
Abstract
Background
Sapropterin dihydrochloride, an EMEA-approved synthetic formulation of BH4, has been available in Europe since 2009 for PKU patients older than 4 years, but its use with younger children is allowed in France based on an expert recommendation. We report the cases of 15 patients treated under the age of 4 years and demonstrate the safety and efficacy of this treatment for patients in this age group.
Patients and method
We report the use of BH4 in 15 PKU patients treated before the age of 4 years.
Results
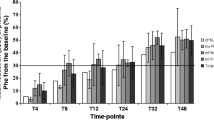
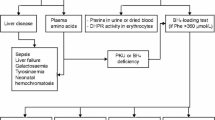
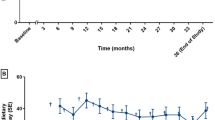
Fifteen patients were enrolled in this retrospective study. Mean phenylalaninemia at diagnosis was 542±164 μM and all patients had mild PKU (maximal phenylalaninemia: 600-1200 μM). BH4 responsiveness was assessed using a 24-hour BH4 loading test (20 mg/kg), performed during the neonatal period (n = 11) or before 18 months of age (n = 4). During the test, these patients exhibited an 80±12% decrease in phenylalaninemia. Long-term BH4 therapy was initiated during the neonatal period (n = 7) or at the age of 13±12 months (n = 8). The median duration of treatment was 23 months [min 7; max 80]. BH4 therapy drastically improved dietary phenylalanine tolerance (456±181 vs 1683±627 mg/day, p < 0.0001) and allowed a phenylalanine-free amino acid mixture to be discontinued or not introduced in 14 patients. Additionally, in the eight patients treated after a few months of diet therapy, BH4 treatment significantly decreased mean phenylalaninemia (352±85 vs 254±64μM, p < 0.05), raised the percentage of phenylalaninemia tests within therapeutic targets [120-300 μM] (35±25 vs 64±16%, p < 0.05), and reduced phenylalaninemia variance (130±21 vs 93±27μM, p < 0.05). No side effects were reported.
Conclusion
BH4-therapy is efficient and safe before the age of 4 years in mild PKU, BH4-responsive patients.




Similar content being viewed by others
Abbreviations
- PKU:
-
Phenylketonuria
- Phe:
-
Phenylalanine
References
Adamczyk P, Morawiec-Knysak A, Pludowski P, Banaszak B, Karpe J, Pluskiewicz W (2011) Bone metabolism and the muscle-bone relationship in children, adolescents and young adults with phenylketonuria. J Bone Miner Metab 29:236–244
Anastasoaie V, Kurzius L, Forbes P, Waisbren S (2008) Stability of blood phenylalanine levels and IQ in children with phenylketonuria. Mol Genet Metab 95:17–20
Antshel KM (2010) ADHD, learning, and academic performance in phenylketonuria. Mol Genet Metab 99(Suppl 1):S52–S58
Belanger-Quintana A, Garcia MJ, Castro M et al (2005) Spanish BH4-responsive phenylalanine hydroxylase-deficient patients: evolution of seven patients on long-term treatment with tetrahydrobiopterin. Mol Genet Metab 86(Suppl 1):S61–S66
Blau N, Erlandsen H (2004) The metabolic and molecular bases of tetrahydrobiopterin-responsive phenylalanine hydroxylase deficiency. Mol Genet Metab 82:101–111
Blau N, Belanger-Quintana A, Demirkol M et al (2009) Optimizing the use of sapropterin (BH(4)) in the management of phenylketonuria. Mol Genet Metab 96:158–163
Boveda MD, Couce ML, Castineiras DE et al (2007) The tetrahydrobiopterin loading test in 36 patients with hyperphenylalaninaemia: evaluation of response and subsequent treatment. J Inherit Metab Dis 30:812
Brumm VL, Grant ML (2010) The role of intelligence in phenylketonuria: a review of research and management. Mol Genet Metab 99(Suppl 1):S18–S21
Brumm VL, Bilder D, Waisbren SE (2010) Psychiatric symptoms and disorders in phenylketonuria. Mol Genet Metab 99(Suppl 1):S59–S63
Burton BK, Bausell H, Katz R, Laduca H, Sullivan C (2010) Sapropterin therapy increases stability of blood phenylalanine levels in patients with BH4-responsive phenylketonuria (PKU). Mol Genet Metab 101:110–114
Burton BK, Adams DJ, Grange DK et al (2011) Tetrahydrobiopterin therapy for phenylketonuria in infants and young children. J Pediatr 158:410–415
Christ SE, Huijbregts SC, de Sonneville LM, White DA (2010) Executive function in early-treated phenylketonuria: profile and underlying mechanisms. Mol Genet Metab 99(Suppl 1):S22–S32
Haute Autorité de Santé (2010) Phénylcétonurie : Protocole National de Diagnostic et de Soins. http://www.has-sante.fr
Enns GM, Koch R, Brumm V, Blakely E, Suter R, Jurecki E (2010) Suboptimal outcomes in patients with PKU treated early with diet alone: revisiting the evidence. Mol Genet Metab 101:99–109
Feillet F, Chery C, Namour F et al (2008) Evaluation of neonatal BH4 loading test in neonates screened for hyperphenylalaninemia. Early Hum Dev 84:561–567
Feillet F, van Spronsen FJ, MacDonald A et al (2010) Challenges and pitfalls in the management of phenylketonuria. Pediatrics 126:333–341
Gassio R, Artuch R, Vilaseca MA et al (2005) Cognitive functions in classic phenylketonuria and mild hyperphenylalaninaemia: experience in a paediatric population. Dev Med Child Neurol 47:443–448
Hennermann JB, Buhrer C, Blau N, Vetter B, Monch E (2005) Long-term treatment with tetrahydrobiopterin increases phenylalanine tolerance in children with severe phenotype of phenylketonuria. Mol Genet Metab 86(Suppl 1):S86–S90
Karacic I, Meili D, Sarnavka V et al (2009) Genotype-predicted tetrahydrobiopterin (BH4)-responsiveness and molecular genetics in Croatian patients with phenylalanine hydroxylase (PAH) deficiency. Mol Genet Metab 97:165–171
Lambruschini N, Perez-Duenas B, Vilaseca MA et al (2005) Clinical and nutritional evaluation of phenylketonuric patients on tetrahydrobiopterin monotherapy. Mol Genet Metab 86(Suppl 1):S54–S60
Modan-Moses D, Vered I, Schwartz G et al (2007) Peak bone mass in patients with phenylketonuria. J Inherit Metab Dis 30:202–208
Muntau AC, Gersting SW (2010) Phenylketonuria as a model for protein misfolding diseases and for the development of next generation orphan drugs for patients with inborn errors of metabolism. J Inherit Metab Dis 33:649–658
Roato I, Porta F, Mussa A et al (2010) Bone impairment in phenylketonuria is characterized by circulating osteoclast precursors and activated T cell increase. PLoS One 5:e14167
Shintaku H, Kure S, Ohura T et al (2004) Long-term treatment and diagnosis of tetrahydrobiopterin-responsive hyperphenylalaninemia with a mutant phenylalanine hydroxylase gene. Pediatr Res 55:425–430
Trefz FK, Burton BK, Longo N et al (2009) Efficacy of sapropterin dihydrochloride in increasing phenylalanine tolerance in children with phenylketonuria: a phase III, randomized, double-blind, placebo-controlled study. J Pediatr 154:700–707
Viau KS, Wengreen HJ, Ernst SL, Cantor NL, Furtado LV, Longo N (2011) Correlation of age-specific phenylalanine levels with intellectual outcome in patients with phenylketonuria. J Inherit Metab Dis 34(4):963–971
Acknowledgements
The authors thank Prof. JL Guéant, CHU Nancy (genotype analyses) and Dr B Giraudeau, CHU Tours (technical assistance for statistical analyses). Part of this work was presented as an oral communication at the Annual Symposium of the Society for the Study of Inborn Errors of Metabolism held in Istanbul, Turkey, 31 August– 3 September 2010.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by: John H. Walter
References to electronic databases: Phenylketonuria, OMIM#261600
Competing interest: None declared.
Rights and permissions
About this article
Cite this article
Leuret, O., Barth, M., Kuster, A. et al. Efficacy and safety of BH4 before the age of 4 years in patients with mild phenylketonuria. J Inherit Metab Dis 35, 975–981 (2012). https://doi.org/10.1007/s10545-012-9464-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10545-012-9464-3