.
Davy JM, Herold M, Hoglund C, et al. ERATO Study Investigators. Dronedarone for the control of ventricular rate in permanent atrial fibrillation: the Efficacy and safety of dRonedArone for the cOntrol of ventricular rate during atrial fibrillation (ERATO) study. Am Heart J. 2008;156:527–9.
Touboul P, Brugada J, Capucci A, Crijns HJ, Edvardsson N, Hohnloser SH. Dronedarone for prevention of atrial fibrillation: a dose-ranging study. Eur Heart J. 2003;24:1481–7.
Singh BN, Connolly SJ, Crijns HJ, et al. EURIDIS and ADONIS Investigators. Dronedarone for maintenance of sinus rhythm in atrial fibrillation or flutter. N Engl J Med. 2007;357:987–99.
Davy JM, Herold M, Hoglund C, Radzik D, Timmermans AJM. Effect of dronedarone on exercise in patients with permanent atrial fibrillation. Eur Heart J. 2006;1:885.
Kober L, Torp-Pedersen C, McMurray JJ, et al. Dronedarone study group. Increased mortality after dronedarone therapy for severe heart failure. N Engl J Med. 2008;358:2678–87.
Zimetbaum PJ. Dronedarone for atrial fibrillation–an odyssey. N Engl J Med. 2009;360:1811–3.
Hohnloser SH, Crijns HJ, van Eickels M, et al. ATHENA Investigators. Effect of dronedarone on cardiovascular events in atrial fibrillation. N Engl J Med. 2009;360:668–78.
MULTAQ (dronedarone) tablets [prescribing information]. http://products.sanofi-aventis.us/multaq/multaq.pdf. Accessed 8 Jan 2010.
World Medical Association Declaration of Helsinki. Ethical principles for medical research involving human subjects. Hong Kong. 1989.
Lipkin DP, Scriven AJ, Crake T, Poole-Wilson PA. Six minute walking test for assessing exercise capacity in chronic heart failure. Br Med J (Clin Res Ed). 1986;292:653–5.
Rocchetti M, Bertrand JP, Nisato D. Cellular electrophysiological study of dronedarone, a new amiodarone-like agent, in guinea pig sinoatrial node. Naunyn-Schmiedebergs Arch Pharmacol. 1998;358:R617.
Fox K, Borer JS, Camm AJ, et al. Heart rate working Group. Resting heart rate in cardiovascular disease. J Am Coll Cardiol. 2007;50:823–30.
Patton KK, Page RL. Pharmacological therapy of atrial fibrillation. Expert Opin Investig Drugs. 2007;16:169–79.
Acknowledgments
We thank Joëlle Sissmann, MD (trial monitor), and Sylvie Bozzi (statistician). The editorial assistance of Vicky Hinstridge is greatly appreciated.
Sources of funding
The trial was sponsored by sanofi-aventis. Funding for editorial support was provided by sanofi-aventis.
See Appendix for list of investigators.
Disclosures/Conflict of interest
T. Barry Levine discloses that he has received a research grant for this study. Thomas D. Giles discloses that he has received honoraria (<$10,000) for participating in a sanofi-aventis advisory board, and that he has received compensation (<$10,000) as a consultant to sanofi-aventis. Jalal Ghali has no relevant disclosures for this manuscript. David Radzik is an employee of sanofi-aventis.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Clinical Trial Registration: Trial conducted between 1996 and 1998 and not included in a clinical trials registration database.
Appendix
Appendix
Study investigators
Douglas Blank, MD, University of Nebraska Medical Center, Omaha, NE, USA; Douglas Chapman, MD, Alegent Heart Institute, Omaha, NE, USA; Clinton N. Corder, MD, Oklahoma Foundation for Cardiovascular Research, Oklahoma City, OK, USA; Jalal Ghali, MD, LSU School of Medicine, Shreveport, LA, USA; Thomas Giles, MD, LSU School of Medicine, New Orleans, LA, USA; Robert Kipperman, MD, Oklahoma Foundation for Cardiovascular Research, Oklahoma City, OK, USA; T. Barry Levine, MD, Michigan Institute for Heart Failure and Transplant Care, Farmington Hills, MI, USA; Roger Marion Mills, MD, University of Florida, Gainesville, FL, USA; Pramod Mohanty, MD, VA Medical Center, Richmond, VA, USA; Joel Michael Neutel, MD, Orange County Research Center, Orange, CA, USA; Imran K. Niazi, MD, Wisconsin Center for Clinical Research, Milwaukee, WI, USA; Maria-Teresa Olivari, MD, University of Nebraska Medical Center, Omaha, NE, USA; Carl Pepine, MD, University of Florida, Gainesville, FL, USA; Jonathan Plehn, MD, Dartmouth Hitchcock Medical Center, Lebanon, NH, USA; Stephen Singh, MD, Institute of Clinical Research, Washington VA Medical Center, Washington, DC, USA.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Levine, T.B., Giles, T., Radzik, D. et al. Effect of Dronedarone on Exercise Capacity and Cardiac Function in Patients With Severe Left Ventricular Dysfunction and Compensated Stable Heart Failure. Cardiovasc Drugs Ther 24, 449–458 (2010). https://doi.org/10.1007/s10557-010-6251-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10557-010-6251-y