
Abstract
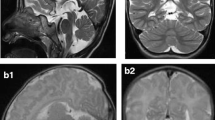
Recessive mutations in the mitochondrial arginyl-transfer RNA synthetase (RARS2) gene have been associated with early onset encephalopathy with signs of oxidative phosphorylation defects classified as pontocerebellar hypoplasia 6. We describe clinical, neuroimaging and molecular features on five patients from three unrelated families who displayed mutations in RARS2. All patients rapidly developed a neonatal or early-infantile epileptic encephalopathy with intractable seizures. The long-term follow-up revealed a virtual absence of psychomotor development, progressive microcephaly, and feeding difficulties. Mitochondrial respiratory chain enzymes in muscle and fibroblasts were normal in two. Blood and CSF lactate was abnormally elevated in all five patients at early stages while appearing only occasionally abnormal with the progression of the disease. Cerebellar vermis hypoplasia with normal aspect of the cerebral and cerebellar hemispheres appeared within the first months of life at brain MRI. In three patients follow-up neuroimaging revealed a progressive pontocerebellar and cerebral cortical atrophy. Molecular investigations of RARS2 disclosed the c.25A>G/p.I9V and the c.1586+3A>T in family A, the c.734G>A/p.R245Q and the c.1406G>A/p.R469H in family B, and the c.721T>A/p.W241R and c.35A>G/p.Q12R in family C. Functional complementation studies in Saccharomyces cerevisiae showed that mutation MSR1-R531H (equivalent to human p.R469H) abolished respiration whereas the MSR1-R306Q strain (corresponding to p.R245Q) displayed a reduced growth on non-fermentable YPG medium. Although mutations functionally disrupted yeast we found a relatively well preserved arginine aminoacylation of mitochondrial tRNA. Clinical and neuroimaging findings are important clues to raise suspicion and to reach diagnostic accuracy for RARS2 mutations considering that biochemical abnormalities may be absent in muscle biopsy.






Similar content being viewed by others


References
Agamy O, Ben-Zeev B, Lev D et al (2010) Mutations disrupting selenocysteine formation cause progressive cerebello-cerebral atrophy. Am J Hum Genet 87:538–544
Antonellis A, Green ED (2008) The role of aminoacyl-tRNA synthetases in genetic diseases. Annu Rev Genomics Hum Genet 9:87–107
Ben-Zeev B, Hoffman C, Lev D et al (2003) Progressive cerebellocerebral atrophy: a new syndrome with microcephaly, mental retardation, and spastic quadriplegia. J Med Genet 40:e96
Brown MV, Reader JS, Tzima E (2010) Mammalian aminoacyl-tRNA synthetases: cell signaling functions of the protein translation machinery. Vasc Pharmacol 52:21–26
Budde BS, Namavar Y, Barth PG et al (2008) tRNA splicing endonuclease mutations cause pontocerebellar hypoplasia. Nat Genet 40:1113–1118
Cassandrini D, Calevo MG, Tessa A et al (2006) A new method for analysis of mitochondrial DNA point mutations and assess levels of heteroplasmy. Biochem Biophys Res Commun 342:387–393
Cassandrini D, Biancheri R, Tessa A et al (2010) Pontocerebellar hypoplasia: clinical, pathologic, and genetic studies. Neurology 75:1459–1464
Chang GG, Pan F, Yeh C, Huang TM (1983) Colorimetric assay for aminoacyl-tRNA synthetases. Anal Biochem 130:171–176
Chang GG, Pan F, Lin YH, Wang HY (1984) Continuous spectrophotometric assay for aminoacyl-tRNA synthetases. Anal Biochem 142:369–372
Chrzanowska-Lightowlers ZM, Horvath R, Lightowlers RN (2011) 175th ENMC International Workshop: mitochondrial protein synthesis in health and disease, 25-27th June 2010, Naarden, The Netherlands. Neuromuscul Disord 21(2):142–147
Dickinson EK, Adams DL, Schon EA, Glerum DM (2000) A human SCO2 mutation helps define the role of Sco1p in the cytochrome oxidase assembly pathway. J Biol Chem 275:26780–26785
DiMauro S (2011) A history of mitochondrial diseases. J Inherit Metab Dis 34:261–276
Edvardson S, Shaag A, Kolesnikova O et al (2007) Deleterious mutation in the mitochondrial arginyl-transfer RNA synthetase gene is associated with pontocerebellar hypoplasia. Am J Hum Genet 81:857–862
Enriquez JA, Attardi G (1996) Analysis of aminoacylation of human mitochondrial tRNAs. Methods Enzymol 264:183–196
Friedman SD, Shaw DW, Ishak G, Gropman AL, Saneto RP (2010) The use of neuroimaging in the diagnosis of mitochondrial disease. Dev Disabil Res Rev 16:129–135
Glamuzina E, Brown R, Hogarth K, Saunders D, Russell-Eggitt I, Pitt M, de Sousa C, Rahman S, Brown G, Grunewald S (2011) Further delineation of pontocerebellar hypoplasia type 6 due to mutations in the gene encoding mitochondrial arginyl-tRNA synthetase, RARS2. J Inherit Metab Dis. Nov 16
Hausmann CD, Ibba M (2008) Aminoacyl-tRNA synthetase complexes: molecular multitasking revealed. FEMS Microbiol Rev 32:705–721
Hom XB, Lavine JE (2004) Gastrointestinal complications of mitochondrial disease. Mitochondrion 4:601–607
Kasher PR, Namavar Y, van Tijn P et al (2011) Impairment of the tRNA-splicing endonuclease subunit 54 (tsen54) gene causes neurological abnormalities and larval death in zebrafish models of pontocerebellar hypoplasia. Hum Mol Genet 20:1574–1584
Ling J, Reynolds N, Ibba M (2009) Aminoacyl-tRNA synthesis and translational quality control. Annu Rev Microbiol 63:61–78
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods 25:402–408
Lloyd AJ, Thomann HU, Ibba M, Söll D (1995) A broadly applicable continuous spectrophotometric assay for measuring aminoacyl-tRNA synthetase activity. Nucleic Acids Res 23:2886–2892
McFarland R, Turnbull DM (2009) Batteries not included: diagnosis and management of mitochondrial disease. J Intern Med 265:210–228
McFarland R, Taylor RW, Turnbull DM (2010) A neurological perspective on mitochondrial disease. Lancet Neurol 9:829–840
Mitochondrial Medicine Society's Committee on Diagnosis, Haas RH, Parikh S, Falk MJ et al (2008) The in-depth evaluation of suspected mitochondrial disease. Mol Genet Metab 94:16–37
Naito HK (1975) Modification of the Fiske and SubbaRow method for total phospholipid in serum. Clin Chem 21:1454–1456
Namavar Y, Barth PG, Kasher PR et al (2011) Clinical, neuroradiological and genetic findings in pontocerebellar hypoplasia. Brain 134:143–156
Nogueira C, Carrozzo R, Vilarinho L, Santorelli FM (2011) Infantile-onset disorders of mitochondrial replication and protein synthesis. J Child Neurol 26(7):866–875
Peleg D, Kennedy CM, Hunter SK (1998) Intrauterine growth restriction:identification and management. Am Fam Physician 58(2):453–460, 466–7
Rankin J, Brown R, Dobyns WB et al (2010) Pontocerebellar hypoplasia type 6: a British case with PEHO-like features. Am J Med Genet A 152A:2079–2084
Renbaum P, Kellerman E, Jaron R et al (2009) Spinal muscular atrophy with pontocerebellar hypoplasia is caused by a mutation in the VRK1 gene. Am J Hum Genet 85:281–289
Scheper GC, van der Klok T, van Andel RJ et al (2007) Mitochondrial aspartyl-tRNA synthetase deficiency causes leukoencephalopathy with brain stem and spinal cord involvement and lactate elevation. Nat Genet 39:534–539
Shammas C, Menne TF, Hilcenko C et al (2005) Structural and mutational analysis of the SBDS protein family. Insight into the leukemia-associated Shwachman-Diamond Syndrome. J Biol Chem 280:19221–19229
Tuppen HA, Fattori F, Carrozzo R et al (2008) Further pitfalls in the diagnosis of mtDNA mutations: homoplasmic mt-tRNA mutations. J Med Genet 45:55–61
Wong LJ (2010) Molecular genetics of mitochondrial disorders. Dev Disabil Res Rev 16:154–162
Acknowledgements
This research was supported in part by grants from the Italian Ministry of Heath, the Telethon Foundation Onlus (Mitocon Project GUP09004 to EB and GGP09207 to LS) Fondazione CARIPARO, Padova (to LS), the European Commission projects FP7 LeukoTreat (to EB), and EUROBFNS (to MRC) and grant from the University of Padova to GS.
Conflict of interest
None.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by: Shamima Rahman
Maria Roberta Cilio and Marzia Bianchi contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Table
(DOC 223 kb)
Supplementary Table
(DOC 40 kb)
Supplementary Fig. 1
(JPEG 45 kb)
Supplementary Fig. 2A
(JPEG 73 kb)
Supplementary Fig. 2B
(JPEG 101 kb)
Supplementary Fig. 3
(JPEG 69 kb)
Rights and permissions
About this article
Cite this article
Cassandrini, D., Cilio, M.R., Bianchi, M. et al. Pontocerebellar hypoplasia type 6 caused by mutations in RARS2: definition of the clinical spectrum and molecular findings in five patients. J Inherit Metab Dis 36, 43–53 (2013). https://doi.org/10.1007/s10545-012-9487-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10545-012-9487-9