
Abstract
Morphotaxonomy based on phenotypic traits of immature hard ticks (Acari: Ixodidae) is a skill challenge and has prompted many inexperienced acarologists to adopt DNA-based methods for identifying and discriminating the species. The aim of this study is therefore to utilize COI gene for verifying the morphological status of Haemaphysalis ticks in Peninsular Malaysia. A total of 19 on-host ticks collected from four localities were first identified using specific illustrated taxonomic keys that lead to the genus of Haemaphysalis. Genotypic traits of tick species were then verified molecularly based on cytochrome oxidase subunit I (COI) gene using polymerase chain reaction and direct sequencing. Clustering analysis was carried out by constructing a phylogenetic tree to determine the genetic variation and diversity of local Haemaphysalis ticks. Based on external morphological characterizations, all immature ticks were successfully identified down to the genus level only. Molecular analysis of the genotypic using COI gene revealed 16 individuals (84%) as Haemaphysalis hystricis, and three individuals as H. humerosa with sequence homology of 97–99 and 86–87%, respectively. Haemaphysalis hystricis were clustered in their respective monophyletic group in the phylogeny trees with a bootstrap of 100%. Furthermore, a low intraspecific variation (<0.3%) was observed among Malaysian H. hystricis but high interspecific value (>15%) recorded. This study morphologically and molecularly confirms the presence of H. hystricis in Malaysia and the findings will add value to the existing knowledge in identification of ticks in this country.
Similar content being viewed by others

Introduction
In tropical countries, ticks are second important arthropods after mosquitoes that have potential to be vectors for transmission of infectious agents including bacteria, viruses and protozoan parasites. Twelve genera comprising 104 species of ticks are found in Southeast Asia with the recent addition of two new species of Dermacentor (Apanaskevich and Apanaskevich 2015). In Malaysia, at least 34 tick species belonging to the genera Amblyomma, Dermacentor, Haemaphysalis, Ixodes and Rhipicephalus have been documented (Hoogstraal et al. 1969; Mariana et al. 2007; Petney et al. 2007; Kolonin 2009). The most species-rich genus in Asia is Haemaphysalis with about 52 species or 31% of the world haemaphysalid fauna (Petney et al. 2007). The genus is distributed globally, though the greatest diversity is found in Southeast Asia (Hoogstraal and Trapido 1966; Kolonin 2009).
Haemaphysalis hystricis Supino is a three-host tick with a relatively broad host spectrum including human, domestic dogs, wild boar, pigs, buffalo and tigers (Mahara 1997; Cao et al. 2000; Parola et al. 2003). Its distribution occurs throughout the Australasian, Oriental, subtropical and temperate belt of Eastern Asia including Malaysia (Yamaguti et al. 1971). The species is a putative vector of pathogens such as Ehrlichia, Coxiella, Trypanosoma and Rickettsia spp., that may cause spotted fever group (SFG), ehrlichiosis and rickettsiosis (Parola et al. 2003; Ando et al. 2010; Arthan et al. 2015; Khoo et al. 2016). Despite its local abundance in Malaysia, most of the information regarding this tick species were published decades ago and little attention has been given to its medical importance and vectorial role. Haemaphysalis hystricis has often been misidentified as H. bispinosa Neumann, H birmaniae Supino, H. semermis Neumann and H. papuana nadchatrami Hoogstraal (Hoogstraal et al. 1965). To date, the tick-borne diseases of this region remain poorly characterized, mainly due to the limited expertise and accurate information on tick species found in Southeast Asia.
Although the morphological approach for tick identification based on phenotypic traits is economic and convenient, it requires solid training and experience in morphology and taxonomy. The approach is less applicable for damaged ticks and inaccurate for close-related species due to incomplete existing keys for immature stages (Well and Stevens 2008). Subsequently, it is significant to develop more relevant characterization methods in order to differentiate subspecies and species while at the same time offers reliable and convenient technique. Molecular approach, mainly based on mitochondrial (mt) and ribosomal DNA (rDNA) fragments, has provided a complementary tool for accurate identification of ticks (Rumer et al. 2011; Brahma et al. 2014) and characterization of their pathogens (Cheng et al. 2013). Furthermore, molecular identification can be the only technique when there are no other obvious means to match adults with immature stages (Frezal and Leblois 2008; Khera and Vohra 2013). Molecular data can also estimate genetic variation of specific genes directly from the examined taxa and discriminate the closely-related species (Lv et al. 2014; Kanduma et al. 2016).
According to Amendt et al. (2004), polymerase chain reaction (PCR) amplification of suitable regions of the genome, sequence analysis of the amplicons obtained and alignment of the data with reference sequence at various life stages of specimens are the usual and recommended methods to identify organisms. The cytochrome oxidase subunit I (COI) is the most frequently used marker and produced highly standard barcode for identification of almost all animal (Hebert et al. 2003). Due to higher mutation rate, maternal inheritance and haploid nature, the mtDNA encoded COI gene has been identified as a species-level marker for phylogenetic and taxonomic studies of arthropods including ticks (Casati et al. 2008; Lv et al. 2014; Ernieenor et al. 2016). Caparole et al. (1995) in their study has proven that mtDNA sequences were useful for unraveling the systematics of Ixodes ticks while Cakic et al. (2014) successfully discriminated and characterized the COI gene of I. ricinus ticks in Serbia.
To date, there is no such study on identification of Haemaphysalis ticks using well defined molecular approach in Malaysia. Therefore, this study is the first attempt to utilize COI gene for verifying the morphological status of Haemaphysalis ticks in Malaysia. The genetic species variations and phylogenetic relationship of local Haemaphysalis ticks were further determined using clustering analysis based on COI sequences.
Materials and methods
Collection of tick and morphological identification
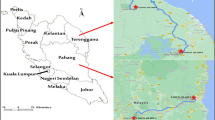
Ticks were collected from vertebrate animals caught by live-trapping in four localities (Fig. 1) namely Hulu Langat (Selangor), Janda Baik (Pahang), Seremban (Negeri Sembilan) and Gunung Tebu (Terengganu) between February 2012 and July 2013. Rodent trapping were carried out for four consecutive nights using banana and oil palm fruits as bait. Caught animals were anesthetized with diethyl-ether before screening and the ticks were collected using soft-forceps or sharpened wooden applicators sticks. All experimental procedures involving animals were approved by Animal Use Committee, Ministry of Health Malaysia [Reference Number: ACUC/KKM/02(6)2009] and conducted in accordance to International Conference of Harmonization Good Clinical Practice Guidelines. The ticks were kept individually in vials containing 100% ethanol. All samples were preliminary identified to genus-level based on external morphological characteristics under a stereo microscope (Model Stemi DV4 Zeiss, Germany) using specific illustrated morphological taxonomic keys (Kohls 1957; Walker et al. 2003).

Map of the tick’s collection study sites in Peninsular Malaysia: 1 Hulu Langat, Selangor; 2 Seremban, Negeri Sembilan; 3 Janda Baik, Pahang; 4 Gunung Tebu, Terengganu
DNA extraction
DNA extraction was performed using whole body of tick after three times of washing with sterile distilled water. Total genomic DNA was extracted using QIAamp DNA Mini Kit (Qiagen, Germany). The ticks were first macerated using sterile tips for 5 min in 80 μl of sterile phosphate buffer saline (PBS) followed by adding 100 μl of ATL lysis buffer. Samples were then incubated at 56 °C for 6 h after adding 20 μl Proteinase K for complete lyses. The following steps were performed according to manufacturer’s protocol. The DNA was then used for subsequent PCR.
PCR for the detection of COI gene of ticks
The first PCR was performed using the cycling parameters and primer pairs (cox-1F and cox-1R) from Chitimia et al. (2010). The amplification program consists of a total of 40 cycles: denaturing at 95 °C for 30 s, annealing at 55 °C for 1 min, and extension at 72 °C for 1 min, with an initial denaturation at 95 °C for 5 min. In the primary amplification, the PCR reaction mix of 50 μl which consisted of 25 μl Taq PCR Master Mix 2X, 10 μl of DNA template, 2.5 μl of 0.5 µM of each primer and 10 μl of nuclease free water. Low amplification rates (<50%) were found and this is a common problem in the recovery of COI fragment from tick specimen (Lv et al. 2014). To solve this issue, the COI of Haemaphysalis ticks was amplified using nested PCR in the present study. For the nested PCR amplification, 5 μl of the first amplification product was used as a template with the primers C1-J-1718 and C1-N-2329 (Shao et al. 2001). In the second amplification, the total reaction volume of 50 μl was made up of 25 μl Taq PCR Master Mix 2X, 5 μl of DNA template, 2.5 μl of 0.5 µM of each primer and 15 μl of nuclease free water. The reamplified PCR followed this modified cycling parameters: 94 °C for 5 min, then 35 cycles of 94 °C for 30 s, annealing temperature at 59 °C for 1 min, and extension at 72 °C for 1 min. For each PCR reaction, a negative control containing double distilled water was included. Both PCR reactions were performed using an Eppendorf Master Cycler Personal machine (Eppendorf, Germany). The PCR amplicons were visualized in 1.5% agarose gels electrophoresis stained with SeeNA II Nucleic Acid Stain DNA (Mbiotech, Korea) and viewed under an ultraviolet trans-illuminator.
DNA sequencing and data analysis
The PCR products was gel-purified using 5 Prime Agarose Gel Extract Mini Kit (Hamburg, Germany) according to manufacturer’s protocols. The purified PCR products were then sent to a commercial sequencing service company, First Base Laboratory Malaysia. Both strands of forward and reverse PCR products were sequenced using the Applied Biosystems BigDye Terminator v3.1 cycle sequencing kit (Applied Biosystem, USA). The obtained sequencing chromatograms were then analyzed and exported as FASTA sequence files. The specimens were molecularly identified by pasting their sequence record in both BLAST (Basic Local Alignment Search Tool) from NCBI’s GenBank and BOLD-IDS tool from BOLD Systems. In GenBank, the nucleotide collection database with MEGABLAST search was used, which is more appropriate for comparing a query to closely related sequences. In BOLD (Barcode of Life Data System), the search was performed with BOLD-IDS tool for animal identification (that use the COI barcode) in “Species Level Barcode Records” search database and then in “All Barcode Records on BOLD” search database if the former failed to identify.
The sequences that allowed the species-level identification were aligned with the corresponding sequences of Haemaphysalis tick species available in GenBank using CLUSTAL W. Clustering analysis was carried out using Phylogenetic Analysis Using Parsimony (PAUP), version 4.0b10. For distance analysis, a Neighbor-joining (NJ) tree was generated from a Kimura’s two-parameter distance matrix. Maximum parsimony (MP) analysis was performed to determine the most parsimonious tree(s) with a heuristic search of 1000 replications using tree bisection and reconnection option for branch-swapping algorithm. Confidence values for individual branches of the resulting trees were determined through bootstrap analysis with 1000 replicate. The TreeViewX version 0.5.1 software was used to visualize the phylograms obtained from all analyses. In this analysis, Ixodes granulatus (GenBank accession no. AB231673) was selected as an outgroup for COI gene and one H. hystricis sequence (GenBank accession no. JX573137) were aligned simultaneously as a species control.
Results
A total of 19 on-host immature ticks were collected from six species of hosts comprising Leopoldamys sabanus, Sundamys muelleri, Rattus tiomanicus, Maxomys rajah, Rhinosciurus laticaudatus and Tupaia glis (Table 1). The hosts were from the family Muridae, Ptilocercidae and Sciuridae. The ticks collected from all localities were correctly identified as Haemaphysalis sp. according to their morphological characters using specific taxonomic keys. Briefly, the unique character of Haemaphysalis ticks is their second segment of palps that were laterally produced beyond the basis capituli (Fig. 2). Their eyes are lacking, festoons are present, no ornamentation on scutum and possessed a distinct anal groove embracing the anus posteriorly (Fig. 2). The limitation of this study was that all immature ticks were only identified to the genus level only due to the lack of their morphological descriptions. Those individual immature ticks were therefore subjected to molecular identification.

External morphological characteristics of adult male Haemaphysalis ticks on dorsal (left) and ventral (right) view
DNA was extracted from ticks prior to PCR and after partial amplification, the PCR products yielded approximately 630 bp from all samples. Blast analysis of 16 (84%) mitochondrial sequences confirmed the morphological identification of the tick specimens processed by revealing 98–99% and 97.45–99.51% sequence nucleotide similarity with COI gene of Haemaphysalis hystricis available from GenBank and BOLD, respectively (Table 2). The other three (16%) sequences namely HL06_6, GT01_13 and GT01_14 matched to available sequence of H. humerosa with very low nucleotide similarity range from 86 to 87% and 86.39–86.54% for both GenBank and BOLD databases. The mean nucleotide content of the COI was 29.4% A, 37.8% T, 18.2% C and 14.5% G. A total of 597 bp fragments were obtained from the multiple alignments of the COI gene. Sequence analysis indicated that 144 (24%) variable sites were detected within the COI gene and 87 (60%) characters were parsimony informative. Additionally, the conserved sites were constituted by 453 (76%) characters showing that COI segment is a very conserved gene in the mtDNA.
Based on clustering analysis, both Neighbor-joining (Fig. 3) and Maximum parsimony (Fig. 4) trees revealed a distinction with high bootstrap value of 100% for H. hystricis which can be easily distinguished from other species. Significant grouping of three H. humerosa ticks sequences in independent monophyletic subclade was obtained with a bootstrap value of 100% in both analyses. Pairwise distance analysis of H. hystricis showed that the local species is genetically different from GenBank species with low genetic distance value ranged from 0.5 to 0.7% (Table 3). Genetic distance analysis of H. hystricis collected from all the four localities also indicated a low level of intraspecific value (< 0.3%). However, interspecific distance analyzed by the pairwise comparison revealed that H. humerosa sequences genetically differ from H. hystricis with high level of genetic variation (15.9–16.1%).

The Neighbor-joining tree generated from 21 sequences (including one outgroup) of Haemaphysalis hystricis and H. humerosa identified in the present study. The numbers at branches stand for bootstrap values of 1000 replications

The MP tree generated from 21 sequences (including one outgroup) of Haemaphysalis hystricis and H. humerosa identified in the present study. The numbers at branches stand for bootstrap values of 1000 replications
Discussion
Some tick genera are associated with various diseases, for example Haemaphysalis are disease agents for rickettsial spotted fever, tick typhus, anaplasmosis and ehrlichiosis (Kang et al. 2016; Khoo et al. 2016). Ticks tend to be localized in specific ecosystems but the increased speed and movement of people, ecotourism, translocation of wildlife and climate change provide risks of pathogen spreading beyond their natural ranges (Muruthi et al. 2016). Thus, accurate identification of ticks is of great significance for the investigation of epidemic disease epidemiology and to develop better control measures.
In this study, ticks collected from different localities far apart were confirmed morphologically as the genus Haemaphysalis. One of the defining morphological features of this genus is the presence of a prominent “blade-like dorsal retrograde process (Nuttall and Warburton 1915) on trochanter I. They also have short and wide palps with the palp femur projecting laterally beyond the rectangular basis capituli (Hoogstraal and Kim 1985). The dominant hosts of Haemaphysalis ticks in our sampling sites were Muridae family comprising of L. sabanus, S. muelleri, M. rajah and R. tiomanicus. This observation is consistent with previous studies that reported the abundance of Haemaphysalis ticks with 166 valid species (Burger et al. 2013) and their prevalence in domestic animals and rodents surrounding South East Asia (Kolonin 2009). Control of these animals need to be considered if local Haemaphysalis ticks were identified as a cause for any potential tick-borne infections.
Findings of the present study have verified the identity of Haemaphysalis ticks with high percentage of similarities to H. hystricis species as supported not only by the genetic clade but also with those from international databases. Our results clearly indicate the advantages of using COI gene that can provide sufficient power in identifying and discriminating species of Haemaphysalis ticks. Both NJ and MP tree topology also revealed close grouping of local H. hystricis and reference species with 100% bootstrap value. The high bootstrap support of this node may be due to amino acid homoplasy of the COI sequences (Burger et al. 2013). Prior to this study, there was only one COI sequence for H. hystricis that have been published in the GenBank and BOLD database. Therefore, sequences in this study only revealed 99% nucleotide similarity compensating for this lack of consistent data on specimens and a few number of populations from other countries in the databases. Small differences were probably caused by intraspecific variation which explains the polymorphism of this marker (Nava et al. 2010). The present study also revealed the distribution of only one tick species of H. hystricis collected from all localities despite the emergence of several known Haemaphysalis ticks in Peninsular Malaysia. It is speculated that the widely distribution of this species around Malaysia is probably due to climate and surrounding ecological conditions such as forests, shrub-undergrowth and presence of river at the sampling locality that might favored the survival (Estrada-Pena et al. 2012) of H. hystricis ticks.
The low percentage similarity value (86–87%) shown by three samples to corresponding accession sequences of H. humerosa species could be associated with the cryptic hybridization factor or geographical separations (Taberlet et al. 1997) which according to Rees et al. (2003) results to nucleotide substitutions. Significant grouping of H. humerosa in independent monophyletic clade also showed that small sample size of this species provides little support for intraspecific genetic diversity and phylogenetic inferences (Low et al. 2015). Furthermore, the high dissimilarity value and failure to cluster together with the rest of H. hystricis could be attributed to the absence of this species recorded in Malaysia and limited representation of their sequence in GenBank and BOLD. Haemaphysalis humerosa ticks have been reported mainly from Australia and can transmit Q fever (Stewart et al. 1987; Hammer et al. 2015).
Regarding to the genetic distance, a low intraspecific variation was observed among H. hystricis ticks collected from different localities (0–0.3%), but a high interspecific value (15.9–16.1%) with other species of the same genus. Thus, these observations suggest that mitochondrial COI gene is usually informative for determination of genetic variation either by interspecies or intraspecies of ticks. Moreover, tree topologies from different clustering analysis clearly indicated that different geographical in the present study had a smaller source of genetic variation by clustering all H. hystricis ticks in one clade. Notably, ecological variables and geographical distance did not explain the local patterns of differentiation observed in H. hystricis. This finding is in agreeable with previous studies which reported that short range movement of on-host ticks could explain for the low intraspecific value and similarity of ticks from some localities in Peninsular Malaysia (Fajs et al. 2012; Ernieenor et al. 2016).
The comparison between GenBank and BOLD databases reveals that GenBank had higher success rate in one time BLAST searches. This may probably due to the fact that GenBank presents a most comprehensive, more recent and specific database than BOLD (Benson et al. 2012). Moreover, the distribution of ticks COI sequences were more numerous in GenBank and some reference sequences were tagged as barcodes fragment (Sonet et al. 2013) for accurate species identification.
Conclusion
In conclusion, this study presents phenotypic identification of local Haemaphysalis ticks were supported by genotypic analysis using COI genetic marker. Our study produced the first COI barcoding sequences for H. hystricis from different localities in Peninsular Malaysia which contribute to the existing of nucleotide database of ticks. Based on clustering analysis, both NJ and MP tree showed very clear grouping of H. hystricis with reference sequences supported by high bootstrap value. Further sampling on a wide geographical of the genus Haemaphysalis, particularly H. hystricis should be considered to improve our understanding of the taxonomic and genetic variation of this species. The presence of H. hystricis species in Malaysia also merits further investigation as a potential vector of tick-borne diseases. The use of COI as a standard genetic marker to differentiate and identify tick species in Malaysia is therefore proposed.
References
Amendt J, Krettek R, Zehner R (2004) Forensic entomology. Naturwissenschaften 91:51–65
Ando S, Kurosawa M, Sakata A, Fujita H, Sakai K, Sekine M, Katsumi M, Saitou W, Yano Y, Takada N, Takano A, Kawabata H, Hanaoka N, Watanabe H, Kurane I, Kishimoto T (2010) Human Rickettsia heilongjiangensis infection, Japan. Emerg Infect Dis 16:1306–1308
Apanaskevich MA, Apanaskevich DA (2015) Description of new Dermacentor (Acari: Ixodidae) species from Malaysia and Vietnam. J Med Entomol 52:156–162
Arthan W, Sumrandee C, Hirunkanokpun S, Kitthawee S, Baimai V, Trinachartvanit W, Ahantarig A (2015) Detection of Coxiella-like endosymbiont in Haemaphysalis tick in Thailand. Ticks Tick Borne Dis 6:63–68
Benson DA, Karsch-Mizrachi I, Clark K, Lipman DJ, Ostell J, Sayers EW (2012) GenBank. Nucleic Acid Res 40(Database issue):D48–D53
Brahma RK, Dixit V, Sangwan AK, Doley R (2014) Identification and characterization of Rhipicephalus (Boophilus) microplus and Haemaphysalis bispinosa tick (Acari: Ixodidae) of North East India by ITS2 and 16S rDNA and morphological analysis. Exp Appl Acarol 62:253–265
Burger TD, Shao R, Barker SC (2013) Phylogenetic analysis of the mitochondrial genomes and nuclear rRNA genes of ticks reveals a deep phylogenetic structure within the genus Haemaphysalis and further elucidates the polophyly of the genus Amblyomma with respect to Amblyomma sphenodonti and Amblyomma elaphense. Ticks Tick Borne Dis 4:265–274
Cakic S, Mojsilovis M, Mihaljica D, Milutinovic M, Petrovic A, Tomanovic S (2014) Molecular characterization of COI gene of Ixodes ricinus (Linnaeus, 1758) from Serbia. Arch Biol Sci 66:683–690
Cao WC, Gao YM, Zhang PH, Zhang XT, Dai QH, Dumler JS, Fang LQ, Yang H (2000) Identification of Ehrlichia chaffeensis by nested PCR in ticks from Southern China. J Clin Microbiol 38:2778–2780
Caparole DA, Rich SM, Spielman A, Telford SR III, Kocher TD (1995) Discriminating between Ixodes ticks by means of mitochondrial DNA sequences. Mol Phylogenet Evol 4:361–365
Casati S, Bernasoni MV, Gern L, Piffaretti JC (2008) Assessment of intraspecific mtDNA variability of European Ixodes ricinus sensu stricto (Acari: Ixodidae). Infect Genet Evol 8:152–158
Cheng WY, Zhao GH, Jia YQ, Bian QQ, Du SZ, Fang YQ, Qi MZ, Yu SK (2013) Characterization of Haemaphysalis flava (Acari: Ixodidae) from qinling subspecies of giant panda (Ailuropoda melanoleuca qinlingensis) in Qinling Mountains (Central China) by morphology and molecular markers. PLoS ONE 8:e69793
Chitimia L, Lin R, Cosoroaba I, Wu XY, Song HQ, Yuan ZG, Zhu XQ (2010) Genetic characterization of ticks from southwestern Romania by sequences of mitochondrial cox1 and nad5 genes. Exp Appl Acarol 52:305–311
Ernieenor FCL, Yaakop S, Mariana A, Ernna G, Shukor MN (2016) Precise identification of different stages of tick, Ixodes granulatus Supino, 1897 (Acari: Ixodidae). Asian Pac J Trop Biomed 6:597–604
Estrada-Pena A, Ayllon N, de la Fuente J (2012) Impact of climate trends on tick-borne pathogen transmission. Front Physiol 3:64
Fajs L, Durmisi E, Knap N, Strle F, Avsic-Zupanc T (2012) Phylogeographic characterization of tick-borne encephalitis virus from patients, rodents and ticks in Slovenia. PLoS ONE 7:e48420
Frezal L, Leblois R (2008) Four years of DNA barcoding: current advances and prospects. Infect Genet Evol 8:727–736
Hammer JF, Emery D, Bogema DR, Jenkin C (2015) Detection of Theileria orientalis genotypes in Haemaphysalis longicornis ticks from southern Australia. Parasites Vectors 8:229
Hebert PDN, Cywinska A, Ball SL, deWard JR (2003) Biological identifications through DNA barcodes. Proc Biol Sci 270:313–321
Hoogstraal H, Kim KC (1985) Tick and mammal co-evolution, with emphasis on Haemaphysalis. In: Kim KC (ed) Coevolution of parasitic arthropods and mammals. Wiley-Inter-Science, New York, pp 505–568
Hoogstraal H, Trapido H (1966) Studies on Southeast Asian Haemaphysalis ticks (Ixodoidea, Ixodidae). Species described by Supino in 1897 from Burma, with special reference to H. (Rhipistma) asiaticus (=H. dentipalpis Warburton and Nuttall). J Parasitol 52:1172–1187
Hoogstraal H, Trapido H, Kohls GM (1965) Studies on Southeast Asian Haemaphysalis ticks (Ixodoidea, Ixodidae). The identity, distribution and hosts of H. (Kaiseriana) hystricis Supino. J Parasitol 51:467–480
Hoogstraal H, Lim BL, Anastos G (1969) Haemaphysalis (Kaiseriana) bispinosa Neumann (Ixodoidea, Ixodidae): evidence for consideration as an introduced species in the Malay Peninsula and Borneo. J Parasitol 55:1075–1077
Kanduma EG, Mwacharo JM, Githaka NW, Kinyanjui PW, Njuguna JN, Kamau LM, Kariuki E, Mwaura S, Skilton RA, Bishop RP (2016) Analyze of mitochondrial genes reveal two sympatric but genetically divergent lineages of Rhipicephalus appendiculatus in Kenya. Parasites Vectors 9:353
Kang JG, Ko S, Smith WB, Kim HC, Lee IY, Chae JS (2016) Prevalence of Anaplasma, Bartonella and Borrelia species in Haemaphysalis longicornis collected from goats in North Korea. J Vet Sci 17:207–216
Khera KS, Vohra P (2013) DNA barcoding: current advances and future prospects—a review. Asian J Biol Life Sci 2:185–189
Khoo JJ, Fezshin C, Kho L, Ahmad Shanizza AI, Lim FS, Tan KK, Chang LY, AbuBakar S (2016) Bacterial community in Haemaphysalis ticks of domesticated animals from the Orang Asli communities in Malaysia. Ticks Tick Borne Dis 7:929–937
Kohl GM (1957) Tick (Ixodidae) of Borneo and Malaya. Stud Inst Med Res Malaya 28:65–94
Kolonin GV (2009) Fauna of the Ixodid ticks of the world (Acari, Ixodidae), Moscow. (http://www.kolonin.org/)
Low VL, Tay ST, Kho KL, Koh FX, Tan TK, Lim YAL, Ong BL, Panchadcharam C, Rashid YN, Sofian-Azirun M (2015) Molecular characterization of the tick Rhipicephalus microplus in Malaysia: new insight into the cryptic diversity and distinct genetic assemblages throughout the world. Parasites Vectors 8:341
Lv J, Wu S, Zhang Y, Chen Y, Feng C, Yuan X, Jia G, Deng J, Wang C, Wang Q, Mei L, Lin X (2014) Assessment of four DNA segments (COI, 16S rDNA, ITS2, 12S rDNA) for species identification of the Ixodida (Acari: Ixodida). Parasites Vectors 7:93–114
Mahara F (1997) Japanese Spotted Fever: report of 31 cases and review of the literature. Emerg Infect Dis 3:105–111
Mariana A, Zuraidawati Z, Mohd Kulaimi B, Saleh I, Ho TM (2007) Fauna Ektoparasit di Bukit Labohan, Ma’ Daerah, Terengganu. Dlm. Sharma et al (pnyt) Biodiversity Expedition in Bukit Labohan and Ma’ Daerah, Terengganu. Petaling Jaya: WWF-Malaysia, pp 50–57
Muruthi CW, Lwande OW, Makumi JN, Runo S, Otiende M, Makori WA (2016) Phenotypic and genotypic identification of ticks sampled from wildlife species in selected conservation sites of Kenya. Vet Sci Technol 7:1–8
Nava S, Venzal JM, Labruna MB, Mastropaolo M, Gonzales EM, Mangold AJ, Guglielmone AA (2010) Hosts, distribution and genetic divergence (16S rDNA) of Amblyomma dubitatum (Acari: Ixodidae). Exp Appl Acarol 51:335–351
Nuttall GHF, Warburton C (1915) Ticks. A monograph of the Ixodida. Part III. The genus Haemaphysalis. Cambridge University Press, London, pp 349–550
Parola P, Cornet JP, Sanogo YO, Miller RS, Thien HV, Gonzales JP, Raoult D, Telford IS, Wongsrichanalai C (2003) Detection of Ehrlichia spp., Anaplasma spp., Rickettsia spp., and other eubacteria in ticks from the Thai-Myanmar border and Vietnam. J Clin Microbiol 41:1600–1608
Petney TN, Kolonin GV, Robbins RG (2007) Southeast Asian ticks (Acari: Ixodida): a historical perspective. Parasitol Res 101:S201–S205
Rees DJ, Diolli M, Kirkendall LR (2003) Molecules and morphology: evidence for cryptic hybridization in African Hyalomma (Acari: Ixodidae). Mol Phylogenet Evol 27:131–142
Rumer L, Sheshukova O, Dautel H, Mantke OD, Niedrig M (2011) Differentiation of medically important Euro-Asian tick species Ixodes ricinus, Ixodes persulcatus, Ixodes hexagonus, and Dermacentor reticulatus by polymerase chain reaction. Vector Borne Zoonotic Dis 11:899–905
Shao R, Campbell NJ, Barker SC (2001) Numerous gene rearrangements in the mitochondria genome of the wallaby louse, Heterodoxus macropus (Phthiraptera). Mol Biol Evol 18:858–865
Sonet G, Jordaens K, Braet Y, Bouruignon L, Dupont E, Backeljau T, de Mayer M, Desmyter S (2013) Utility of GenBank and the Barcode of Life Data Systems (BOLD) for the identification of forensically important Diptera from Belgium and France. Zookeys 365:307–328
Stewart NP, de Vos AJ, McGregor W, Shiels I (1987) Haemaphysalis humerosa, not H. longicornis, is the likely vector of Theileria buffeli in Australia. Aust Vet J 64:280–282
Taberlet P, Meyer A, Bouvet J (1997) Unusual mitochondrial DNA polymorphism in two local population of blue tit (Parus caeruleus). Mol Ecol 1:27–36
Walker AR, Bouattour A, Camicas JL, Estrada-Pena A, Horak IG, Latif A, Pegram RG, Preston PM (2003) Ticks of domestic animals in Africa: a guide to identification of species. Biosciences Report, London, pp 74–221
Well JD, Stevens JR (2008) Application of DNA-based methods in forensic entomology. Annu Rev Entomol 53:103–120
Yamaguti N, Tipton VJ, Keegan HL, Toshioka S (1971) Ticks of Japan, Korea and the Ryuku Islands. Brigham Young Univ Sci Bull 15:77–83
Acknowledgements
The authors would like to thank the Director-General of Health, Malaysia for his permission to publish this article. We wish to thank staff of Acarology, Institute for Medical Research for their assistance in the field. The study was supported by National Institute of Health Grant (Code: JPP-IMR 11-010) from the Ministry of Health, Malaysia.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Ernieenor, F.C.L., Ernna, G. & Mariana, A. Phenotypic and genotypic identification of hard ticks of the genus Haemaphysalis (Acari: Ixodidae) in Peninsular Malaysia. Exp Appl Acarol 71, 387–400 (2017). https://doi.org/10.1007/s10493-017-0120-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10493-017-0120-3