
Abstract
The phylum Neocallimastigomycota contains eight genera (about 20 species) of strictly anaerobic fungi. The evolutionary relationships of these genera are uncertain due to insufficient sequence data to infer their phylogenies. Based on morphology and molecular phylogeny, thirteen isolates obtained from yak faeces and rumen digesta in China were assigned to Neocallimastix frontalis (nine isolates), Orpinomyces joyonii (two isolates) and Caecomyces sp. (two isolates), respectively. The phylogenetic relationships of the eight genera were evaluated using complete ITS and partial LSU sequences, compared to the ITS1 region which has been widely used in this phylum in the past. Five monophyletic lineages corresponding to six of the eight genera were statistically supported. Isolates of Caecomyces and Cyllamyces were present in a single lineage and could not be separated properly. Members of Neocallimastigomycota with uniflagellate zoospores represented by Piromyces were polyphyletic. The Piromyces-like genus Oontomyces was consistently closely related to the traditional Anaeromyces, and separated the latter genus into two clades. The phylogenetic position of the Piromyces-like genus Buwchfawromyces remained unresolved. Orpinomyces and Neocallimastix, sharing polyflagellate zoospores, were supported as sister genera in the LSU phylogeny. Apparently ITS, specifically ITS1 alone, is not a good marker to resolve the generic affinities of the studied fungi. The LSU sequences are easier to align and appear to work well to resolve generic relationships. This study provides a comparative phylogenetic revision of Neocallimastigomycota isolates known from culture and sequence data.
Similar content being viewed by others


Introduction
Since their first recognition as Fungi in the mid-1970s (Orpin 1975), the anaerobic fungi have been widely recognised as active and major contributors to the degradation of plant fibre within the rumen and hindgut of larger herbivorous animals (Bauchop 1981; Wood et al. 1986; Joblin et al. 1989; Trinci et al. 1994). They are not only crucially involved in the rumen function and animal nutrition, but also have great potential for improving the conversion of lignocellulose into bioenergy products (Dashtban et al. 2009; Youssef et al. 2013; Haitjema et al. 2014; Solomon et al. 2016). These organisms were initially described only as a group of chytridiomycetous fungi (Orpin 1975) due to the attributes of their life cycle (with a vegetative structure from which zoospores are produced) and biochemistry (chitin-containing cell walls). Later, based on the type species Neocallimastix frontalis, Heath et al. (1983) formally classified them into a new family Neocallimastigaceae in the chytridiomycetous order Spizellomycetales mainly due to the similarities of zoospore ultrastructure to some members of this order. Their taxonomic position in Spizellomycetales was then contended by molecular data (Li and Heath 1992), and the order Neocallimastigales was proposed for the family based on the comparison of multiple morphological, ultrastructural, and developmental characters among the Chytridiomycota (Li et al. 1993). Further phylogenetic analyses with nrDNA (18S + 5.8S + 28S) from an extensive range of chytridiomycetous fungi revealed Neocallimastigales to be a monophyletic group within the polyphyletic Chytridiomycota (James et al. 2006). Combined with morphological, ecological and ultrastructural data, the phylum Neocallimastigomycota was subsequently erected (Hibbett et al. 2007).
On the basis of the thallus morphology and the number of flagella per zoospore, six genera were described (Ho and Barr 1995; Ozkose et al. 2001; Gruninger et al. 2014). There are four genera possessing rhizoidal thalli composed of a branched rhizoidal system and sporangia: Anaeromyces has polycentric thalli (exhibiting multiple centers of reproduction with more than one sporangia in common) and uni-flagellate zoospores; Neocallimastix possesses monocentric thalli (with one reproductive body or sporangium) and multi-flagellate zoospores; Orpinomyces has polycentric thalli and multi-flagellate zoospores; Piromyces possesses monocentric thalli and uni-flagellate zoospores. Two other genera, Caecomyces and Cyllamyces, produce thalli composed of bulbous vegetative cell (or holdfast) and sporangia. Caecomyces normally possesses only one, or a limited number of, zoosporangia formed directly on the surface of the bulbous holdfast or on the end of a simple sporangiophore (Wubah et al. 1991a; Ho and Barr 1995; Fig. 1 in Gruninger et al. 2014); while multiple zoosporangia are produced on branched sporangiophores with nuclei for Cyllamyces (Ozkose et al. 2001). In addition, uni-flagellate zoospores are defined by predominantly one flagellum per spore, with up to 10 % of zoospores having 2–4 flagella; while multi-flagellate zoospores are characterised by more than four flagella per spore.

Morphology of anaerobic fungi from yak. Neocallimastix sp.: a thallus, b zoospore; Orpinomyces sp.: c thallus, d zoospore; Caecomyces sp.: e, g thalli, f, h zoospores. (bars a, c, e, g = 20 µm; b, d, f, h = 10 µm)
From the 1970s to 1990s, nearly 20 species of anaerobic fungi were defined primarily by zoospore ultrastructure (Heath et al. 1983; Orpin and Munn 1986; Munn et al. 1988; Webb and Theodorou 1991), including four species in the genus Neocallimastix, three species in the genus Orpinomyces and two species in the genus Caecomyces. However these ultrastructural criteria have been questioned, not only due to the complicated techniques used for ultrastructural study, but also to the fact that these features tend to vary with culture conditions, age, as well as technique used for sample preparation (Ho and Barr 1995; Brookman et al. 2000).
Several studies attempted to delimit the species morphology under the light microscope. Wubah et al. (1991b) compared two cultures of N. frontalis from New Zealand with a culture of Neocallimastix patriciarum from the UK. They then concluded that these cultures belonged to the same species. In 1995, a monographic study by Ho and Barr critically examined the thallus morphology of all available species using a light microscope. A broad range of morphological variation of a single species was frequently observed. It was also found that media, stage of growth, etc. could easily influence thallus shape and size. For example, when the medium is rich in glucose or contains filter paper, the sporangia often became abnormally large and abort. Fourteen species were re-described by Ho and Barr (1995). In the genus Neocallimastix, the type species N. frontalis was observed to produce endogenous or exogenous sporangia quite variable in shape (spherical, ellipsoidal, broad ellipsoidal, ovoid, broadly ovoid or pyriform, occasionally angular, tubular or irregular) and in size (8.5–170 µm diam, or 10 to over 100 µm long), zoospores were also variable in shape (ovoid to globose) and size (7–22 µm diam.). Based on their morphological investigation, they treated both N. patriciarum and N. variabilis as synonyms of N. frontalis because both of them fell within the morphological limits of N. frontalis. The status of the third species, Neocallimastix hurleyensis, as a separate species was also questioned. N. hurleyensis was originally reported to have minor ultrastructural differences from N. frontalis (Webb and Theodorou 1988), and then was considered to be different in having zoospores discharging through a clearly defined apical pore in the sporangium from those of N. frontalis which released through an apical pore accompanied by dissolution and rupture of the sporangium wall (Webb and Theodorou 1991). But Wubah et al. (1991b) did not accept zoospore discharge as an unequivocal difference between N. hurleyensis and N. frontalis. In the genus Orpinomyces, O. joyonii and O. intercalaris were recognised, and O. bovis was reduced to a synonym of O. joyonii. Orpinomyces joyonii (originally described by Li et al. 1991) was distinguished by the terminal formation of sporangia on the simple or branched sporangiophores, while O. intercalaris (described by Ho et al. 1994) was characterised by sporangia developing intercalarily from expansion of hyphae or as a lateral outgrowth of a hypha, rarely terminal. In the genus Caecomyces, C. equi was described, on the basis of a cultured material embedded in plastic, as monocentric thallum producing a sporangium on a single bulbous vegetative cell with attached fibrillar or coralloid rhizoids; and C. communis was described as one, two or more sporangia on single or multiple bulbous vegetative cells (Ho and Barr 1995). The authors found that, in culture, the single sporangium form of C. communis predominated in the first day of culture, and that thalli with several bulbous vegetative cells and two or more sporangia occurred frequently in older cultures. Consequently, the status of C. equi as a distinct species was questioned (Ho and Barr 1995). Since no culture of C. equi was available for further study, it is difficult to determine whether the two species are synonymous or not. Additionally, six Piromyces species and two Anaeromyces species were recognised by Ho and Barr (1995). Apparently, the delimitation of some species remained contentious, which gave rise to the publication of a large number of hitherto-unnamed isolates (Brookman et al. 2000; Fliegerová et al. 2004; Sirohi et al. 2013a; Youssef et al. 2013).
Li and Heath (1992) made the first attempt to reveal the relationships among the anaerobic fungi using DNA sequence data. Based on the less conserved ITS1 (first internal transcribed spacer region of the nrDNA cistron) sequence data from single Anaeromyces, Neocallimastix, Orpinomyces and Piromyces isolates, the authors showed Anaeromyces to be more distant from the other three genera and the relationships of the other genera remained unresolved. Brookman et al. (2000) expanded the ITS1 sequence analysis to include a larger number of isolates and successfully differentiated Neocallimastix from Piromyces. Their results also revealed the genus Piromyces as being divergent. Later, the nrDNA sequences from the type material of the Cyllamyces type species (Cyllamyces aberensis) were made publicly available (James et al. 2006), and the D1/D2 domain of the 28S nrDNA gene (LSU) was used to differentiate the species of Orpinomyces (Dagar et al. 2011). Using molecular data, two more genera, Oontomyces (Dagar et al. 2015) and Buwchfawromyces (Callaghan et al. 2015) were proposed recently. Both genera morphologically fit with the genus Piromyces, but genetically appeared much closer to the genus Anaeromyces. However, the evolutionary relationships of different genera remain open for discussion, and the phylogenetic affiliations of species were challenged more frequently not only because the highly variable nature of the ITS region in this phylum makes it difficult to get a reliable phylogenetic tree, but also because most described species of anaerobic fungi lack sequence data from their type specimens (Griffith et al. 2010; Gruninger et al. 2014). On the other hand, ITS1 sequences have been widely employed in the studies of both diversity and ecology of the anaerobic fungi in the environment (Griffith et al. 2009; Nicholson et al. 2010; Fliegerová et al. 2010; Liggenstoffer et al. 2010; Kittelmann et al. 2012; Sirohi et al. 2013b; Koetschan et al. 2014). These cultivation-independent investigations have indicated the existence of many putative novel lineages (candidate genera) in this phylum (Nicholson et al. 2010; Liggenstoffer et al. 2010; Gruninger et al. 2014). At the same time, the generic identification of a large number of released ITS1 sequences from anaerobic fungal isolates or clones is doubtful (Nicholson et al. 2010; Fliegerová et al. 2010, Kittelmann et al. 2012). Koetschan et al. (2014) proposed the use of the common core secondary structure of the ITS1 region to reconstruct a more reliable phylogenetic tree of Neocallimastigomycota. Over 1100 of complete neocallimastigomycete ITS1 sequences were used, most of which were unclassified and obtained from uncultured samples. The integration of the additional structural information into the phylogenetic analyses allowed the authors to recognise the six known genera, as well as nine other defined monophyletic groups from uncultured sequences waiting for morphological characterisation. In their analysis, ten sequences failed to cluster into any of the defined groups (Fig. 4 in Koetschan et al. 2014); of these, two sequences (GenBank accessions AF170205 and AF170206) were from two isolates morphologically identified as members of Piromyces (Brookman et al. 2000), further confirming the polyphyletic nature of the morphologically defined Piromyces. Although there has been much progress in both the morphological and phylogenetic study of anaerobic fungi, it is clear that a stable taxonomic backbone complying with the Botanical Code of Nomenclature is urgently required for a better understanding the evolution, ecology and functions of the organisms in the Neocallimastigomycota (Ho and Barr 1995; Griffith et al. 2010; Gruninger et al. 2014).
Yak (Bos grunniens), as one kind of large ruminants existing on the Qinghai-Tibetan Plateau at elevations of 3000–5500 m, possesses unusual physiological adaptations to the extreme conditions such as low temperature (as low as −40 °C) and full-grazing style with grasses, sedges, and fobs as their sole nutritional source (Wiener et al. 2003; Zhong et al. 2006; Leslie and Schaller 2009). The prokaryotic populations in yak rumen have been reported to be distinct, with more than half of the species belonging to hitherto uncultured groups of prokaryotes (An et al. 2005). Neocallimastix, Orpinomyces and Piromyces were reported from yak in recent years (Feng 2005; Wei et al. 2016). However, our understanding of the fungal population diversity inside the yak digestive track is still limited.
In this study, as part of our survey on anaerobic fungi in yak rumen, we isolated fungi from both yak faeces and rumen digesta in Qinghai province, where yaks accounting for nearly half of the total Chinese yak population can be found. The aim of the present study was to examine the diversity of the obtained anaerobic fungal isolates in a phylogenetic context by assessing the phylogenetic relationships among the anaerobic fungi at the genus level based on additional sequences from the GenBank nucleotide database.
Materials and methods
Isolation and cultivation
The isolates were obtained from naturally grazing, rumen-fistulated or slaughtered yaks. Two rumen samples were taken directly from an animal carcas at local yak slaughterhouses in Qinghai province, China: one in Jiegu county of Yushu, and the other in Xining city. Five other rumen samples were taken at different time points from the two nonlactating rumen-fistulated yaks housed at the Academy of Animal Science and Veterinary Medicine affiliated to Qinghai University in Xining, China. All the rumen samples were promptly put into anaerobic Hungate tubes containing 10 ml modified Orpin’s medium C (Theodorou et al. 2005), and then used directly for purification in order to get the maximum coverage of the fungal diversity in the rumen using the rolling tube method described below. All animal procedures were approved by the Committee on the Ethics of Animal Experiments of the Institute of Microbiology, Chinese Academy of Sciences, China (permit number: PZIMCAS2008001) and every effort was devoted to minimising suffering of the rumen-fistulated yaks. For the naturally grazing yaks, fresh faeces were collected from the pastures in Qinghai province. The faecal samples were air-dried for transportation and were used for isolation within 1 week after collection. Isolation and four cycles of purification by the rolling tube method were performed as described by Theodorou et al. (2005). All the isolates were maintained in modified Orpin’s medium C with 8 g milled wheat straw l−1 as carbon source. Detailed information about all novel isolates are listed in Table 1. The morphological features of thalli and zoospores were examined by phase contrast observation using a Zeiss Axioskop 2 microscope or by DIC observation using a Nikon ECLIPSE 80i microscope.
DNA extraction and PCR amplification
Total genomic DNA was extracted from cultures using the E.Z.N.A.™ High Performance (HP) Fungal DNA Kit (Omega Bio-tek, USA) following the manufacturer’s instructions. The complete ITS1/5.8S nrRNA gene/ITS2 regions (ITS) and the D1/D2 domains of 28S nrDNA (LSU) were amplified in a Hybaid Px2 Thermal Cycler (Thermo Scientific, USA) using the primer combination ITS5 (White et al. 1990) and NL4 (O’Donnell 1993). The PCR reaction (50 μl total volume) contained 0.25 μM of each primer, 0.5 mM dNTP, 0.8 U of Taq DNA polymerase in 1× reaction buffer containing 2 mM MgCl2 (Transgen, China), and with 2 μL of template gDNA. The PCR program consisted of an initial denaturation of 5 min at 95 °C followed by 30 cycles of denaturation at 94 °C for 1 min, annealing at 55 °C for 1 min and elongation at 72 °C for 2 min and a final extension of 72 °C for 10 min. After purification using a QIAquick PCR Purification Kit (Qiagen, USA), the PCR products of all strains were cloned into the pGEM®-T Vector System I (Promega, USA). The ligated products were transformed into competent E. coli DH5α cells. One of the recombinant colonies of each isolate was randomly selected and sent to SinoGenoMax Co., Ltd. (http://www.sinogenomax.com) for sequencing using a standard M13 primer set in an Applied Biosystems 3730xl DNA Analyzer (Thermo Fisher Scientific, USA).
Sequence alignment and phylogenetic analyses
The novel sequences obtained for all the isolates were deposited in GenBank under the accession numbers shown in Table 1. The final data sets incorporated all representative sequences publicly available from pure cultures, covering each of the eight known genera (Tables 1, S1). In addition, our sequences were also checked against the expanded dataset of Koetschan et al. (2014) to confirm that none of our sequences fits with any of the uncultured lineages of their phylogeny. As different numbers of reference sequences are available for the ITS1 fragment, the complete ITS region and the partial LSU respectively, the phylogenetic analyses were performed separately using three data sets. Two Monoblepharella strains, as representatives of Monoblepharidales, were used as outgroup based on its relatively close relationship to anaerobic fungi of Neocallimastigales (James et al. 2006). The sequence datasets were initially aligned using MAFFT v. 7 (Katoh and Standley 2013), and were manually optimised using MEGA v. 6 (Tamura et al. 2013). For the ITS data sets, the alignment was obtained with the Q-INS-i method of iterative refinement as implemented in MAFFT to consider their secondary structure due to the highly variable nature of these sequences.
Phylogenetic analyses of individual locus datasets were based on Bayesian inference (BI), Maximum Likelihood (ML) and Maximum Parsimony (MP) analyses. For BI, the best evolutionary model for each partition was determined using MrModeltest2 (Nylander 2004) and incorporated into the analyses. A Markov Chain Monte Carlo (MCMC) algorithm was used to generate phylogenetic trees using MrBayes v. 3.2.1 (Ronquist and Huelsenbeck 2003; Ronquist et al. 2012) under the optimal criteria for each locus. The MCMC analysis lasted until the average standard deviation of split frequencies came below 0.01 with trees saved every 1000 generations. The first 25 % of saved trees were discarded as the ‘burn-in’ phase and posterior probabilities (PP) were determined from the remaining trees. The MP analysis was performed using PAUP v. 4.0b10 (Phylogenetic Analysis Using Parsimony; Swofford 2003). Phylogenetic relationships were estimated by heuristic searches with 1000 random addition sequences. Tree bisection-reconnection was used, with the branch swapping option set on ‘best trees’ only with all characters weighted equally and alignment gaps treated as fifth state. The tree length (TL), consistency index (CI), retention index (RI) and rescaled consistence index (RC) were calculated for the MP phylogenies and the bootstrap analysis (Hillis and Bull 1993) was based on 1000 replications. The ML analysis was performed under the GTR-GAMMA model of evolution using RAxML-VI-HPC v. 7.0.3 (Stamatakis 2006) with nonparametric bootstrapping using 1000 replicates. Trees were viewed in FigTree v. 1.1.2 (Rambaut 2009). The alignments used in the phylogenetic analyses, and resulting phylogenetic trees, were deposited in TreeBASE (submission ID S20001; https://treebase.org/).
Results
Morphology
Thirteen fungal isolates (Table 1) were obtained from yak rumen and faeces in Qinghai province, representing three morphological types. Nine of these isolates produced rhizoidal and monocentric thalli (Fig. 1a) with multi-flagellate zoospores (Fig. 1b) releasing through an irregularly ruptured apex of the mature sporangium, fitting with the morphology of N. frontalis. Two isolates possessed rhizoidal and polycentric thalli producing several sporangia terminally (Fig. 1c) with multi-flagellate zoospores (Fig. 1d), fitting with the morphology of Orpinomyces joyonii. The last two isolates possessed monocentric and bulbous thalli (Fig. 1e, g) with uni-flagellate zoospores (Fig. 1h) or bi-flagellate zoospores (Fig. 1f), fitting with the morphology of the genus Caecomyces.
Phylogenetic analyses
Thirteen sequences of approximately 1500 bp long were generated for each of the isolates in this study, spanning from the end of the 18S nrRNA gene, the complete ITS1 and ITS2 region with intervening 5.8S nrRNA gene to the D1/D2 domain of the 28S nrRNA gene. The matrix statistics and related indices resulting from the phylogenetic analyses of the ITS1, the complete ITS and the partial LSU (D1/D2 domains) datasets are summarised in Table 2. Each of the three datasets covered all eight known genera, and included the ex-type sequences of Oontomyces anksri and Buwchfawromyces eastonii, as well as two common reference sequences in the Neocallimastix clade (isolates GE13 and SR4), two common reference sequences in the Orpinomyces clade (isolates OUS1 and KF2) and one in the Anaeromyces clade (isolate K9). None of our sequences fitted into any of the remaining clades published by Koetschan et al. (2014).
ITS1 phylogeny
The BI consensus tree of ITS1 is presented in Fig. 2 with the respective PP, maximum parsimony bootstrap (MP-BS) and maximum likelihood bootstrap (ML-BS) support values indicated at the nodes. The ITS1 phylogenetic tree (Fig. 2) resolved only Orpinomyces (MP-BS = 100 %; ML-BS = 90 %; PP = 1.0), Buwchfawromyces (MP-BS = 100 %; ML-BS = 91 %; PP = 1.0), Cyllamyces (MP-BS = 92 %; ML-BS = 53 %; PP = 0.95) and Neocallimastix (MP-BS = 98 %; ML-BS = 63 %; PP < 0.95) as monophyletic lineages. In the Neocallimastix clade, the representatives of N. patriciarum and N. hurleyensis cluster together with isolate GE13 and 9 isolates from yak clustered closely with two representatives of N. frontalis, indicating that they represent the same species, N. frontalis. Two other isolates, NCS2 and NMG2, although on longer branches, also clustered in that clade. The genus Anaeromyces was divided into two sister lineages, one of which is designated here as the core Anaeromyces clade (MP-BS = 96 %; ML-BS = 79 %; PP = 1.0) and included the majority of the Anaeromyces isolates together with the isolate of Oontomyces on a long branch; the second Anaeromyces clade (MP-BS = 100 %; ML-BS = 92 %; PP = 1.0) contained two isolates: FFEX4 from cow faeces in UK (Griffith et al. 2009) and AF-CTS-RUA1 from rhinoceros faeces in India (Dagar et al. 2015). Koetschan et al. (2014) referred to this second clade as a distinct lineage “DT1”, which remains to be characterised morphologically as a novel genus. The two sister Anaeromyces clades had low statistic support (MP-BS = 65 %; ML-BS = 66 %; PP = 0.95) at their shared node. Two Caecomyces isolates from China formed a well-supported clade (MP-BS = 100 %; ML-BS = 77 %; PP = 1.0), but this clade did not include Caecomyces isolate A GRL-11. All three Caecomyces isolates clustered with the genus Cyllamyces (MP-BS < 50 %; ML-BS = 94 %; PP = 1.0). Isolates representing the genus Piromyces were polyphyletic and clustered at several positions throughout the phylogenetic tree. The backbone of the ITS1 phylogeny was collapsed to a basal polytomy and therefore no evidence was provided in the ITS1 tree to show the evolutionary relationships between any of the genera.

Consensus phylogram resulting from a Bayesian analysis of the ITS1 region, with the confidence values of bootstrap (BS) proportions from the MP analysis (before the backslash), the ML analysis (after the backslash) above branches, and the posterior probabilities (PP) from the Bayesian analysis below branches. The “-” indicates lacking statistical support (<50 % for ML-BS and MP-BS analyses; <0.90 for PP from Bayesian analyses). The branches with statistical support (MP-BS >50 %; ML-BS >50 %; PP >0.90) are highlighted with thickened branches. The tree is rooted to two isolates of Monoblepharella. Each genus clade is distinguished with boxes in different colours and lineages indicated with a solid square on the right represent isolates distinct from the eight known genera. Chinese isolates from yak are indicated with a star on the right side. The scale bar shows the expected number of changes per site
Complete ITS phylogeny
The BI consensus tree of the complete ITS is presented in Fig. 3 with the respective PP, MP-BS and ML-BS values indicated at the nodes. With the addition of phylogenetic information from ITS2, the complete ITS tree (Fig. 3) confirmed the monophyly of Cyllamyces (MP-BS = 100 %; ML-BS = 92 %; PP = 0.98), Buwchfawromyces (MP-BS = 100 %; ML-BS = 96 %; PP = 1.0) and Neocallimastix (MP-BS = 100 %; ML-BS = 95 %; PP = 1.0), and also resolved Caecomyces (MP-BS = 100 %; ML-BS = 78 %; PP = 0.99) as a monophyletic lineage. The highly supported Neocallimastix clade (MP-BS = 100 %; ML-BS = 95 %; PP = 1.0) included nine isolates from yak and the reference isolate (SR4) of N. frontalis, providing further evidence that our Neocallimastix isolates belong to the species N. frontalis. Similar to the ITS1 phylogeny, the genus Anaeromyces was again divided into two clades, with Oontomyces represented by a single lineage intermediate between the two clades. The node joining all three clades was poorly supported (MP-BS <50 %; ML-BS = 60 %; PP = 0.97), whereas the core clade (MP-BS = 100 %; ML-BS = 89 %; PP = 1) and smaller clade (MP-BS = 100 %; ML-BS = 100 %; PP = 1) containing isolates identified morphologically as Anaeromyces were both well-supported. O. anksri was placed basal to the core Anaeromyces clade with statistical support (MP-BS = 96 %; ML-BS = 83 %; PP = 0.98). With limited sampling due to lack of full-length sequences, only three Piromyces isolates could be included in this analysis and these clustered together with low support (MP-BS < 50 %; ML-BS = 78 %; PP = 0.96) and showed a close relationship (MP-BS < 50 %; ML-BS = 88 %; PP = 0.95) to the Cyllamyces/Caecomyces clade. The Cyllamyces clade (MP-BS = 100 %; ML-BS = 92 %; PP = 0.98) and Caecomyces clade (MP-BS = 100 %; ML-BS = 78 %; PP = 0.99) were both well-supported and cluster as sister clades with variable support (MP-BS < 50; ML-BS = 88 %; PP = 0.98), but no species of Caecomyces identified to species level were available. The Orpinomyces clade in the complete ITS phylogeny was only fully supported in the maximum parsimony analysis (MP-BS = 100 %; ML-BS < 50 %; PP < 0.90) but no species identified to species level were available.

Consensus phylogram resulting from a Bayesian analysis of the complete ITS region, with the confidence values of bootstrap (BS) proportions from the MP analysis (before the backslash), the ML analysis (after the backslash) above branches, and the posterior probabilities (PP) from the Bayesian analysis below branches. The “-” indicates lacking statistical support (<50 % for ML-BS and MP-BS analyses; <0.90 for PP from Bayesian analyses). The branches with statistical support (MP-BS >50 %; ML-BS >50 %; PP >0.90) are highlighted with thickened branches. The tree is rooted to two isolates of Monoblepharella. Each genus clade is distinguished with boxes in different colours. Chinese isolates from yak are indicated with a star on the right side. The scale bar shows the expected number of changes per site
LSU phylogeny
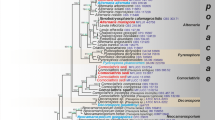
The BI consensus tree of LSU is presented (Fig. 4) with the respective PP, MP-BS and ML-BS values indicated at the nodes. In the LSU phylogeny (Fig. 4), the monophyletic clades of Neocallimastix (MP-BS = 87 %; ML-BS = 67 %; PP = 0.97) and Orpinomyces (MP-BS = 84 %; ML-BS = 99 %; PP = 1) cluster as sister lineages (MP-BS = 81 %; ML-BS = 97 %; PP = 1). The reference isolate (SR4) of N. frontalis, and isolates GE13 and 9 from yak again clustered in a well-supported clade (MP-BS = 97 %; ML-BS = 84 %; PP = 1). Two yak Orpinomyces isolates grouped with the representative isolates of O. joyonii (MP-BS = 88 %; ML-BS = 96 %; PP = 1), a close sister to the O. intercalaris clade (MP-BS = 87 %; ML-BS = 95 %; PP = 1). O. anksri clustered again basal to the core Anaeromyces with high support (MP-BS = 97 %; ML-BS = 99 %; PP = 1); the core Anaeromyces clade is well-supported (MP-BS = 100 %; ML-BS = 90 %; PP = 1). No LSU data were available for the second smaller Anaeromyces clade present in both ITS trees. The position of Buwchfawromyces was poorly supported in the LSU phylogeny (MP-BS < 50 %; ML-BS = 72 %; PP < 0.9). The two Chinese Caecomyces isolates clustered together with high support (MP-BS = 93 %; ML-BS = 99 %; PP = 0.99) in a clade containing C. aberensis and Caecomyces isolate GRL-12 (MP-BS = 86 %; ML-BS = 67 %; PP = 0.96). Similar to the ITS1 and complete ITS phylogenies, Piromyces was again polyphyletic. Two of the Piromyces isolates (BRL3 and GRL9) formed a clade (MP-BS = 99 %; ML-BS = 82 %; PP = 0.95 %), but the third Piromyces isolate (Pr1) clustered basal to all other species included in the phylogeny. The morphologies of the eight genera are illustrated to the right of the LSU tree to match the core morphological features of the genera with their phylogenetic relationships (Fig. 4).

Consensus phylogram resulting from a Bayesian analysis of the D1/D2 domain of LSU, with the confidence values of bootstrap (BS) proportions from the MP analysis (before the backslash), the ML analysis (after the backslash) above branches, and the posterior probabilities (PP) from the Bayesian analysis below branches. The “-” indicates lacking statistical support (<50 % for ML-BS and MP-BS analyses; <0.90 for PP from Bayesian analyses). The branches with statistical support (MP-BS >50 %; ML-BS >50 %; PP >0.90) are highlighted with thickened branches. The tree is rooted to two isolates of Monoblepharella. Each genus clade is distinguished with boxes in different colours and the lineage indicated with a solid square on the right represents an isolate distinct from the eight known genera. Chinese isolates from yak are indicated with a star on the right. The scale bar on the phylogeny shows the expected number of changes per site. Morphological characteristics of each of the known genera are illustrated to the right of the tree. The illustrations of the zoospores and the thalli of Neocallimastix and Orpinomyces were derived from this study (Fig. 1d h, a, c, respectively); the thallus of Anaeromyces was derived from Fliegerová et al. (2004); the thallus of Oontomyces was derived from Dagar et al. (2015); the thallus of Buwchfawromyces was derived from Callaghan et al. (2015); the thallus of Cyllamyces was derived from Ozkose et al. (2001); and the thalli of Caecomyces and Piromyces were derived from Gruninger et al. (2014). Thalli are shown on the left and zoospores are on the right (scale bars zoospores = 10 µm; sporangium 1, 2, 7 = 20 µm; 3 = 100 µm; 4, 5, 6, 8 = 50 µm). Thalli of Neocallimastix (1) and Orpinomyces (2) are illustrated with a sporangium filled with zoospores. Thalli of the other genera are illustrated with nuclei as black dots inside the sporangia or mycelia, for example those in Anaeromyces (3)
Discussion
Yak (Bos grunniens) exhibits unique adaptations to its fibre-rich diet, representing a promising reservoir of enzymes for degrading plant biomass. Over 95 % of yak populations exist in China, but only limited information is available about the composition of its anaerobic fungal population (Feng 2005; Wei et al. 2016). Our morphological investigation and phylogenetic analyses show the presence of a Neocallimastix species (N. frontalis) and an Orpinomyces species (O. joyonii), and also showed the existence of the bulbous-type genus Caecomyces in yak for the first time.
Unlike all the other members in the kingdom Fungi, their strict anaerobic nature and strict temperature requirement make it extremely difficult to retain anaerobic fungi in a viable and healthy state for study. The lack of a reliable method for long-term preservation of pure cultures of anaerobic fungi (Gruninger et al. 2014; Haitjema et al. 2014) resulted in the loss of the cultures of most described species in Neocallimastigomycota. The discovery of high variation in morphology further challenged the taxonomic study of Neocallimastigomycota (Barr et al. 1989; Ho and Barr 1995). Application of molecular technique has provided a route towards a more robust reappraisal of taxa in this group of fungi (Gruninger et al. 2014). As noted in the Introduction, genera of anaerobic fungi (Ho and Barr 1995; Ozkose et al. 2001; Gruninger et al. 2014) were traditionally defined by the thallus morphology (rhizoidal/bulbous and polycentric/monocentric) and the number of flagella per zoospore (uni-flagellate/multi-flagellate). A reappraisal of the morphologically-defined generic concept based on the phylogenetic analyses of three loci in this study (Figs. 2, 3, 4) shows that when two taxa have different combinations of thallus and zoospore morphology they indeed belong to different genera, while also showing that taxa with the same morphological combination can in fact belong to different genera. For example, Oontomyces, Buwchfawromyces and isolates that are morphologically Piromyces-like but phylogenetically distinct from Piromyces share the same morphological combination (rhizoidal and monocentric thalli with uni-flagellate zoospores) but these four groups are phylogenetically not congeneric. Isolates FFEX4 and AF-CTS-RUA1 share the same morphological combination (rhizoidal and monocentric thalli with uni-flagellate zoospores) with the core Anaeromyces clade, but based on the molecular data should represent a separate genus (“DT1”, Koetschan et al. 2014). Unfortunately, we did not have access to LSU sequences of these isolates to determine their position in the LSU phylogeny. Species identification is quite problematic because of the difficulty in comparative investigation of all samples using consistent criteria, with the exception of the genera Neocallimastix and Orpinomyces. After the monographic study of Ho and Barr (1995), only O. joyonii and O. intercalaris were recognised in the genus Orpinomyces; and only N. frontalis and N. hurleyensis in the genus Neocallimastix. The D1/D2 domain of LSU was reported to be capable of distinguishing the two species of Orpinomyces based on the analysis of a dataset without representatives of any other genera (Dagar et al. 2011). Our LSU analyses supported the distinction between O. joyonii and O. intercalaris, although they seemed genetically very close to each other (Fig. 4). Phylogenetic analyses of three loci in this study (Figs. 2, 3, 4) indicated that N. hurleyensis should be a synonym of N. frontalis, which implies that N. frontalis is the only species successfully isolated and cultured in the genus Neocallimastix. On the other hand, recent evidence from cultivation-independent surveys and sequence analyses suggest the presence of several novel lineages, as well as a much higher diversity of taxa in each of the known genus clades, including the Neocallimastix clade and the Orpinomyces clade (Nicholson et al. 2010; Liggenstoffer et al. 2010; Koetschan et al. 2014), implying that potential candidates of novel species and genera exist. The development of advanced culturing techniques is thus encouraged in order to attempt to isolate representatives of these uncharacterised taxa (Gruninger et al. 2014).
A polycentric growth habit was once believed to be of significance in the evolution of zoosporic fungi, representing a potential route for the evolution of more advanced fungal forms (Barr 1983; Ozkose et al. 2001). Gruninger et al. (2014) suggested that Neocallimastix was closely related with Orpinomyces, both genera having polyflagellate zoospores. In our analyses, both ITS1 and the complete ITS phylogenies failed to provide credible support for the relationship of Neocallimastix and Orpinomyces, but they were strongly supported to be sister lineages in our LSU phylogeny. This study also supports the belief that Caecomyces and Cyllamyces are closely related, sharing bulbous thalli (Gruninger et al. 2014). Caecomyces is generally defined by having monocentric thalli compared to Cyllamyces defined by having polycentric thalli (Gruninger et al. 2014). However, the morphological variability of the genus Caecomyces confused the delimitation of these two bulbous genera. It has been observed that isolates of Caecomyces produced several bulbous holdfasts and several sporangia, especially in old (more than 20 h) cultures (Wubah et al. 1991a; Ho and Barr 1995), and even exhibited multisporangiate thalli with sporangia sympodially arising from unbranched sporangiophores (Chen et al. 2007). In this study, all of the analyses (except for the parsimony bootstrap analyses of ITS1 and the complete ITS), supported a shared ancestry for Caecomyces and Cyllamyces. The ITS1 phylogeny did not resolve all isolates of Caecomyces as monophyletic. This could be explained by a lack of resolution when ITS1 alone is used to delimit these two genera. Although the LSU phylogeny failed to resolve the two genera, it is quite possible that sampling could be playing a role since only one LSU sequence of Cyllamyces was available to be included. The complete ITS sequences, which represent a wider sampling than LSU, suggest that Caecomyces and Cyllamyces could be distinct genera. More LSU sequences representing these two genera are needed to reach a final conclusion about whether the difference between Cyllamyces and Caecomyces should be at genus level or at species level. The complete ITS and LSU phylogenies supported Oontomyces as a sister lineage to Anaeromyces, whereas the phylogenetic position of Buwchfawromyces could not be consistently fixed by all analyses and alignments. In all analyses and alignments, isolates of Piromyces were polyphyletic, indicating that this genus requires a more detailed revision utilising longer sequences from more cultures and critical morphological examination of their microscopic characteristics. Care should therefore be taken to assign next-generation sequencing data from environmental samples to this genus.
Our phylogenetic analyses based on different loci allow us to evaluate the phylogenetic value of the complete ITS, the ITS1 region, and the D1/D2 region of LSU. The complete ITS region has been designated as official barcode for fungi (Schoch et al. 2012). Since Brookman et al. (2000) proposed the use of ITS1 for the classification of the gut fungi, it has become the most widely used amplicon for the study of taxonomy and community composition of Neocallimastigomycota. However, the highly variable nature of the ITS1 makes it difficult to align obtained sequences to get a reliable phylogenetic tree. In order to solve this issue, an ITS1 secondary structure prediction approach was proposed by Koetschan et al. (2014). Their revised ITS1 phylogenetic tree (Fig. 4 in Koetschan et al. 2014) showed better relationships between the known genera as well as several uncultured lineages, which is similar to the phylogenetic relationships revealed in the complete ITS tree here (Fig. 3) to some extent. For example, the genus Orpinomyces was located at the basis of the whole tree rather than forming a sister to Neocallimastix, and the Piromyces clade was close to the Cyllamyces/Caecomyces clade. On the other hand, intragenomic variation within the ITS region has been noted which would cause problems for direct sequencing PCR products (Li and Heath 1992; Brookman et al. 2000). A recent study has shown significant differences between ITS1 sequences obtained from different transformed colonies after cloning of a PCR amplicon of one isolate (GE09) of Buwchfawromyces eastonii. This recently described species was isolated from buffalo faeces in west Wales, but the ITS1 phylogeny (Fig. 3 in Callaghan et al. 2015) showed that clone A2 (GenBank EU414756) and clone A1 (GenBank EU414755) were more distant to clone A6 (EU414758) than to several uncultured sequences obtained from red deer in New Zealand. For this reason, further study is strongly needed to clarify how high the intragenomic variation in the ITS region of isolates in different genera is before utilising ITS1 in future studies classifying and identifying anaerobic fungi. On the other hand, no intragenomic variation was shown within the D1/D2 region of LSU in the study of B. eastonii (Fig. 2 in Callaghan et al. 2015). Furthermore, the LSU data obtained in this study included only a few gaps after sequence alignment using MAFFT v.7 (Katoh and Standley 2013); therefore the phylogeny based on this region is less sensitive to artefacts introduced by the sequence alignment. Except for Cyllamyces and Caecomyces, the LSU alignment provided a better resolved and supported phylogeny of the different genera.
Based on the LSU phylogeny, a hypothetical evolution can be proposed here that a Piromyces-like organism with monocentric thalli and uniflagellate zoospores might have been the ancestral lineage, from which members of the Cyllamyces/Caecomyces clade (with bulbous thalli and uniflagellate zoospores) were derived first, followed by Anaeromyces (with uniflagellate zoospores but polycentric thalli) and then Neocallimastix/Orpinomyces (with polyflagellate zoospores). The morphologically Piromyces-like genera Oontomyces and Buwchfawromyces are basal to the Anaeromyces clade, implying that the evolutionary process associated with the combination of monocentric thalli and uniflagellate zoospores happened more than once in the order.
With the great progress in both morphological and molecular study of anaerobic fungi, it is a good time now to establish a stable and reliable classification system in Neocallimastigomycota. It is necessary to recollect new isolates for all the known species for which type cultures have not been available. Form these fresh isolates, more inclusive morphological data and sequence data could be generated promptly by critical examination of morphology under light microscope together with sequencing of multiple DNA loci before the isolates are threatened by the lack of a reliable method for their long-term preservation. These data will be used to epi- or neotypify all the old names of the known species without type cultures. The release of genomic data could provide suitable protein-coding genes to supplement nrDNA sequence data, which would help to further resolve the phylogenetic relations between each of the known genera, and to more accurately delimit species within each genus, paving the way to the discovery of new taxa.
Conclusions
This study presents the first summary of the current molecular knowledge of rumen fungi of Neocallimastigomycota known from culture. The phylogenetic informativity of the ITS1 region, which is commonly used in DNA analyses of this group of fungi, was compared to phylogenies derived from the complete ITS region as well as the D1/D2 of the LSU region. Our data show that ITS1, or the complete ITS, alone do have limitations in the generic phylogeny and classification of rumen fungi, for example for Anaeromyces/Oontomyces in the ITS1 tree and for Orpinomyces in the ITS tree. However, in general the use of LSU does provide molecular support for most of the currently accepted genera of rumen fungi, and it does resolve the relationships between the genera much clearer. The only exception being members of Piromyces, which we hypothesise based on the presented LSU phylogeny to represent the ancestral morphology of the rumen fungi. This could also explain why this morphology occurs at multiple positions in the phylogeny; for some of those occurrences, novel genera with a Piromyces-like morphology were introduced in recent years, e.g. Oontomyces and Buwchfawromyces. To further stabilise the phylogeny of rumen fungi, a multi-locus approach may be useful. By both morphology and molecular phylogeny, the 13 isolates obtained from yak faeces and rumen digesta in China could be assigned to N. frontalis (9 isolates), O. joyonii (2 isolates) and an unclassified species of Caecomyces (2 isolates), giving more insight into the anaerobic fungal diversity in yak. Our Caecomyces isolates were genetically different from other taxa published in this genus with molecular evidence. Since all the described species of Caecomyces lack sequence data from their ex-type or authentic cultures, no new taxa were proposed in this study.
References
An D, Dong X, Dong Z (2005) Prokaryote diversity in the rumen of yak (Bos grunniens) and Jinnan cattle (Bos taurus) estimated by 16S rDNA homology analyses. Anaerobe 11:207–215. doi:10.1016/j.anaerobe.2005.02.001
Barr DJS (1983) The zoosporic grouping of plant pathogens. In: Buczacki ST (ed) Zoosporic plant pathogens. Academic Press, London, pp 161–192
Barr DJS, Kudo H, Jakober KD, Cheng K-J (1989) Morphology and development of rumen fungi: Neocallimastix sp., Piromyces communis, and Orpinomyces bovis gen. nov., sp. nov. Can J Bot 67:2815–2824. doi:10.1139/b00-067
Bauchop T (1981) The anaerobic fungi in rumen fibre digestion. Agric Environ 6:339–348. doi:10.1016/0304-1131(81)90021-7
Brookman JL, Mennim G, Trinci AP, Theodorou MK, Tuckwell DS (2000) Identification and characterization of anaerobic gut fungi using molecular methodologies based on ribosomal ITS1 and 18S rRNA. Microbiology 146:393–403. doi:10.1099/00221287-146-2-393
Callaghan TM, Podmirseg SM, Hohlweck D, Edwards JE, Puniya AK, Dagar SS, Griffith GW (2015) Buwchfawromyces eastonii gen. nov., sp. nov.: a new anaerobic fungus (Neocallimastigomycota) isolated from buffalo faeces. MycoKeys 9:11–28. doi:10.3897/mycokeys.9.9032
Chen YC, Tsai SD, Cheng HL, Chien CY, Hu CY, Cheng TY (2007) Caecomyces sympodialis sp. nov., a new rumen fungus isolated from Bos indicus. Mycologia 99:125–130. doi:10.3852/mycologia.99.1.125
Dagar SS, Kumar S, Mudgil P, Singh R, Puniya AK (2011) D1/D2 domain of large-subunit ribosomal DNA for differentiation of Orpinomyces spp. Appl Environ Microbiol 77:6722–6725. doi:10.1128/AEM.05441-11
Dagar SS, Kumar S, Griffith GW, Joan E, Edwards JE, Callaghan TM, Singh R, Nagpal AK, Puniya AK (2015) A new anaerobic fungus (Oontomyces anksri gen. nov., sp. nov.) from the digestive tract of the Indian camel (Camelus dromedarius). Fungal Biol 119:731–737. doi:10.1016/j.funbio.2015.04.005
Dashtban M, Schraft H, Qin W (2009) Fungal bioconversion of lignocellulosic residues; opportunities & perspectives. Int J Biol Sci 5:578–595. doi:10.7150/ijbs.5.578
Feng YL (2005) Ruminant nutrition. Scientific Press, Beijing
Fliegerová K, Hodrová B, Voigt K (2004) Classical and molecular approaches as a powerful tool for the characterization of rumen polycentric fungi. Folia Microbiol 49:157–164. doi:10.1007/BF02931392
Fliegerová K, Mrázek J, Hoffmann K, Zábranská J, Voigt K (2010) Diversity of anaerobic fungi within cow manure determined by ITS1 analysis. Folia Microbiol 55:319–325. doi:10.1007/s12223-010-0049-y
Griffith GW, Ozkose E, Theodorou MK, Davies DR (2009) Diversity of anaerobic fungal populations in cattle revealed by selective enrichment culture using different carbon sources. Fungal Ecol 2:87–97. doi:10.1016/j.funeco.2009.01.005
Griffith GW, Scott B, Fliegerová K, Liggenstoffer AS, van der Giezen M, Voigt K, Beakes G (2010) Anaerobic fungi: Neocallimastigomycota. IMA Fungus 1:181–185
Gruninger RJ, Puniya AK, Callaghan TM, Edwards JE, Youssef N, Dagar SS, Fliegerová K, Griffith GW, Forster R, Tsang A, McAllister T, Mostafa S, Elshahed MS (2014) Anaerobic fungi (phylum Neocallimastigomycota): advances in understanding their taxonomy, life cycle, ecology, role and biotechnological potential. FEMS Microbiol Ecol 90:1–17. doi:10.1111/1574-6941.12383
Haitjema CH, Solomon KV, Henske JK, Theodorou MK, O’Malley MA (2014) Anaerobic gut fungi: advances in isolation, culture, and cellulolytic enzyme discovery for biofuel production. Biotechnol Bioeng 111:1471–1482. doi:10.1002/bit.25264
Heath IB, Bauchop T, Skipp RA (1983) Assignment of the rumen anaerobe Neocallimastix frontalis to the Spizellomycetales (Chytridiomycetes) on the basis of its polyflagellate zoospore ultrastructure. Can J Bot 61:295–307. doi:10.1139/b83-033
Hibbett DS, Binder M, Bischoff JF, Blackwell M, Cannon PE, Eriksson OE, Huhndorf S, James T, Kirk PM, Lücking R, Thorsten Lumbsch H, Lutzoni F, Matheny PB, McLaughlin DJ, Powell MJ, Redhead S, Schoch CL, Spatafora JW, Stalpers JA, Vilgalys R, Aime MC, Aptroot A, Bauer R, Begerow D, Benny GL, Castlebury LA, Crous PW, Dai YC, Gams W, Geiser DM, Griffith GW, Gueidan C, Hawksworth DL, Hestmark G, Hosaka K, Humber RA, Hyde KD, Ironside JE, Kõljalg U, Kurtzman CP, Larsson KH, Lichtwardt R, Longcore J, Miadlikowska J, Miller A, Moncalvo JM, Mozley-Standridge S, Oberwinkler F, Parmasto E, Reeb V, Rogers JD, Roux C, Ryvarden L, Sampaio JP, Schüssler A, Sugiyama J, Thorn RG, Tibell L, Untereiner WA, Walker C, Wang Z, Weir A, Weiss M, White MM, Winka K, Yao YJ, Zhang N (2007) A higher-level phylogenetic classification of the Fungi. Mycol Res 111:509–547. doi:10.1016/j.mycres.2007.03.004
Hillis DM, Bull JJ (1993) An empirical test of bootstrapping as a method for assessing confidence in phylogenetic analysis. Syst Biol 42:182–192. doi:10.1093/sysbio/42.2.182
Ho YW, Barr DJS (1995) Classification of anaerobic fungi from herbivores with emphasis on rumen fungi from Malaysia. Mycologia 87:655–677. doi:10.2307/3760810
Ho YW, Abdullah N, Jalaludin S (1994) Orpinomyces intercalaris, a new species of polycentric anaerobic rumen fungus from cattle. Mycotaxon 50:139–150
James TY, Letcher PM, Longcore JE, Mozley-Standridge SE, Porter D, Powell MJ, Griffith GW, Vilgalys R (2006) A molecular phylogeny of the flagellated fungi (Chytridiomycota) and description of a new phylum (Blastocladiomycota). Mycologia 98:860–871. doi:10.3852/mycologia.98.6.860
Joblin KN, Campbell GP, Richardson AJ, Stewart CS (1989) Fermentation of barley straw by anaerobic rumen bacteria and fungi in axenic culture and in co-culture with methanogens. Lett Appl Microbiol 9:195–197. doi:10.1111/j.1472-765X.1989.tb00323.x
Katoh K, Standley DM (2013) MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30:772–780. doi:10.1093/molbev/mst010
Kittelmann S, Naylor GE, Koolaard JP, Janssen PH (2012) A proposed taxonomy of anaerobic fungi (Class Neocallimastigomycetes) suitable for large-scale sequence-based community structure analysis. PLoS ONE 7(5):e36866. doi:10.1371/journal.pone.0036866
Koetschan C, Kittelmann S, Lu J, Al-Halbouni D, Jarvis GN, Müller T, Wolf M, Janssen PH (2014) Internal transcribed spacer 1 secondary structure analysis reveals a common core throughout the anaerobic fungi (Neocallimastigomycota). PLoS ONE 9(3):e91928. doi:10.1371/journal.pone.0091928
Leslie DM, Schaller GB (2009) Bos grunniens and Bos mutus (Artiodactyla: Bovidae). Mamm Spec 836:1–17. doi:10.1644/836.1
Li J, Heath IB (1992) The phylogenetic relationships of the anaerobic chytridiomycetous gut fungi (Neocallimasticaceae) and the Chytridiomycota. I. Cladistic analysis of rRNA sequences. Can J Bot 70:1738–1746. doi:10.1139/b92-215
Li J, Heath IB, Cheng K-J (1991) The development and zoospore ultrastructure of a polycentric chytridiomycete gut fungus, Orpinomyces joyonii comb. nov. Can J Bot 69:580–589. doi:10.1139/b91-079
Li J, Heath IB, Packer L (1993) The phylogenetic relationships of the anaerobic chytridiomycetous gut fungi (Neocallimasticaceae) and the Chytridiomycota. II. Cladistic analysis of structural data and description of Neocallimasticales ord. nov. Can J Bot 71:393–407. doi:10.1139/b93-044
Liggenstoffer AS, Youssef NH, Couger MB, Elshahed MS (2010) Phylogenetic diversity and community structure of anaerobic gut fungi (phylum Neocallimastigomycota) in ruminant and non-ruminant herbivores. ISME J 4:1–11. doi:10.1038/ismej.2010.49
Munn EA, Orpin CG, Greenwood CA (1988) The ultrastructure and possible relationships of four obligate anaerobic chytridiomycete fungi from the rumen of sheep. Biosystems 21:67–82. doi:10.1016/0303-2647(88)90051-2
Nicholson MJ, McSweeney CS, Mackie RI, Brookman JL, Theodorou MK (2010) Diversity of anaerobic gut fungal populations analysed using ribosomal ITS1 sequences in faeces of wild and domesticated herbivores. Anaerobe 16:66–73. doi:10.1016/j.anaerobe.2009.05.003
Nylander JAA (2004) MrModeltest v. 2. Programme distributed by the author. Evolutionary Biology Centre, Uppsala University
Orpin CG (1975) Studies on the rumen flagellate Neocallimastix frontalis. J Gen Microbiol 91:249–262. doi:10.1099/00221287-91-2-249
Orpin CG, Munn EA (1986) Neocallimastix patriciarum sp. nov., a new member of the Neocallimasticaceae inhabiting the rumen of sheep. Trans Br Mycol Soc 86:178–181. doi:10.1016/S0007-1536(86)80138-3
Ozkose E, Thomas BJ, Davies DR, Griffith GW, Theodorou MK (2001) Cyllamyces aberensis gen. nov. sp. nov., a new anaerobic gut fungus with branched sporangiophores isolated from cattle. Can J Bot 79:666–673. doi:10.1139/b01-047
O'Donnell K (1993) Fusarium and its near relatives. In: Reynolds R, Taylor JW (eds) The fungal holomorph: mitotic, meiotic and pleomorphic speciation in fungal systematics. CAB International, Wallingford, pp 225–233
Rambaut A (2009) FigTree v. 1.3.1. Computer program and documentation distributed by the author at http://tree.bio.ed.ac.uk/software/
Ronquist F, Huelsenbeck JP (2003) MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19:1572–1574. doi:10.1093/bioinformatics/btg180
Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Höhna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP (2012) MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol 61:539–542. doi:10.1093/sysbio/sys029
Schoch CL, Seifert KA, Huhndorf S, Robert V, Spouge JL, Levesque CA, Chen W, Fungal Barcoding Consortium (2012) Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc Natl Acad Sci USA 109:6241–6246. doi:10.1073/pnas.1117018109
Sirohi SK, Choudhury PK, Dagar SS, Puniya AK, Singh D (2013a) Isolation, characterization and fibre degradation potential of anaerobic rumen fungi from cattle. Ann Microbiol 63:1187–1194. doi:10.1007/s13213-012-0577-6
Sirohi SK, Choudhury PK, Puniya AK, Dheer Singh D, Dagar SS, Singh N (2013b) Ribosomal ITS1 sequence-based diversity analysis of anaerobic rumen fungi in cattle fed on high fiber diet. Ann Microbiol 63:1571–1577. doi:10.1007/s13213-013-0620-2
Solomon KV, Haitjema CH, Henske JK, Gilmore SP, Borges-Rivera D, Lipzen A, Brewer HM, Purvine SO, Wright AT, Theodorou MK, Grigoriev IV, Regev A, Thompson DA, O’Malley MA (2016) Early-branching gut fungi possess a large, comprehensive array of biomass-degrading enzymes. Science 351:1192–1195. doi:10.1126/science.aad1431
Stamatakis A (2006) RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22:2688–2690. doi:10.1093/bioinformatics/btl446
Swofford DL (2003) PAUP*: phylogenetic analysis using parsimony (*and other methods), version 4. Sinauer Associates, Sunderland
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30:2725–2729. doi:10.1093/molbev/mst197
Theodorou MK, Brookman J, Trinci APJ (2005) Anaerobic fungi. In: Makkar HPS, McSweeney CS (eds) Methods in gut microbial ecology for ruminants. Springer, Dordrecht, pp 55–66
Trinci APJ, Davies DR, Gull K, Lawrence MI, Nielsen BB, Rickers A, Theodorou MK (1994) Anaerobic fungi in herbivorous animals. Mycol Res 98:129–152. doi:10.1016/S0953-7562(09)80178-0
Webb J, Theodorou MK (1988) A rumen anaerobic fungus of the genus Neocallimastix: ultrastructure of the polyflagellate zoospore and young thallus. BioSystem 21:393–401
Webb J, Theodorou MK (1991) Neocallimastix hurleyensis sp. nov., an anaerobic fungus from the ovine rumen. Can J Bot 69:1220–1224. doi:10.1139/b91-156
Wei YQ, Yang HJ, Luan Y, Long RJ, Wu YJ, Wang ZY (2016) Isolation, identification and fibrolytic characteristics of rumen fungi grown with indigenous methanogen from yaks (Bos grunniens) grazing on the Qinghai-Tibetan Plateau. JAppl Microbiol 120:571–587. doi:10.1111/jam.13035
White TJ, Bruns T, Lee S, Taylor J (1990) Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: Innis MA, Gelfand DH, Sninsky JJ, White TJ (eds) PCR protocols: a guide to methods and applications. Academic Press, New York, pp 315–322
Wiener G, Han JL, Long RJ (2003) The yak, 2nd edn. Regional Office for Asia and the Pacific, Food and Agriculture Organization of the United Nations, Bangkok
Wood TM, Wilson CA, McCrae SI, Joblin KN (1986) A highly active extracellular cellulase from the anaerobic rumen fungus Neocallimastix frontalis. FEMS Microbiol Lett 34:37–40
Wubah DA, Fuller MS, Akin DE (1991a) Studies on Caecomyces communis: morphology and development. Mycologia 83:303–310
Wubah DA, Fuller MS, Akin DE (1991b) Neocallimastix: a comparative morphological study. Can J Bot 69:835–843. doi:10.1139/b91-109
Youssef NH, Couger MB, Struchtemeyer CG, Liggenstoffer AS, Prade RA, Najar FZ, Atiyeh HK, Wilkins MR, Elshahed MS (2013) The genome of the anaerobic fungus Orpinomyces sp. strain C1A reveals the unique evolutionary history of a remarkable plant biomass degrader. Appl Environ Microbiol 79:4620–4634. doi:10.1128/AEM.00821-13
Zhong JC, Chen ZH, Zhao SJ, Xiao YP (2006) Classification of ecological types of the Chinese yak. Acta Ecol Sin 26:2068–2072. doi:10.1016/S1872-2032(06)60032-2
Acknowledgments
The authors acknowledge Jiangpu Meng for the morphological illustrations of eight genera in Fig. 4. This work was jointly supported by the National Basic Research Program of China (grant No. 2011CB100804) and the Natural Science Foundation of Beijing in China (No. 5092016).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Wang, X., Liu, X. & Groenewald, J.Z. Phylogeny of anaerobic fungi (phylum Neocallimastigomycota), with contributions from yak in China. Antonie van Leeuwenhoek 110, 87–103 (2017). https://doi.org/10.1007/s10482-016-0779-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10482-016-0779-1