
Abstract
Benzene adsorption from the gaseous phase at 298 K and the thermodynamics of this process using Monte Carlo methods is studied. A series of closed single-walled carbon nanotube (SWCNT) bundles (triangular packing arrangement) with various distance between nanotubes is investigated. Isolated closed nanotube is treated as the reference system. Benzene is considered as a model compound to represent aromatics. Simulation results show the significant effect of the geometric heterogeneity on benzene adsorption as well as the enthalpy and entropy at low surface coverages. For insignificantly separated nanotubes and at low benzene uptakes, the entropy of the adsorbed phase is close to the entropy of solid benzene. Therefore, C6H6 molecules strongly interacting with walls are ordered in quasi-solid structures. At higher surface coverages, the ordering and packing of benzene molecules is liquid-like. The geometry of bundles (mutual position of SWCNTs) significantly influences the position and orientation of adsorbed molecules against the wall.
Similar content being viewed by others


1 Introduction
Industrial processes are the main sources of benzene in the environment. C6H6 and many of its substituted analogues belong to a class of several hundreds compounds designated by environmental protection agencies as “hazardous air pollutants” (Wood et al. 2008; Shah et al. 2011). Given the increasing stringency of environmental standards, new technologies for effective removal of these compounds are in demand. Organic molecules have been shown to have a particularly high affinity for the interiors of carbon pores (Terzyk 2004). Membranes composed of carbon nanotubes (so-called carbon nanotube forests) have been recently synthesized and found to function well as environmental sensors or superior adsorbents for removing many kinds of organic pollutants such as dioxin and volatile aromatic organic compounds from air streams and aqueous solutions, and of course C6H6 (Agnihotri et al. 2005; Bittner et al. 2003; Castillejos et al. 2012; Eswaramoorthy et al. 1999; Jing et al. 2004; Long and Yang 2001; Peng et al.2003; Penza et al.2004; Podkościelny et al. 2014; Star et al.2003; Sumanasekera et al. 2002; Wiśniewski et al. 2015a). The problem of adsorption of organic compounds inside carbon nanotube (CNT) type materials has been also discussed on the basis of theoretical studies (Bhide and Yashonath 2000; Irving et al. 2004; Menon et al. 2000; Pańczyk et al. 2014; Podkościelny et al. 2014; Takaba et al. 1995; Tournus and Charlier 2005; Tournus et al. 2005).
Benzene adsorption/desorption porosimetry is a traditional and well-established technique used for characterization of crystalline and amorphous porous materials, especially ill-defined activated carbons. Moreover, this technique has been used for analysis of CNTs—see for example (Carrott et al. 2000; Eswaramoorthy et al. 1999; Gauden et al. 2006; Jing et al. 2004; Stoeckli and Centeno 2005; Wiśniewski et al. 2015a). In addition, CNT functionalization with biological molecules (such as proteins, DNA, nucleic acids, etc.), which contain repetitions of many aromatic rings, provides potential for developing drug delivery systems, molecule immobilization, and sensors (Werengowska-Ciećwierz et al. 2015). Therefore, a fundamental theoretical and systematic understanding of carbon nanotube interaction with benzene derivatives, which appear as building blocks in large organic and biological systems, is still necessary. Summing up, this simple aromatic molecule can be treated as the target one.
It is generally accepted that there are various possible types of adsorption centers for nanotube bundles: centers distributed over the internal porous volume of the bundle [(i) intratubular void space and (ii) interstices] but also over its external surface [(iii) grooves and (iv) exposed surfaces of the peripheral tubes] (Agnihotri et al. 2005; Arai et al. 2009; Calvaresi and Zerbetto 2014; Cruz and Mota 2009; Furmaniak et al. 2012; Harris 2009; Ho et al. 2014; Wiśniewski et al. 2013) as shown in Fig. S1(a) in the Electronic Supplementary Material. The existence of distinct types of adsorption sites is the fundamental property of bundles. Unfortunately, since the contribution of various adsorption centers of bundles depends mainly on the preparation procedure it is obvious that it can be different for different nanotubes. The efficiency of multi-walled carbon nanotubes (MWCNTs) and traditional activated carbons for benzene adsorption from the gaseous phase was studied by Gauden et al. (2006). Obtained experimental results show that in the case of MWCNTs adsorption of this organic compound between nanotubes is very important stage determining the mechanism of studied process. In the next paper the studies of benzene adsorption were used by Wiśniewski et al. (2013) for the explanation of the mutual position of nanotubes forming the so-called CNT bundles. The authors investigated a series of eight commercial closed carbon nanotubes. These materials were taken from three producers, and in consequence, they possess various diameters and different number of walls (single-, double, and multi-walled). On the basis of experimental measurements (nitrogen and benzene vapour adsorption isotherms or kinetics of benzene adsorption from aqueous solution on CNTs under and/or without compression) and computer simulations (nitrogen adsorption on tubes with different geometry of bundles and the distance between them) Wiśniewski et al. (2013) stated that generally widely accepted model of tube bundles, where the tubes are osculating and highly oriented, is valid only at the contact places. Moreover, in their opinion, isolated nanotubes dominate in the structure of a bundle and this type of nanotubes is the most important during adsorption process.
Therefore, in our opinion it is interesting to extend previous experimental and theoretical investigations (Furmaniak et al. 2009; Gauden et al. 2006; Gauden et al. 2013; Wiśniewski et al. 2013; Wiśniewski et al. 2015a) to the analysis of the influence of geometry of carbon bundles (related to the change of the distance between nanotubes) on benzene adsorption from the gaseous phase. In the current paper adsorption isotherms, enthalpies and entropies of adsorption on a series of carbon nanotube bundles are simulated using Monte Carlo method. These theoretical calculations allow to investigate the position and orientation of molecules against the wall and the degree of order of adsorbed molecules on the basis of the collected configurations. We expect that the use of simulation can help to advance our understanding of the adsorption mechanism of aromatics on carbon nanotube bundles from the gaseous phase at the microscopic level, taking benzene as a model compound representing aromatics.
2 Simulations details
The molecular models of CNT bundles used in this study consist of single-walled carbon nanotube (SWCNTs) with (13,0) chiral vector and theoretical diameter (defined as the center-to-center) equal to 1.016 nm. The respective atomic structure was obtained using the Nanotube Modeller software v. 1.7.3 (JCrystalSoft. Nanotube modeler. Version 1.7.3. Copyright JCrystalSoft, 2005–2012. (http://www.jcrystal.com)) assuming distance between carbon atoms as equal to 0.141 nm. All the systems of closed bundles are similar to material A-0 (Nanostructured & Amorphous Materials, Nanoamor, Houston, TX, USA) which has been extensively studied by us previously (Pacholczyk et al. 2011; Terzyk et al. 2012; Wiśniewski et al. 2013). The following values of parameters characterizing nanotubes A-0 were provided by the producer: the diameter and number of walls are equal to 1–2 nm and 1–2, respectively (Pacholczyk et al. 2011; Terzyk et al. 2012; Wiśniewski et al. 2013). On the other hand, the same quantities were also estimated by us from the position of the maximum of the diameter distribution plots based on high-resolution transmission electron microscopy images (diameter equal 1 nm and two walks, respectively) (Pacholczyk et al. 2011; Terzyk et al. 2012; Wiśniewski et al. 2013). Three mentioned above model carbon nanotubes were placed parallel (triangular packing arrangement––see Fig. S1 of the supplementary material) at the middle of the simulation box with the sizes of it along x, y and z axes equal to L box,x , L box,y , and L box,z , respectively. The respective values are collected in Table 1. The periodic boundary conditions (PBC) in all three directions were considered. We assumed L box,z = 8.52 nm (length along axes of tubes). However, due to PBC the model systems are equivalent to infinite SWCNTs. It should be noted that similar geometry of bundles was analysed previously (Arai et al. 2009; Furmaniak et al. 2009; Furmaniak et al. 2012; Gauden et al. 2013; Ho et al. 2014; Pacholczyk et al. 2011, Wiśniewski et al. 2013). Taking into account the experimental knowledge, we assumed that the internal space of our model carbon nanotube is inaccessible for benzene molecules. In order to take into account for the geometric heterogeneity of model nanotube bundles, we changed the effective distance between CNTs, ∆l tube . Precisely defining ∆l tube was the shortest distance between the center of carbon atoms of the neighboring nanotubes along the vector joining the axes of them. This distance was equal 0.34 nm in the case of osculating CNTs (b1, Table 1). Carbon nanotubes were increasingly spaced and; consequently, we considered also the following values of ∆l tube viz., ∆l tube = 0.80 (b2), 1.20 (b3), 1.60 (b4), 2.00 (b5), and 2.40 (b6) nm (Table 1 and Figs. 1–3). Additionally, isolated closed tube (b0) was investigated, and this system was treated as the reference one for all the theoretical considerations. For all the systems we considered the comparable distances between tubes from neighboring simulation boxes in the x and y directions (full periodic conditions). It should be noted that these distances are large enough to treat three-tube bundles as independent systems. Summing up, with the gradual increase in the distance between CNTs, we generated a series of model bundle systems that are characterized by various degree of geometric heterogeneity, as is presented in Figs. 2, 3.

The changes in shapes of benzene adsorption isotherms (298 K) simulated for bundles of carbon nanotubes with the progressive increase in the separation between tubes. Additionally, the data related to isolated tube (b0) are presented

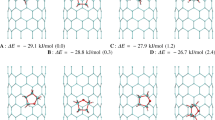
Snapshots showing the configurations of adsorbed benzene molecules for selected relative pressures on the single-walled carbon nanotubes bundles. We show respective scaled simulation boxes (their sizes are marked as solid black lines). A view along the principal axis of the cylinder is shown. Molecules in monolayer are marked in the following way: C atoms—red and H atom—grey, contrary to the other molecules, where carbon and hydrogen atoms are dark and light blue, respectively. Nanotubes are drawn as bold tubes or thin one to guide the eye. This figure and Fig. 3 were created using the VMD program (Humphrey et al. 1996) (Color figure online)

The same as Fig. 2 but the configurations are shown for a view perpendicular to the axis of model carbon nanotube
Benzene adsorption isotherms on generated model nanotubes were simulated using the hyper parallel tempering Monte Carlo (HPTMC) method proposed by Yan and de Pablo (1999). The simulations were performed for ambient temperature, i.e. T = 298 K. For each system, 117 replicas (corresponding to the relative pressures from 1.0 × 10−10 to 1.0) were considered. The HPTMC simulations utilized 5 × 106 cycles. One cycle equal to 20 attempts of the change of each replica state by (i) creation, (ii) annihilation or (iii) rotation and/or displacement of a randomly chosen molecule with equal probabilities, and one attempt of a configuration swap between a pair of randomly chosen replicas. The first 1 × 106 cycles were discarded to guarantee equilibration (Furmaniak 2015; Wiśniewski et al. 2012).
12-centre model of C6H6 was studied (Jorgensen and Severance 1990). Benzene molecules were modelled as the rigid ones (with the fixed bond lengths l C-C = 0.140 nm and l C-H = 0.107 nm). Each centre was the (12, 6) Lennard-Jones one (LJ) as well as the point charge (q). The energy of interaction of C6H6 molecules pair with the centres separated by a distance r mc-mc (and defined angular orientation–ω 1 , ω 2 ) may be calculated as the sum of LJ and electrostatic terms:
where \( U_{LJ}^{ij} \left( {r_{ij} } \right) \) is the energy of dispersion interactions between a pair of centres i and j (in the interacting molecules) located at the distance r ij , and calculated from the truncated LJ potential (Frenkel 1999):
where σ ij and ε ij are the collision diameter and the depth of the potential well for interactions between the centres of the type i and j. The second term in Eq. (1) [U electr (r mc-mc , ω1, ω2)] describes electrostatic interactions of a pair of molecules, described by:
where q i and q j are the values of the charges of the centres, and ε 0 (8.8543 × 10−12 C2 J−1 m−1) is the dielectric permittivity of free space. One can observe that, we used the cut-offs for electrostatic interactions but this is used for whole molecules i.e. if the centres of mass of two molecules are located at a distance smaller than r mc-mc,cut,C the sum of interactions between all pairs of charges occurring in both molecules is calculated, otherwise the electrostatic interactions are neglected. In this study this distance (r mc-mc,cut,C ) was assumed as equal to 1.5 nm. At this distance the energy of interactions of a pair of molecules is negligibly small.
The energy of interactions between adsorbed molecule and the structure of adsorbent is given as the sum of centre–centre LJ contributions:
where N C denotes the number of carbon atoms in the simulation box. For all LJ–type interactions the cut-off is assumed as equal to r cut,ij = 5 × σ ij . Table 2 collects all the values of applied interaction parameters. Cross-interaction parameters were calculated using the following mixing rules: σ ij = (σ ii × σ jj )1/2, ε ij = (ε ii × ε jj )1/2 (Hess et al. 2008; Jorgensen and Severance 1990).
The isosteric enthalpies of adsorption were calculated from the theory of fluctuations (Nicholson and Parsonage 1982):
where R is the universal gas constant, <…> is the ensemble average, N is the number of benzene molecule in the simulation box and U is the configuration energy of the system.
In order to characterize the ordering of C6H6 molecules during adsorption on bundles we computed the entropy of the adsorbed phase (S a ) for each value of pressure (each loading) from the following expression (see the supplementary material for its derivation):
where \( S_{g}^{o} \) is the standard entropy of the gaseous phase at the given temperature (T) and standard pressure (p o = 101,325 Pa) and p is equilibrium benzene pressure.
In order to analyze equilibrium configurations of C6H6 adsorbed on CNT bundles, we generated 45 000 representative equilibrium configurations for the selected points on the isotherm (Table S1 of the supplementary material) (Wiśniewski et al. 2015b). We start from the final configuration for each replica, and simulation was continued according the scheme described above. Actual configurations from the end of each fiftieth cycle were collected. On the basis of these results, we calculated (averaging over all of them) the density concentration ρ N (molecules per nm3) and orientation of C6H6 (the ring plane), \( S_{ \bot } \) against nanotube. The mass density profiles of benzene (ρ N ) were determined assuming that the centers of molecules were located on a center of benzene ring. In the case of orientation we have calculated the structural (order) parameter (the value of squared cosine of the angle) using the following relationship (Ori et al. 2014; Striolo et al. 2005):
where a part with brackets is the mean and α is the angle between the normal to the benzene ring of the i-th molecule and the main axis of a nanotube wall (Fig. S2 of the supplementary material). Thus, for example, the structural parameter represents the orientation of benzene rings perpendicular to the radial direction (\( S_{ \bot } \left( \alpha \right)\,\sim\,1.0 \), α = 90o) and parallel to the CNT surface (\( S_{ \bot } \left( \alpha \right)\,\,\sim\, - 0.5 \), α = 0o), while an isotropic distribution yields \( S_{ \bot } \left( \alpha \right)\,\sim\,0 \). Taking into account the monolayer adsorption it was easy to calculate ρ N (β) (where β is the angle around any particular nanotube as shown in Fig. S1(a) of the supplementary material). In the case of ρ N (r) and \( S_{ \bot } \left( r \right) \) (where r is the distance to the center of nanotube axis, see Fig. S1(a) of the supplementary material) the analysis of the system was more complicated. In natural way this attempt caused the division the simulation box into three separate but equivalent parts due to the symmetry of the system. This division runs along planes that, for example, can be lead through the center of the simulation box, z-axis and symmetrically between appropriate nanotubes. Moreover, these considerations should take into account the prevalence of monolayer coverage and remaining of the adsorbed molecules within interaction with a isolated tube. The further details of the division procedure were described in the supplementary material.
3 Results and discussion
Figure 1 shows benzene adsorption isotherms simulated on the mentioned above series of bundles on the basis of the HPTMC method. The number of molecules is converted into the absolute adsorption amount expressed as milimoles per gram of the adsorbent in order to compare data obtained for three CNT bundles (b1-b6) and single tube (b0). The simulated C6H6 adsorption isotherms show the type II behaviour of the IUPAC classification, which is fully consistent with experimental measurements (Wiśniewski et al. 2012). From the analysis of this figure one can easily observe the progressive increase in maximum adsorption (for p/p s \( \approx \,1 \)), i.e. the increase in total pore volume between CNTs of the given bundle under separation of these nanotubes. From the comparison of adsorption data simulated on b1 and b2 with the other adsorbents the differences between curves are also visible for low range of relative pressures (up to p/p s = 10−4). This result indicates the existence of high-energy centres for adjoining tubes or for adsorbent, where the distance between CNTs is equal to 0.80 nm (b2). This value of ∆l tube is sufficient so that the molecules absorbed between the tubes to form complete monolayer. Simultaneously, remarkable changes in the adsorption for monolayer (i.e. at the B-point) are observed for all systems excepting b5 and b6 bundles. The distances between these tubes are so large that CNTs can be treated as separated; and therefore, the same adsorption is detected for the given relative pressures after the B-point in comparison with reference b0 tube. For b5 and b6 the abrupt adsorption is also observed associated with the pore filling of the available space between tubes after monolayer completing by benzene molecules of the three nanotubes. Similar mechanism is observed for b3 and b4, however, before B-point and the pore filling of the available pore space. Summing up, the separation of CNTs leads the change of microporosity and influences on the monolayer region of adsorption isotherms and pore filling mechanism.
It is well-known that in the case of insignificantly separated and closed CNT bundles several stages for benzene adsorption can be observed: in grooves (or other high-energy centers), in interstitial channel, creation of monolayer on the pore walls, pore filling, and finally multilayer adsorption (Furmaniak et al. 2009). Of course, the order of their appearance may be different depends on the given system. Therefore, one can easy state that the mechanism of C6H6 (and other compounds) on non-ideal carbon nanotubes forming bundles is complex and it is still not fully known (Calvaresi and Zerbetto 2014; Wiśniewski et al. 2015a; Wiśniewski et al. 2015b). Snapshots of aromatics in carbon nanotube arrays (various intertube distances) may shed some light on the relationship between adsorption mechanism and the geometric heterogeneity of CNT bundles (Figs. 2, 3). For the case of osculating nanotubes (b1) we observe adsorption in corners, next monolayer formation, and finally multilayer adsorption. The increase in the distance between nanotubes makes it possible to create initially full layers on inner surface of nanotubes and the absence of adsorption on external tube walls-b2. Next a monolayer adsorption is visible, and finally multilayer adsorption is observed. For larger distance between tubes (b3) two layers of C6H6 are created at the space between tubes. The further stages are similar as for b2. In the case of b4 system the creation of monolayer on the walls of tubes is accompanied by a partial condensation between CNTs. Finally, the complete filling of the interstitial channel is detected. Further rise in the distance between nanotubes (b5 and b6) leads to the appearance of condensation for higher values of relative pressure after monolayer adsorption. The tendency of the existence of monolayer on all the nanotubes guarantees the similarity of the mechanism of adsorption for the region of adsorption isotherm related to B-point. In other words, the contribution of multilayer adsorption to the total adsorption is insignificantly for relative pressures lower than p/p s ≅ 0.1. The derogation from this rule is only observed for b4, where the filling of the available pore space is observed after monolayer formation for this range of relative pressures. From the analysis of snapshots showing the final configuration of benzene molecules on CNTs (perpendicular view to the central axis of nanotubes) it is seen that molecules do not form clusters and the surface is evenly covered.
Figures 4, 5 present the comparison of theoretical enthalpy and entropy versus adsorption or relative pressure, respectively. At low surface coverages, i.e. a <2 mmol/g (p/p s < 10−5) simulations show regular changes of these two fundamental state functions with the change of distance between nanotubes. From the analysis of data shown in Fig. 4 it is seen that the highest energies are observed for b2 system. Next, lower values of qst are visible for b1. Finally, the same values of isosteric enthalpy are detected for other bundle systems and also for isolated tube. This trend corresponds to the presence of high-energy centers between tubes (b2) and in grooves (b1). For higher coverages (~10−2 p/p s ) the enthalpy of C6H6 for all the systems tending to comparable values. What is more interesting, we found that the state of C6H6 molecules adsorbed on b0 and b3–b6 nanotubes at low surface coverages (p/p s < 10−5) corresponds to the gaseous phase (S a values are higher than \( S_{g}^{o} \)) as shown in Fig. 5.

The comparison of isosteric enthalpy of benzene adsorption at 298 K for all the studied carbon nanotube bundles (enthalpy of C6H6 condensation, L c , = 33.54 kJ/mol—dashed line)

The comparison of entropy of adsorbed benzene at 298 K for all the studied carbon nanotubes (the standard entropy of solid, liquid, and gaseous C6H6 (\( S_{s}^{o} \),\( S_{l}^{o} \), and \( S_{g}^{o} \), respectively) are equal to 136.5, 173.2, and 269.2 J/mol/K, respectively and marked as the dashed lines)
Therefore, following the simulation results, the first adsorbed C6H6 molecules are quite mobile. This seems reasonable because for defect-free graphene surfaces the motion of C6H6 in the monolayer is not restricted (i.e., adsorbate molecules move freely parallel to the nanotube surface). On the other hand, in the case b1 system the values of S a are between the entropies of the gaseous and the liquid phases. In this case the mobility of adsorbed molecules is limited due to the adsorption in grooves. Finally, for b2 system the entropy is the lowest in comparison with other systems. For zero coverages S a is close to \( S_{l}^{o} \) and tending to the differential entropy of solid C6H6 for higher adsorption values. This observation indicates that C6H6 molecules interact with high-energetic adsorption centres between CNTs of the investigated bundle. Adsorbed molecules are strongly localized and strongly ordered in solid-like structures. With further adsorption, the entropy of the adsorbed phase approaches \( S_{l}^{o} \) for all systems. This simply means that adsorbed C6H6 molecules are arranged into liquid-like structures. This is the behaviour expected for short-range dispersion forces. From the analysis of thermodynamic data it is seen that the change in the shape of both function corresponds the mechanism of the adsorption processes. These correlations are more visible in Figs. S3 and S4 of the supplementary material. In these figures the curves of benzene adsorption, enthalpy and entropy of adsorbed C6H6 at 298 are compared in the same plot for the given system. For example, the decreases/increases of entropy/enthalpy are connected with the pore filling the pore space between nanotubes.
In order to investigate more systematically the properties of adsorbed molecules we take into account the analysis of the molecular concentration and the their orientation against tubes as shown in Figs. 6–9. The investigations are limited to three regions of the available space for C6H6 molecules in the simulation box: (a) whole space (b) only external tube walls, and (c) the intertubular space. In the third case, the analysis is limited to monolayer coverages. The details of the division of the adsorption space are discussed in the supporting information (see also Fig. S1). Representative molecular concentrations, ρ N versus the distance to the center of nanotube axis (r) are displayed in Fig. 6. In the next figure (Fig. 7) plots of molecular concentration of benzene (ρ N ) as the function of the angle (β) around any particular nanotube are plotted. Additionally, the results of the systematic calculations of the orientation of benzene molecules, \( S_{ \bot } \left( r \right) \) on the external tube walls or in interstitial channel with grooves are shown in Figs. 8, 9, respectively.

Molecular concentration of benzene molecules (ρ N ) calculated as the function of the distance to the center of nanotube (r). The label of curve corresponds to the value of relative pressure and the average number of benzene molecules taken from adsorption isotherms and from Table S1 of the supplementary material. The arrows show the direction of changes related to the rise in the number of benzene molecules

Molecular concentration of benzene (ρ N ) as the function of the angle around nanotubes (β) calculated for monolayer adsorption only. All the values are averaged over one (b0) or three (b1–b6) CNTs. The curves shown in the same scale

Orientation of benzene molecules, \( S_{ \bot } \) as the function of the distance to the center of nanotube (r) calculated on the basis of Eq. (7). The dashed vertical lines denote the position of the nanotube wall and the monolayer region, respectively. The calculations are limited to the exposed space. All the values are averaged over one (b0) or three (b1–b6) CNTs

The same as Fig. 8 but the presented curves are calculated for the intertubular space for monolayer coverages. All the values are averaged over one (b0) or three (b1–b6) CNTs
From the analysis of data collected in Fig. 6 it is seen that the second solvation shell centered around 1.35 nm from the axis of the respective tube shows a single-peak structure. By contrast, the first external solvation shell has two peaks. It suggests that the different behavior of adsorbed molecules can be detected in the monolayer up to r = 1.15 nm. Probably this result is the effect of various orientations of adsorbed molecules against the wall, however, the confirmation and the explanation of this observation will be given later (see Figs. 8, 9). For low coverages of organic molecules on the walls maximum of the first peak (0.89 nm) predominates in comparison to the second one (0.99 nm). The ratio of intensity of the above mentioned two peaks vanishes with the rise of amount of adsorbed molecules for all three cases shown in Fig. 6a–c. Greater differences between plots (the comparison of three mentioned above regions of the available space) are detected for bundles with narrow pores, i.e. b1–b3, where the contribution of monolayer adsorption between nanotube walls to the total benzene accumulation is dominant. Additionally, as it can be expected the enhancement of intermolecular potential between nanotubes strongly influences on ρ N in the case of interstitial channel (Fig. 6c). In the case of separated CNTs (b6) the mass density profiles have the same shapes for all the regions and the similar plots as for b0 system are observed.
In Fig. 7 the relationship between average ρ N (calculated for monolayer coverage) and the angle around nanotube is presented. The analysis of plots for b2 system shows that the behavior of the adsorbed phase is significantly affected by the neighborhood of the nanotube surface for in the intertubular region for β from 120o to 240o. C6H6 molecules are strongly ordered. For b1 system (the adjoining nanotubes) the intertubular space is unavailable for benzene molecules (β from 110o to 250o). Two types of adsorption centres, i.e. higher-energy grooves and external tube walls are detected for this bundle. In consequence, the main peak of ρ N is observed for β = 105o (or 255o) and the position of maximum is related to the location of grooves. The effect of gradually formation of monolayer is also shown in this figure; increase of the adsorption leads to the appearance of more peaks together with their decrease for β tending to 0o (360o). From the comparison of b2 and b3 systems the similarity respective curves for low coverage is seen. This situation is the consequence of the comparable influence of overlapping adsorption potential from opposite walls on the adsorbed phase. However, for higher values of adsorption the differences are more visible due to the less impact of groves for b3 bundle. In the case of higher values of ∆l tube (b6) the differences in the intensity of peaks related to interstitial channels and external tube walls are smaller comparing to previously analyzed systems. The following tendency can be deduced: the larger is the distance between opposite tubes the smaller are differences between peaks for two investigated regions of the available pore space.
The results for \( S_{ \bot } \left( r \right) \) are shown in Fig. 8. The investigations are limited to the exposed space. Analysis of these plots reveals that benzene molecules in the first peak (located about 0.89 nm from the nanotube surface) of the first external solvation shell form mainly π-stack on its exterior wall with their rings parallel to the surface (i.e., \( S_{ \bot } \left( r \right)\, \cong \,1 \)). In the case of the second peak (0.99 nm) the reorientation of molecules is observed. Majority of C6H6 are close to perpendicular arrangement to the surface (\( S_{ \bot } \left( r \right)\, \cong \, - 0.37 \)). Summing up, we detect two predominate positions of center of benzene rings: parallel (closer tube) and perpendicular (at a greater distance from the tube, however, in monolayer). It should be pointed out that the similar trends were observed by Shim et al. (2011) from the analysis of the structure and dynamics of benzene outside of single-walled carbon nanotubes in the (n,n) armchair configuration immersed in liquid C6H6 (molecular dynamics computer simulations). If we consider the influence of benzene adsorption amount on the intensity and position of peaks on \( S_{ \bot } \) versus r curves one can see the insignificant differences between bundles and isolated nanotubes (b0). In the case of low concentration of adsorbed molecules identical first peaks are detected. On the other hand, one can see decrease in the values of structural parameter for the second peak and increase for the third one. This result suggests that during adsorption process the parallel orientation of benzene molecules is preferred at the initial stage. The orientation of next adsorbed molecules is close to perpendicular arrangement against the carbon wall (middle coverages). Finally, \( S_{ \bot } \) tends again to dominant parallel orientation in the second layer, however, in this layer also the other orientations are also observed (see peak for r > 1.35 nm). For this stage the change in orientation of organic molecules is larger, tending for higher coverages to less ordered arrangement in higher layers than monolayer. Similar tendencies are observed for interstitial channel with grooves (Fig. 9). Dominant behaviour of parallel orientation of molecules located in monolayer can be detected independently of the degree of wall coverage. No preferential orientation is also observed (\( S_{ \bot } \left( r \right)\, \cong \,0 \)). This effect is more visible for grooves in b1 bundle, where the differences between maxima of the both peaks are lower in comparison with other systems. As one might expect that the increase of the distance between CNTs leads to the similarity of the relationship \( S_{ \bot } \) versus r for the external tube and intertubular space [b5 (Fig. 8), b4 and b6 (Fig. 9)]. Summing up, from the analysis of all the results it is seen that the geometry of bundles (mutual position of SWCNTs) significantly influenced the position and orientation of adsorbed molecules against the wall for intertubular space. Moreover, we detected two predominate positions of center of benzene rings: parallel (closer tube) and perpendicular (at a greater distance from the tube, however, in monolayer). These different orientations are the result of the curvature of the nanotubes and the intermolecular adsorbate–adsorbate and adsorbate–adsorbent interactions.
4 Conclusions
We have studied the effect of the distance between single-walled carbon nanotubes forming bundles (in the other words the separation of CNTs) on the thermodynamics of C6H6 adsorption at 298 K. The performed Monte Carlo simulations show that geometric heterogeneity affects the C6H6 adsorption enthalpy and entropy. The entropy of C6H6 adsorption at low surface coverages is significantly reduced and reaches the entropy of solid. On consequence the mobility of adsorbed molecules is limited due to the restricted pore space. This result can be explained by the enhanced interactions between C6H6 molecules and high-energetic adsorption centres on carbon nanotubes. At higher surface coverages, the ordering and packing of C6H6 molecules is liquid-like, which results from the short-range character of dispersion forces. These results suggest the essential role of the arrangement of CNTs in the bundle. The similar effect were observed for isolated and closed CNT with high-energy carbon atoms (surface heterogeneities) (Wiśniewski et al. 2012). This type of study will be continued in future for nanotubes characterizing by lower and higher values of outer diameters. Additionally, some calorimetric measurements of benzene adsorption enthalpy from the gaseous phase for various nanotubes should be reported, compared and discussed together with theoretical calculations, and the results will be reported in near future.
References
Agnihotri, S., Rood, M.J., Rostam-Abadi, M.: Adsorption equilibrium of organic vapors on single-walled carbon nanotubes. Carbon 43, 2379–2388 (2005)
Arai, M., Utsumi, S., Kanamaru, M., Urita, K., Fujimori, T., Yoshizawa, N., Noguchi, D., Nishiyama, K., Hattori, Y., Okino, F., Ohba, T., Tanaka, H., Kanoh, H., Kaneko, K.: Enhanced hydrogen adsorptivity of single-wall carbon nanotube bundles by one-step C60-pillaring method. Nano Lett. 9, 3694–3698 (2009)
Bittner, E.W., Smith, M.R., Bockrath, B.C.: Characterization of the surfaces of single-walled carbon nanotubes using alcohols and hydrocarbons: a pulse adsorption technique. Carbon 41, 1231–1239 (2003)
Bhide, S.Y., Yashonath, S.: Structure and dynamics of benzene in one-dimensional channels. J. Phys. Chem. B 104, 11977–11986 (2000)
Calvaresi, M., Zerbetto, F.: Atomistic molecular dynamics simulations reveal insights into adsorption, packing, and fluxes of molecules with carbon nanotubes. J. Mater. Chem. A. 2, 12123–12135 (2014)
Carrott, P.J.M., Ribeiro Carrott, MMl, Cansado, I.P.P., Nabais, J.M.V.: Reference data for the adsorption of benzene on carbon materials. Carbon 38, 465–474 (2000)
Castillejos, E., Bachiller-Baeza, B., Rodríguez-Ramos, I., Guerrero-Ruiz, A.: An immersion calorimetry study of the interaction of organic compounds with carbon nanotube surfaces. Carbon 50, 2731–2740 (2012)
Cruz, F.J.L., Mota, J.P.B.: Thermodynamics of adsorption of light alkanes and alkenes in single-walled carbon nanotube bundles. Phys. Rev. B 79, 165426 (2009)
Eswaramoorthy, M., Sen, R., Rao, C.N.R.: A study of micropores in single-walled carbon nanotubes by the adsorption of gases and vapors. Chem. Phys. Lett. 304, 207–210 (1999)
Frenkel, D., Smit, B.: Understanding Molecular Simulation. Academic Press, San Diego (1999)
Furmaniak, S., Terzyk, A.P., Gauden, P.A., Wesołowski, R.P., Kowalczyk, P.: Ar, CCl4, and C6H6 adsorption outside and inside of the bundles of multi-walled carbon nanotubes—simulation study. Phys. Chem. Chem. Phys. 11, 4982–4995 (2009)
Furmaniak, S., Terzyk, A.P., Gauden, P.A., Kowalczyk, P.: Simulation of SF6 adsorption on the bundles of single walled carbon nanotubes. Microporous Mesoporous Mater. 154, 51–55 (2012)
Furmaniak, S.: Influence of activated carbon porosity and surface oxygen functionalities’ presence on adsorption of acetonitrile as a simple polar volatile organic compound. Environ. Technol. 36, 1984–1999 (2015)
Gauden, P.A., Terzyk, A.P., Rychlicki, G., Kowalczyk, P., Lota, K., Raymundo-Pinero, E., Frąckowiak, E., Béguin, F.: Thermodynamic properties of benzene adsorbed in activated carbons and multi-walled carbon nanotubes. Chem. Phys. Lett. 421, 409–414 (2006)
Gauden, P.A., Terzyk, A.P., Furmaniak, S., Wiśniewski, M., Kowalczyk, P., Bielicka, A., Zieliński, W.: Porosity of closed carbon nanotubes compressed using hydraulic pressure. Adsorption 19, 785–793 (2013)
Harris, P.J.F.: Carbon Nanotube Science. Cambridge University Press, Cambridge (2009)
Hess, B., Kutzner, C., van der Spoel, D., Lindahl, E.: GROMACS 4: algorithms for highly efficient, and scalable molecular simulation. J. Chem. Theory Comput. 4, 435–447 (2008)
Ho, T.M., Howes, T., Bhandari, B.R.: Encapsulation of gases in powder solid matrices and their applications: a review. Powder Technol. 259, 87–108 (2014)
Humphrey, W.F., Dalke, A., Schulten, K.: VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–38 (1996)
Irving, D.L., Sinnott, S.B., Lindner, A.S.: Interaction of functionalized benzene molecules with carbon nanopores. Chem. Phys. Lett. 389, 96–100 (2004)
JCrystalSoft. Nanotube modeler. Version 1.7.3. Copyright JCrystalSoft, 2005–2012. [http://www.jcrystal.com]
Jing, F., Yudasaka, M., Kasuya, Y., Kasuya, D., Iijima, S.: Influence of water on desorption rates of benzene adsorbed within single-wall carbon nanohorns. Chem. Phys. Lett. 397, 5–10 (2004)
Jorgensen, W.L., Severance, D.L.: Aromatic-aromatic interactions: free energy profiles for the benzene dimer in water, chloroform, and liquid benzene. J. Am. Chem. Soc. 112, 4768–4774 (1990)
Long, R.Q., Yang, R.T.: Carbon nanotubes as superior sorbent for dioxin removal. J. Am. Chem. Soc. 123, 2058–2059 (2001)
Menon, M., Andriotis, A.N., Froudakis, G.E.: Curvature dependence of the metal catalyst atom interaction with carbon nanotubes walls. Chem. Phys. Lett. 320, 425–434 (2000)
Nicholson, D., Parsonage, N.G.: Computer Simulation and the Statistical Mechanics of Adsorption. Academic Press, London (1982)
Ori, G., Villemot, F., Viau, L., Vioux, A., Coasne, B.: Ionic liquid confined in silica nanopores: molecular dynamics in the isobaric-isothermal ensemble. Mol. Phys. 112, 1350–1361 (2014)
Pacholczyk, A., Terzyk, A.P., Wiśniewski, M., Gauden, P.A., Wesołowsk, R.P., Furmaniak, S., Szcześ, A., Chibowski, E., Kruszka, B.: Phenol adsorption on closed carbon nanotubes. J. Coll. Interf. Sci. 361, 288–292 (2011)
Pańczyk, T., Wolski, P., Jagusiak, A., Drach, M.: Molecular dynamics study of Congo red interaction with carbon nanotubes. RSC Adv. 4, 47304–47312 (2014)
Peng, X.J., Li, Y.H., Luan, Z.K., Di, Z.C., Wang, H.Y., Tian, B.H., Jia, Z.P.: Adsorption of 1, 2- dichlorobenzene from water to carbon nanotubes. Chem. Phys. Lett. 376, 154–158 (2003)
Penza, M., Antolini, F., Antisari, M.V.: Carbon nanotubes as SAW chemical sensors materials. Sens. Actuators, B 100, 47–59 (2004)
Podkościelny, P., Toth, A., Berke, B., Laszlo, K., Nieszporek, K.: Heterogeneity of multiwalled carbon nanotubes based on adsorption of simple aromatic compounds from aqueous solutions. Adsorption 20, 789–800 (2014)
Shah, M., Santiso, E.E., Trout, B.L.: Computer simulations of homogeneous nucleation of benzene from the melt. J. Phys. Chem. B 115, 10400–10412 (2011)
Shim, Y., Jung, Y.J., Kim, H.J.: Carbon nanotubes in benzene: internal and external solvation. Phys. Chem. Chem. Phys. 13, 3969–3978 (2011)
Star, A., Han, T.-R., Gabriel, J-ChP, Bradley, K., Gruner, G.: Interaction of aromatic compounds with carbon nanotubes: correlation to the Hammett parameter of the substituent and measured carbon nanotube FET response. Nano Lett. 3, 1421–1423 (2003)
Stoeckli, F., Centeno, T.A.: On the determination of surface areas in activated carbons. Carbon 43, 1184–1190 (2005)
Striolo, A., Chialvo, A.A., Gubbins, K.E., Cummings, P.T.: Water in carbon nanotubes: adsorption isotherms and thermodynamic properties from molecular simulation. J. Chem. Phys. 122, 234712 (2005)
Sumanasekera, G.U., Pradhan, B.K., Romero, H.E., Adu, K.W., Eklund, P.C.: Giant thermopower effects from molecular physisorption on carbon nanotubes. Phys. Rev. Lett. 89, 166801 (2002)
Takaba, H., Katagiri, M., Kubo, M., Vetrivel, R., Miyamoto, A.: Molecular design of carbon nanotubes for the separation of molecules. Microporous Mater. 3, 449–455 (1995)
Terzyk, A.P.: Further insights into the role of carbon surface functionalities in the mechanism phenol adsorption. J. Colloid Interface Sci. 268, 301–329 (2004)
Terzyk, A.P., Gauden, P.A., Furmaniak, S., Wesołowski, R.P., Harris, P.J.F., Kowalczyk, P.: Adsorption from aqueous solutions on opened carbon nanotubes-organic compounds speed up delivery of water from inside. Phys. Chem. Chem. Phys. 11, 9341–9345 (2009)
Terzyk, A.P., Pacholczyk, A., Wiśniewski, M., Gauden, P.A.: Enhanced adsorption of paracetamol on closed carbon nanotubes by formation of nanoaggregates: carbon nanotubes as potential materials in hot-melt drug deposition-experiment and simulation. J. Colloid Interface Sci. 376, 209–216 (2012)
Tournus, F., Charlier, J.-C.: Ab initio study of benzene adsorption on carbon nanotubes. Phys. Rev. B 71, 165421 (2005)
Tournus, F., Latil, S., Heggie, M.I., Charlier, J.-C.: π-stacking interaction between carbon nanotubes and organic molecules. Phys. Rev. B 72, 075431 (2005)
Walther, J.H., Jaffe, R., Halicioglu, T., Koumoutsakos, P.: Carbon nanotubes in water: structural characteristics and energetics. J. Phys. Chem. B 105, 9980–9987 (2001)
Werengowska-Ciećwierz, K., Wiśniewski, M., Terzyk, A.P., Furmaniak, S.: The chemistry of bioconjugation in anoparticles-based drug delivery system. Adv. Condens. Matter Phys. 2015, 198175 (2015)
Wesołowski, R.P., Furmaniak, S., Terzyk, A.P., Gauden, P.A.: Simulating the effect of carbon nanotube curvature on adsorption of polycyclic aromatic hydrocarbons. Adsorption 17, 1–4 (2011)
Wiśniewski, M., Furmaniak, S., Kowalczyk, P., Werengowska, K.M., Rychlicki, G.: Thermodynamics of benzene adsorption on oxidized carbon nanotubes—experimental and simulation studies. Chem. Phys. Lett. 538, 93–98 (2012)
Wiśniewski, M., Gauden, P.A., Terzyk, A.P., Kowalczyk, P., Pacholczyk, A., Furmaniak, S.: Detecting adsorption space in carbon nanotubes by benzene uptake. J. Colloid Interface Sci. 391, 74–85 (2013)
Wiśniewski, M., Werengowska-Ciećwierz, K., Terzyk, A.P.: New findings on the influence of carbon surface curvature on energetics of benzene adsorption from aqueous solutions. Chem. Phys. Lett. 619, 219–222 (2015a)
Wiśniewski, M., Furmaniak, S., Terzyk, A.P., Gauden, P.A., Kowalczyk, P.: Properties of phenol confined in realistic carbon micropore model: experiment and simulation. J. Phys. Chem. C 119, 19987–19995 (2015b)
Wood, E.C., Herndon, S., Miake-Lye, R., Nelson, D., Seeley, M.: Aircraft and airport-related hazardous air pollutants: research needs and analysis (ACRP Project 02-03). Airport Cooperative Research Program, Transportation Research Board. Report 7 (2008)
Yan, Q., de Pablo, J.J.: Hyper-parallel tempering Monte Carlo: application to the Lennard-Jones fluid and the restricted primitive model. J Chem Phys. 111, 9509–9516 (1999)
Acknowledgments
P.A.G, S.F., A.P.T., and W.Z. acknowledge the use of the computer cluster at Poznań Supercomputing and Networking Centre (Poznań, Poland) as well as the Information and Communication Technology Centre of the Nicolaus Copernicus University (Toruń, Poland).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Gauden, P.A., Furmaniak, S., Włoch, J. et al. The influence of geometric heterogeneity of closed carbon nanotube bundles on benzene adsorption from the gaseous phase-Monte Carlo simulations. Adsorption 22, 639–651 (2016). https://doi.org/10.1007/s10450-015-9746-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10450-015-9746-9