
Abstract
Early detection of mucopolysaccharidosis (MPS) is an important factor in treatment success; therefore, good disease biomarkers are vital. We evaluate heparin cofactor II-thrombin complex (HCII-T) as a biomarker in serum and dried blood spots (DBS) of MPS patients. Serum HCII-T and urine dermatan sulphate:chondroitin sulphate (DS:CS) ratio are also compared longitudinally against clinical outcomes in MPSI, II and VI patients following treatment. Samples were collected from MPS patients at the Royal Manchester Children’s Hospital. DS:CS ratio was obtained by measuring the area density of spots from 2D electrophoresis of urinary glycosaminoglycans. Serum and DBS HCII-T was measured by sandwich ELISA. Serum HCII-T is elevated approximately 25-fold in MPS diseases that store DS, clearly distinguishing untreated MPSI, II and VI patients from unaffected age-matched controls. Serum HCII-T is also elevated in MPSIII, which leads to storage of heparan sulphate, with an increase of approximately 4-fold over unaffected age-matched controls. Urine DS:CS ratio and serum HCII-T decrease in response to treatment of MPSI, II and VI patients. HCII-T appears to respond rapidly to perturbations in treatment, whilst DS:CS ratio responds more slowly. HCII-T is a suitable biomarker for MPSI, II and VI, and it may also be informative for MPS diseases storing HS alone, such as MPSIII, although the elevation observed is smaller. In treated MPS patients, HCII-T and DS:CS ratio appear to measure short-term and long-term treatment outcomes, respectively. The potential value of HCII-T measurement in DBS for newborn screening of MPS diseases warrants further investigation.
Similar content being viewed by others

Introduction
Mucopolysaccharide (MPS) diseases are lysosomal storage disorders in which deficiencies of glycosaminoglycan (GAG)-degrading enzymes lead to excessive GAG accumulation in cells and tissues. Various combinations of the GAGs heparan sulphate (HS), dermatan sulphate (DS), keratan sulphate (KS) and chondroitin sulphate (CS) are accumulated, depending on the lysosomal enzyme deficiency. Phenotypic variability between types of MPS is caused by mutations in different lysosomal enzymes, and variability in symptoms within each type of MPS is due to different levels of residual enzyme activity. Progressive cognitive impairment is a significant feature of severe MPSI Hurler (IH), II, III and VII, whilst dysostosis multiplex and facial dysmorphia can be presenting features of MPSI, II, IV, VI and VII. Attenuated MPSI, II, III and VII may not show significant neurological involvement until late into the course of the illness. Organomegally, respiratory insufficiency, cardiac compromise and joint problems can greatly affect daily life of MPSI, II and VI patients whilst behavioural disturbances are common in severe MPSII and III patients (Wraith et al. 2004; Meyer et al. 2007; Montano et al. 2007; Jones et al. 2009; Decker et al. 2010).
Treatment is available for some MPS diseases: enzyme replacement therapy (ERT) is currently used to treat MPSI patients with non-neurological manifestations, as well as for MPSII and VI (Wraith et al. 2004; Harmatz et al. 2008; Wraith 2008), whilst haematopoietic stem cell transplantation (HSCT) has been successfully used to treat MPSIH, VI and VII (Krivit et al. 1984; Peters et al. 1998; Yamada et al. 1998; Peters and Steward 2003). However, treatments are less successful in patients with severe neurodegenerative disease and it is unclear whether treatment can reverse existing organ damage. It is known that early diagnosis within the first few months of life and rapid treatment delivery, such as HSCT before the age of 2 years in MPSIH patients, is advantageous for successful outcomes in lysosomal storage disorders (Vellodi et al. 1997; Krivit 2002; Meikle and Hopwood 2003; Escolar et al. 2005; Boelens et al. 2009). Therefore, biomarkers that can be used for early diagnosis of MPS and monitoring treatment outcome are important in effective disease management.
Currently, in the clinic, a diagnosis of MPS is confirmed using both urinary GAG analysis and blood levels of the deficient enzyme. However, these methods are time consuming and expensive. Blood enzyme levels are inappropriate for monitoring disease progression once the deficient enzyme is being supplied by treatment with ERT as these levels can fluctuate considerably, whilst urinary GAG levels can give variable outcomes as results vary with age and hydration state of the patient. In Manchester, it has been determined that, for MPS diseases in which DS accumulates, the ratio of DS to a GAG that is not stored, such as CS in the urine (the DS:CS ratio), is a good predictor of metabolic outcome (Church et al. 2007), and is a superior biomarker to measurement of total urinary GAG. Urine DS:CS ratio has been shown to closely correlate with blood enzyme level in MPSI patients following treatment, and indicates a better metabolic outcome in patients receiving HSCT compared to those receiving ERT alone (Wynn et al. 2009).
Heparin cofactor II-thrombin complex (HCII-T) was identified as a biomarker for MPS diseases by Randall et al. (2006, 2008), and we recently evaluated this biomarker in murine models of MPS (Langford-Smith et al. 2010). In mice, HCII-T is a reliable biomarker for MPSI, but not MPSIIIA and IIIB (Langford-Smith et al. 2010). In vitro and in vivo data suggest that this is because HCII-T formation is increased in the presence of DS, which is stored in MPSI, but not in the presence of HS, which is stored in MPSIII. Here, we extend our investigation to patient samples of MPSI, II, IIIA, IIIB, IIIC, IV and VI, showing that HCII-T is indeed a reliable biomarker for those MPS diseases in which DS accumulates, and to a lesser extent also for those that accumulate HS. We also compare the HCII-T biomarker against urine DS:CS ratio for monitoring treatment outcomes and show that both biomarkers decrease during successful treatment, but that HCII-T responds more rapidly to perturbations in treatment.
Materials and methods
Patient diagnosis
Diagnosis of MPS was achieved by demonstration of elevated urinary GAGs, and enzyme activity below 10% of the lower limit of the normal range. Clinical symptoms and mutation analysis were used to diagnose severity of the disease.
Blood sample collection
Blood samples were collected by clean venepuncture and were not taken from a central line. Serum was collected from blood without anticoagulant which was allowed to clot for 30 min at room temperature, and then centrifuged at 3,000g for 10 min at room temperature. Serum was stored at −80°C. Dried blood spots (DBS) were collected on Guthrie cards (Whatman), allowed to dry for 4 h at room temperature, and then stored at 4°C. Serum and DBS samples were collected from MPS patients undergoing blood sampling at the Royal Manchester Children’s Hospital and from other MPS patients within the UK. Control serum and DBS samples were collected from patients undergoing blood tests for unrelated investigations at the Royal Manchester Children’s Hospital. The study was ethically approved under REC 08H101063.
Measurement of heparin cofactor II-thrombin (HCII-T) complex
The HCII-T complex was quantified using a sandwich ELISA as described previously (Randall et al. 2008; Langford-Smith et al. 2010). A polyclonal anti-thrombin capture antibody and polyclonal anti-HCII detection antibody (Affinity Biologicals) were used. DBS samples were prepared by punching a 3-mm-diameter sample from the Guthrie card using a Wallac DBS Puncher (DELFIA) and were eluted with 200 μl General Purpose Serum Diluent (Serotec) for 80 min at room temperature. Standards, serum and DBS samples were diluted in General Purpose Serum Diluent; serum up to 25,000-fold, DBS up to 20-fold. All standards and samples were assayed in duplicate, with serum samples tested at three different dilutions each and DBS samples tested at two or three dilutions each. A 1-μM HCII-T standard was made by reaction of human thrombin and human heparin cofactor II (Enzyme Research Laboratories) in the presence of 0.05 U/ml heparin. This was used to make a series of standards from 1 to 1,500 pM, the linear range of which was used as the standard curve.
Measurement of urinary glycosaminoglycans (GAGs)
Total urinary GAGs were measured using dimethyl methylene blue. Urine substrate was measured as the DS:CS ratio, as described previously (Church et al. 2007). Urine DS:CS ratio was obtained by measuring area density of spots from two-dimensional electrophoresis of extracted GAGs.
Delivery of KS to mice and serum sampling
Wild-type (WT) mice from an MPSIIIB colony were maintained as described (Canal et al. 2010; Malinowska et al. 2009). KS (160 ug) (AMS Biotechnology) was delivered via tail vein injection to WT mice and serum sampled after 30 min as described in a similar series of experiments conducted previously with HS and DS (Langford-Smith et al. 2010).
Statistical analysis
For statistical analysis, one extreme outlier from the unaffected serum group was formally removed using the boxplot tool in SPSS (extreme outliers are defined as those more than 3× the interquartile range outside of the interquartile box). No other groups were affected. Data were analysed using one-way ANOVA, applying Tukey’s multiple comparisons test as a post hoc comparator. Where appropriate, data were log transformed to normalise data prior to statistical analysis. Significance was assumed where p values were less than 0.05 of a studentised range of Q.
Ethical approval
Ethical review for these studies was performed and approved by The University of Manchester, Central Manchester University Hospitals NHS Foundation Trust, and the North West Research Ethics Committee on REC 08H101063. Written informed consent was obtained from the parents or guardians of the children involved for all samples taken.
Results
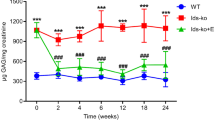
Serum HCII-T is elevated in MPSI, II, III and VI patients
Serum samples were collected from untreated MPS patients and unaffected age-matched controls as well as HSCT-treated MPSI patients who received transplants 1–16 years ago. HCII-T is very significantly increased in MPSI, II and VI patients compared to unaffected individuals or treated MPS I patients (p < 0.001) (Fig. 1a). The measurement ranges of HCII-T for MPSI (61.1–325.0 nM), MPSII (156.7–252.0 nM) and MPSVI patients (149.3–230.3 nM) do not overlap with unaffected age matched controls (5.0–11.0 nM) or HSCT treated MPSIH patients (6.2–37.9 nM). On average, MPSI, II and VI patients have HCII-T levels that are approximately 25-fold higher than unaffected controls. There is no significant difference between the unaffected group and the treated MPSI patient group, though some treated patients still lie above the range of the unaffected group (Table 1). This suggests that serum HCII-T levels are normalised in some MPSI patients receiving HSCT, and are close to the normal range in others.

Ability of the HCII-T biomarker to distinguish between MPS patients and unaffected age matched controls. a Serum HCII-T was significantly higher (*p < 0.001) in MPSI (n = 9) (■), MPSII (n = 3) (◊) and MPSVI (n = 2) (●) patients compared to unaffected controls (n = 8) (Δ) and treated MPSI patients (n = 20) (□). HCII-T is significantly elevated, albeit to a lesser extent in MPSIII (n = 17) (○) and only slightly in MPSIV (n = 1) (▲). b DBS HCII-T levels in all MPS samples that had been stored at −20°C were below levels of unaffected DBS samples that had been stored at 4°C, demonstrating that freezing DBS samples is likely to lead to degradation of the HCII-T complex. c DBS HCII-T levels decrease over time when DBS are stored at room temperature in both samples from MPSI patients (n = 2) (■) and unaffected controls (n = 1) (Δ). d DBS HCII-T levels in MPSI patients (n = 4) (■) were significantly greater (*p < 0.005) than those of unaffected controls (n = 6) (Δ) and treated MPSI patients (n = 6) (□). A small number of samples indicate that DBS HCII-T is only slightly elevated in MPSIII (n = 2) (○) and MPSIV patients (n = 1) (▲). Error bars represent standard deviations about the mean
In serum samples from patients with MPSIIIA (n = 8), IIIB (n = 7) and IIIC (n = 2) there are no significant differences between HCII-T levels of the individual subtypes. HCII-T levels of all MPSIII patients are significantly elevated compared to unaffected controls or treated MPSIH patients (p < 0.02), although the elevation is not as extreme as MPSI, II and VI samples, with average HCII-T levels being approximately 4-fold higher than unaffected controls (Fig. 1a). All but one of the MPSIII values (7.3–91.5 nM) lie outside the range of the unaffected group. Based on our previous work, which showed that injection of DS, but not HS, leads to elevation of HCII-T in WT mice, it is clear that DS has a greater effect on HCII-T complex formation than HS. However, storage of HS in MPSIII patients does have an effect on HCII-T, which is sufficient to distinguish MPSIII patients from unaffected controls.
In a single sample from an untreated MPSIV patient storing KS, the serum HCII-T value (14.8 nM) lies slightly above the unaffected range (Fig. 1a). Further patients would be necessary to determine if this slight elevation would reliably allow distinction of MPSIV patients using this method.
DS affects HCII-T complex formation much more than HS or KS
In our murine work, we determined the effect of intravenous infusion of HS or DS to normal WT mice, showing that only DS produced a significant serum elevation of HCII-T (Langford-Smith et al. 2010). To determine if KS, stored in MPSIV, could mediate HCII-T complex formation, we also injected equivalent amounts of KS (160 μg) intravenously into WT mice and monitored serum and DBS HCII-T levels 30 min after infusion. No changes in HCII-T levels were seen after infusion of exogenous KS, suggesting that KS does not mediate significant HCII-T complex formation and thus that HCII-T may not reliably diagnose MPSIV.
HCII-T deteriorates in improperly stored dried blood spots
We previously determined that measurement of HCII-T in fresh DBS samples from MPSI mice gave better separation between the MPSI and unaffected groups compared to serum HCII-T (Langford-Smith et al. 2010). We therefore measured HCII-T in archived patient DBS samples stored at −20°C to determine if it was possible. HCII-T levels are near the lower limit of detection for the ELISA, and HCII-T was not elevated above the unaffected range in any patient samples suggesting that the complex has deteriorated by freezing (Fig. 1b). We then tested DBS samples before and after storage at room temperature for several months, and show that HCII-T values decline, giving much lower values than the original HCII-T levels (Fig. 1c). In all subsequent work, we have only used DBS stored at 4°C as we have previously shown that HCII-T can be detected reliably in DBS samples that have been stored for up to a year at 4°C (Langford-Smith et al. 2010).
Dried blood spot HCII-T is significantly elevated in MPSI patients
HCII-T was measured in DBS samples from MPS patients and age matched unaffected controls that had been stored at 4°C. No samples were available for MPSII or VI. DBS HCII-T is significantly elevated in untreated MPSI patients compared to unaffected controls or HSCT treated MPSI patients (p < 0.005) (Fig. 1d). This suggests that measurement of HCII-T in appropriately stored DBS can be used to distinguish MPSI patients, although more patient samples would be necessary to be sure of this trend. As with the serum data, the mean and range of the unaffected group (71 pM, 22–100 pM) and treated MPSI patients (78 pM, 37–128 pM) are very similar, with no significant difference between the groups. The range of the MPSI group (132–1272 pM) does not overlap with either the unaffected group or treated MPSI patients. The single MPSIIIA and MPSIV DBS samples showed slight HCII-T elevation over baseline, although with only one patient it is hard to know if this is artefact or real effect.
HCII-T and DS:CS ratio both respond to treatment and correlate with clinical outcomes
Serum HCII-T and urine DS:CS ratio were measured longitudinally in four MPSI, two MPSII and two MPSVI patients (Table 2) and compared against clinical measures of treatment response. In these patients, both biomarkers are elevated above unaffected controls before treatment, and both biomarkers decrease gradually following treatment with ERT alone for MPSII and VI patients, or with ERT and HSCT for MPSI patients (Fig. 2a–g). HCII-T values fluctuate more over time than the urine DS:CS values, though the overall trend in both biomarkers is a decrease towards normal levels. In some cases, the serum HCII-T biomarker reaches the normal range of 5.0–11.0 nM, whilst the urine DS:CS biomarker decreases but remains above baseline in treated MPS patients compared to unaffected individuals who do not have detectable DS in their urine, making the unaffected DS:CS ratio 0. The greater fluctuation of the HCII-T biomarker suggests a rapid response to changes in treatment, which may indicate short-term effects such as minor illness, antibody responses or perturbations in the course of treatment. We suggest that the steadier decline of urinary DS:CS ratio is more indicative of long-term treatment outcomes. Both biomarkers appear to be useful in assessing a patient’s response to treatment; however, the following case reports highlight some of the advantages of the HCII-T biomarker (Table 2 details presenting symptoms).

Response of serum HCII-T and urine DS:CS ratio biomarkers to treatment. Grey shading represents periods of ERT. The dashed line represents the upper limit of the range of serum HCII-T values in unaffected age-matched controls. DS is not detected in the urine of unaffected individuals; therefore, the urine DS:CS ratio for unaffected controls is 0. Patient presentation data is detailed in Table 2. a Patient A has MPSIH. Serum HCII-T and urine DS:CS decrease in response to treatment, although following withdrawal of ERT there is a large increase in HCII-T and DS:CS levels off (1). The patient has improved development but worsening thoracolumbar kyphosis (2). The patient has stopped wearing hearing aids, developed the ability to walk unaided and has shown improvements in speech and language (3). After HSCT, the patient’s alpha-iduronidase activity is within the normal range (3). b Patient B has MPSIH and received 2 courses of ERT and 2 transplants due to failure of the first HSCT. During the unsuccessful transplant, autologous rescue and ERT, serum HCII-T levels fluctuated, but following the report of successful engraftment (4), HCII-T and urine DS:CS ratio have decreased. c Patient C has MPSIH and received ERT, leading to a decrease in both serum HCII-T and urine DS:CS levels. d Patient D has MPSIH and has been treated with ERT, resulting in a decrease in both biomarkers. e Patient E has MPSII and has been receiving ERT since 1 month after diagnosis, leading to an overall decrease in serum HCII-T and urine DS:CS levels. A large increase in serum HCII-T occurred when the patient was experiencing cheek flushing during enzyme infusions (5). This patient still has mild mitral regurgitation (6), but physiotherapy scores have improved from the moderate range (6) to the mild range (7). f Patient F has MPSII and has decreasing levels of serum HCII-T and urine DS:CS following treatment with ERT. A small increase in HCII-T occured when cheek flushing was observed during enzyme infusions (8). Speech and language were mildly delayed (9), but over time this improved (10), as well as increased mobility, exercise tolerance, and a lack of hepatosplenomegaly or joint stiffness (10). g Patient G has MPSVI and has been receiving ERT since diagnosis, but the initial decrease in serum HCII-T has not been maintained. The patient had desaturation during a sleep study, nasal obstruction and large tonsils (11). Mitral valve regurgitation has progressed from mild–moderate to moderate–severe (12). However, posture and walking ability have improved, and the patient has received cast management for the curved spine (13). h Patient H has MPSVI and received ERT, leading to a decrease in both serum HCII-T and urine DS:CS levels
Patient A presented with MPSIH and was treated with ERT and subsequently HSCT. Serum HCII-T and urine DS:CS biomarkers decreased, but when treatment with ERT ended we observe a large increase in HCII-T (Fig. 2a-1). This suggests that in this patient the withdrawal of ERT was followed by short-term reduction in enzyme, leading to increased circulating DS, whilst the transplanted cells were established. Clinical assessments at 11 and 16 months post-transplant showed improvement in several areas, including mobility, hearing and speech (Fig. a-2). By this stage, serum HCII-T was within the normal range and urine DS:CS continued to decrease (Fig. 2a-3). Both biomarkers appear to correlate with an improvement in clinical symptoms.
Patient B presented with MPSIH and was treated with ERT, HSCT, autologous HSCT rescue, followed by further ERT and a second HSCT. The HCII-T biomarker fluctuates, with increases in HCII-T at the end of each phase of ERT. Both serum HCII-T and urine DS:CS ratio decrease after the second HSCT when successful engraftment had been reported (Fig. 2b-4).
Patients C and D presented with MPS IH and have received ERT for 3–4 months to date. During this time, their initially high serum HCII-T and urine DS:CS levels have decreased, correlating with clinical improvement (Fig. 2c, d).
Patient E presented with MPSII and received ERT treatment. No significant infusion associated reactions were observed apart from occasional cheek flushing. Cheek flushing during infusion may indicate a hypersensitivity response and corresponds with marked increases in the HCII-T levels. Although the patient tested negative for IgG and IgE antibodies against the enzyme, something is interfering with the success of ERT at clearing blood DS at this time (Fig. 2e-5). HCII-T levels soon returned to near normal and a clear clinical improvement was noted, including increased energy levels, alertness, and ease of movement, as well as improved hearing, motor development and no sleep apnoea (Fig. 2e-6, 7). Both biomarkers decreased over this time, correlating with an improvement in clinical symptoms (Fig. 2e).
Patient F presented with MPSII and received ERT treatment. Acute elevations in serum HCII-T also appear to correlate with illness and observations of cheek flushing during enzyme infusions (Fig. 2f-8). The patient still had mildly delayed speech and language 7 months after ERT began (Fig. 2f-9), but this is consistent with hearing loss, and after 1 year of treatment with ERT speech was reported to be improved (Fig. 2f-10) correlating with decreased serum HCII-T and urine DS:CS.
Patient G presented with MPSVI and received ERT treatment. Initially, a decrease in serum HCII-T and urine DS:CS was observed, but then HCII-T began to increase and has remained high up to 1 year after treatment began (Fig. 2g). This patient has an increasing trend of anti-Naglazyme IgG antibodies, with a titre of 2,430 at 78 days post-diagnosis and a titre of 7,290 at 177 days post-diagnosis (Figs. 2g-11, 12). Further antibody titres are not available at this time, though if anti-Naglazyme IgG production continues these antibodies may inhibit treatment success. The patient is routinely treated with paracetamol for high temperatures during enzyme infusions, suggesting that a hypersensitivity response may be present when enzyme is delivered.
Patient H presented with MPSVI and received ERT treatment. During the first 24 weeks of ERT, serum HCII-T and urine DS:CS ratio have decreased, with serum HCII-T reaching normal levels (Fig. 2h). This correlates with improved clinical condition including a return to normal hearing and cardiac function.
Discussion
We have demonstrated that serum and DBS HCII-T is elevated in patients storing dermatan sulphate—namely those with MPSI, II and VI, which is very much in agreement with our previous observations in the mouse models of MPS disease (Langford-Smith et al. 2010). We also expect that HCII-T would be elevated in MPSVII patients who store DS, but this disease is rare, and patient samples were not available to test this hypothesis. Our serum HCII-T values for unaffected age-matched controls, as well as MPSI and MPS II patients, fall within the ranges observed by Randall et al. (2008), supporting the overall conclusion that HCII-T is elevated in these diseases.
We have also shown that HCII-T is significantly elevated above unaffected controls in serum from MPSIII patients. This agrees with work by Randall et al. (2008), who showed a small elevation above age matched controls in serum of two MPSIII patients. However, the elevated values that we observed (7.3–91.5 nM), and those reported by Randall et al. (32–91 nM) are not very far above the unaffected controls, particularly when compared to the elevation observed in MPSI and MPSII patients. Therefore, HCII-T may not be as reliable a biomarker for monitoring treatment outcome in MPSIII. In a single sample from an MPSIV patient, we also see slight elevation above the unaffected control range (14.8 nM), as do Randall et al. in one MPSIV patient (27.9 nM). A larger sample size would be necessary to draw a firm conclusion, but HCII-T may also be slightly elevated in MPSIV.
This differs from our previous data showing that intravenous delivery of pathological levels of HS or KS into WT mice do not result in substantial elevation of the HCII-T complex in serum or DBS (Langford-Smith et al. 2010). This may be because the mice live a shorter life-span than patients and thus accumulate less GAGs than patients, or it could be due to specificity of the antibodies used, which are directed to human thrombin and human HCII. It is interesting to note that the average HCII-T values obtained in patient samples are higher than those in mice for MPSI (mouse 130.8 nM, human 176.2) and MPSIII (mouse 16.1 nM, human 32.1 nM), which would support either theory.
Both serum HCII-T and urine DS:CS ratio are elevated prior to treatment of MPSI, II and VI patients and decrease in response to treatment, making both suitable biomarkers for diagnosis and for monitoring treatment outcomes. However, the HCII-T biomarker appears to be more responsive to transient changes such as infusion-related reactions and withdrawal of ERT following HSCT and correlates well with clinical observations. This may make HCII-T more informative when assessing immediate success of treatment whilst urine DS:CS ratio measurements correlate well with long-term clinical outcomes. The rapid response of serum HCII-T is likely to be because HCII-T complex formation responds directly to an increase in circulating DS, so when enzyme levels decrease or enzyme is inhibited by antibodies, the amount of soluble DS in the blood will increase, having an immediate effect on HCII-T levels. In comparison, urine DS:CS ratio is likely to be indicative of renal involvement in the disease pathology and will respond more slowly to levels of DS secreted by cells in the renal pathway.
In MPSIH patient A, the increase in HCII-T post HSCT, immediately following ERT withdrawal, is likely to have been caused by transient DS elevation in the bloodstream whilst the donor cells became established and began to produce sufficient enzyme. Clearly this will vary from patient to patient and with the enzyme produced from the donor graft, as we did not observe significant HCII-T elevations in patient B post-HSCT and ERT withdrawal, but HCII-T could be used to monitor this response. Our HCII-T treatment data also agree with Randall et al. (2008) who observed a decrease in serum HCII-T in one MPSI patient receiving ERT followed by HSCT. Interestingly, they observed an MPSVI patient whose HCII-T levels dropped initially following ERT, but increased again after 65 weeks of treatment (Randall et al. 2008). This is very similar to the trend we observed in MPSVI patient G whose serum HCII-T levels began rising again at 78 days post-diagnosis following an initial decrease in response to ERT. The clinical history of our MPSVI patient suggests that the high HCII-T levels correlate with elevated IgG antibody levels and indicate a less satisfactory response to ERT than that observed in the MPSII patients. It should be noted that, even if inhibitory antibodies are not detected, it has been demonstrated in a mouse model of the lysosomal storage disorder metachromatic leukodystrophy that non-inhibitory antibodies can have a negative effect on ERT by blocking uptake of the recombinant enzyme (Matzner et al. 2008).
Our investigations of HCII-T measurement in DBS samples have shown promising initial data, but further investigation of DBS HCII-T measurement will be required in a larger group of patients to determine if these measurements are as reliable as serum HCII-T for diagnosis. This could in time lead to development of HCII-T for newborn screening in MPSI, II and VI, which all have available treatments.
In conclusion, serum HCII-T is a reliable biomarker for MPS diseases in which DS is stored, including MPSI, II, and VI and probably also MPSVII, and HS also has a smaller but still significant effect on HCII-T formation, with elevation above unaffected controls in MPSIII patients. The ability to measure HCII-T in DBS may provide a suitable method for newborn screening for MPS diseases for which treatments exist. Both serum HCII-T and urine DS:CS are suitable to track progress of MPSI, II, and VI patients. Serum HCII-T decreases rapidly in response to successful treatment with HSCT and/or ERT and elevations appear to correlate with enzyme infusion reactions or rising anti-enzyme antibody levels. However, MPSI patients who have received HSCT 1–16 years ago have HCII-T levels within or near the normal range; therefore, serum HCII-T is likely to be a good biomarker for monitoring short-term treatment outcomes, but may not be suitable to measure long-term treatment effects. Urine DS:CS is able to discriminate between different therapies (Wynn et al. 2009) and appears to be more suited for measuring long-term treatment effects in HSCT patients, but does not respond as rapidly as HCII-T to perturbations in treatment.
References
Boelens, J. J., Rocha, V., Aldenhoven, M., et al. (2009). Risk factor analysis of outcomes after unrelated cord blood transplantation in patients with hurler syndrome. Biol Blood Marrow Transplant, 15(5), 618–625.
Canal MM, Wilkinson FL, Cooper JD, Wraith JE, Wynn R, Bigger BW (2010) Circadian rhythm and suprachiasmatic nucleus alterations in the mouse model of mucopolysaccharidosis IIIB. Behav Brain Res 209(2): 212-20.
Church, H., Tylee, K., Cooper, A., et al. (2007). Biochemical monitoring after haemopoietic stem cell transplant for Hurler syndrome (MPSIH): implications for functional outcome after transplant in metabolic disease. Bone Marrow Transplant, 39(4), 207–210.
Decker, C., Yu, Z. F., Giugliani, R., et al. (2010). Enzyme replacement therapy for mucopolysaccharidosis VI: Growth and pubertal development in patients treated with recombinant human N-acetylgalactosamine 4-sulfatase. J Pediatr Rehabil Med, 3(2), 89–100.
Escolar, M. L., Poe, M. D., Provenzale, J. M., et al. (2005). Transplantation of umbilical-cord blood in babies with infantile Krabbe's disease. N Engl J Med, 352(20), 2069–2081.
Harmatz, P., Giugliani, R., Schwartz, I. V., et al. (2008). Long-term follow-up of endurance and safety outcomes during enzyme replacement therapy for mucopolysaccharidosis VI: Final results of three clinical studies of recombinant human N-acetylgalactosamine 4-sulfatase. Mol Genet Metab, 94(4), 469–475.
Jones, S. A., Almassy, Z., Beck, M., et al. (2009). Mortality and cause of death in mucopolysaccharidosis type II-a historical review based on data from the Hunter Outcome Survey (HOS). J Inherit Metab Dis, 32(4), 534–543.
Krivit, W. (2002). Stem cell bone marrow transplantation in patients with metabolic storage diseases. Adv Pediatr, 49, 359–378.
Krivit, W., Pierpont, M. E., Ayaz, K., et al. (1984). Bone-marrow transplantation in the Maroteaux-Lamy syndrome (mucopolysaccharidosis type VI). Biochemical and clinical status 24 months after transplantation. N Engl J Med, 311(25), 1606–1611.
Langford-Smith, K., Arasaradnam, M., Wraith, J. E., Wynn, R., & Bigger, B. W. (2010). Evaluation of heparin cofactor II-thrombin complex as a biomarker on blood spots from mucopolysaccharidosis I, IIIA and IIIB mice. Mol Genet Metab, 99(3), 269–274.
Malinowska, M., Wilkinson, F. L., Bennett, W., et al. (2009). Genistein reduces lysosomal storage in peripheral tissues of mucopolysaccharide IIIB mice. Mol Genet Metab, 98(3), 235–242.
Matzner, U., Matthes, F., Weigelt, C., et al. (2008). Non-inhibitory antibodies impede lysosomal storage reduction during enzyme replacement therapy of a lysosomal storage disease. J Mol Med, 86(4), 433–442.
Meikle, P. J., & Hopwood, J. J. (2003). Lysosomal storage disorders: emerging therapeutic options require early diagnosis. Eur J Pediatr, 162(Suppl 1), S34–S37.
Meyer, A., Kossow, K., Gal, A., et al. (2007). Scoring evaluation of the natural course of mucopolysaccharidosis type IIIA (Sanfilippo syndrome type A). Pediatrics, 120(5), e1255–e1261.
Montano, A. M., Tomatsu, S., Gottesman, G. S., Smith, M., & Orii, T. (2007). International Morquio A Registry: clinical manifestation and natural course of Morquio A disease. J Inherit Metab Dis, 30(2), 165–174.
Peters, C., Shapiro, E. G., Anderson, J., et al. (1998). Hurler syndrome: II. Outcome of HLA-genotypically identical sibling and HLA-haploidentical related donor bone marrow transplantation in fifty-four children. The Storage Disease Collaborative Study Group. Blood, 91(7), 2601–2608.
Peters, C., & Steward, C. G. (2003). Hematopoietic cell transplantation for inherited metabolic diseases: an overview of outcomes and practice guidelines. Bone Marrow Transplant, 31(4), 229–239.
Randall, D. R., Colobong, K. E., Hemmelgarn, H., et al. (2008). Heparin cofactor II-thrombin complex: a biomarker of MPS disease. Mol Genet Metab, 94(4), 456–461.
Randall, D. R., Sinclair, G. B., Colobong, K. E., Hetty, E., & Clarke, L. A. (2006). Heparin cofactor II-thrombin complex in MPS I: a biomarker of MPS disease. Mol Genet Metab, 88(3), 235–243.
Vellodi, A., Young, E. P., Cooper, A., et al. (1997). Bone marrow transplantation for mucopolysaccharidosis type I: experience of two British centres. Arch Dis Child, 76(2), 92–99.
Wraith, J. E. (2008). Enzyme replacement therapy with idursulfase in patients with mucopolysaccharidosis type II. Acta Paediatr Suppl, 97(457), 76–78.
Wraith, J. E., Clarke, L. A., Beck, M., et al. (2004). Enzyme replacement therapy for mucopolysaccharidosis I: a randomized, double-blinded, placebo-controlled, multinational study of recombinant human alpha-L-iduronidase (laronidase). J Pediatr, 144(5), 581–588.
Wynn, R. F., Wraith, J. E., Mercer, J., et al. (2009). Improved metabolic correction in patients with lysosomal storage disease treated with hematopoietic stem cell transplant compared with enzyme replacement therapy. J Pediatr, 154(4), 609–611.
Yamada, Y., Kato, K., Sukegawa, K., et al. (1998). Treatment of MPS VII (Sly disease) by allogeneic BMT in a female with homozygous A619V mutation. Bone Marrow Transplant, 21(6), 629–634.
Acknowledgements
Funding was provided by the National Specialist Commissioning Advisory Group of the UK Department of Health and grants from the UK Society for Mucopolysaccharide diseases and Canadian Society for Mucopolysaccharide and Related Diseases. R.W., J.E.W. and B.W.B. acknowledge support of the NIHR Manchester Biomedical Research Centre and the Academic Health Sciences Centre.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated: by Frits Wijburg
Competing interest: None declared.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Langford-Smith, K.J., Mercer, J., Petty, J. et al. Heparin cofactor II-thrombin complex and dermatan sulphate:chondroitin sulphate ratio are biomarkers of short- and long-term treatment effects in mucopolysaccharide diseases. J Inherit Metab Dis 34, 499–508 (2011). https://doi.org/10.1007/s10545-010-9254-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10545-010-9254-8