
Abstract
This review summarizes the achievements and novel perspectives that our fish mitochondrial genome (mitogenome) project has brought to molecular phylogenetics and evolution of fishes during the last 15 years (1999–2014). To date, we have assembled ≈1,340 whole mitogenome sequences from fishes, publishing 83 mitogenomic papers on all major fish lineages (except agnathans). Those papers have been cited 5,303 times in total as of 30 September 2014 and have been featured in many textbooks and scientific articles as well as various media. These results have not only had a significant impact on the scientific community, but also attracted considerable attention from the general public. The success of the project largely owes to our own development of a novel, PCR-based approach for sequencing whole mitogenomes (ca. 16,500 bp), which opened a new avenue toward addressing higher-level relationships of fishes based on longer DNA sequences from a number of taxa. Shortly after the development of the method, we explicitly demonstrated the phylogenetic utility of mitogenomic data and actually resolved a long-standing issue in basal teleostean relationships. On the basis of those encouraging results from the initial studies, we published a series of four mitogenomic papers in 2003, which together encompass the whole of actinopterygian diversity and provided a “big picture” phylogenetic framework for the group. Those four studies offered a useful phylogenetic basis for subsequent studies (i.e., with a different choice of outgroups and targeted taxa) and have facilitated massive sequencing efforts for a wide variety of fishes from chondrichthyians to higher teleosts living in diverse habitats from freshwaters to the deep sea. We highlight some of the 83 mitogenomic papers by subject and briefly refer to the phylogenetic and evolutionary significances of those studies. Finally, we argue that intensive taxonomic sampling from an interface between species and populations together with the massive character sampling from mitogenome sequences using next-generation sequencing (NGS) technologies would enable simultaneous attempts to delimit species boundaries and to reconstruct evolutionary relationships at much finer scale, eventually unraveling the fish part of the Tree of Life in a bottom-up manner with more accurate estimations of species diversity.
Similar content being viewed by others

Introduction
Fifteen years have passed since the publication of a novel approach for sequencing the whole mitochondrial genome (mitogenome) of fishes (ca. 16,500 bp) using a combination of the long PCR technique (Cheng et al. 1994) and a number of fish-versatile primers (Miya and Nishida 1999). During this period (1999–2014), our research group has assembled nucleotide sequences for ≈1340 whole mitogenomes from fishes using this method (including unpublished ones) and published 83 peer-reviewed papers on mitochondrial genomics (mitogenomics) of fishes (including those papers based on partial mitogenome sequences >2,000 bp; Fig. 1). Most of those papers (77 papers) are more or less phylogeny oriented with the exception of five reports of single mitogenomes from commercially or zoologically important species (e.g., Japanese sardine, Inoue et al. 2000b), a paper addressing the highly conservative gene arrangement in vertebrate mitogenomes based on a comparative genomic approach (Satoh et al. 2010), and a paper describing a database specifically designed for fish mitogenomes (MitoFish) with a novel gene annotation pipeline (MitoAnnotator) (Iwasaki et al. 2013). Apparently, the course of this direction towards phylogenetics is due to a successful demonstration of the utility of the mitogenomic data for resolving higher-level relationships of fishes at an earlier stage of the study (Miya and Nishida 2000b). Consequently, the “mitogenomic era” was born (Broughton 2010) and an unprecedented number of contributions to molecular phylogenetics and evolution of fishes have been published from this single research group. Although members of this research group comprise mainly Japanese professional scientists, postdocs and graduate students, the affiliations of the 151 coauthors of these papers are distributed across 17 different countries, representing truly international collaborative efforts.

Approximate phylogenetic coverage of the 83 mitogenomic papers published from our research group. More taxonomically comprehensive studies, such as higher teleostean phylogenies of Miya et al. (2003), are placed on internal nodes, while those studies dealing with specific taxa (e.g., lophiiform, Miya et al. 2010) are placed on terminal nodes. Topology of the tree is based mostly on our recent studies (Miya et al. 2005; Nakatani et al. 2011; Campbell et al. 2013b; Chen et al. 2013; Miya et al. 2013) supplemented by those studies based on multiple nuclear genes (Near et al. 2012; Wainwright et al. 2012; Betancur et al. 2013a; Broughton et al. 2013; Near et al. 2013). Terminal taxa are ordinal names taken from Nelson (2006) for actinopterygians supplemented by our recent studies (Miya et al. 2005; Miya et al. 2007; Lavoué et al. 2008b; Miya et al. 2013). Exceptions are those of the Percomorpha, in which all terminal taxa are taken from a revised classification of bony fishes adopted by DeepFin (http://deepfin.org). Numerals indicate those 83 studies according to the temporal sequence of the publications: 1 Miya and Nishida (1999); 2 Inoue et al. (2000b); 3 Miya and Nishida (2000b); 4 Inoue et al. (2000a); 5 Inoue et al. (2001a); 6 Ishiguro et al. (2001); 7 Inoue et al. (2001c); 8 Kawaguchi et al. (2001); 9 Inoue et al. (2001b); 10 Miya et al. (2001); 11 Lee et al. (2001); 12 Inoue et al. (2003a); 13 Miya et al. (2003); 14 Saitoh et al. (2003); 15 Ishiguro et al. (2003); 16 Inoue et al. (2003b); 17 Simmons et al. (2004); 18 Simmons and Miya (2004); 19 Inoue et al. (2004); 20 Yamanoue et al. (2004); 21 Mabuchi et al. (2004); 22 Minegishi et al. (2005); 23 Inoue et al. (2005); 24 Miya et al. (2005); 25 Ishiguro et al. (2005); 26 Lavoué et al. (2005); 27 Miya et al. (2006); 28 Yamanoue et al. (2006); 29 Mabuchi et al. (2006); 30 Satoh et al. (2006); 31 Saitoh et al. (2006); 32 Mabuchi et al. (2007); 33 Lavoué et al. (2007); 34 Mayden et al. (2007); 35 Yamanoue et al. (2007); 36 Miya et al. (2007); 37 Kawahara et al. (2008); 38 Mayden et al. (2008); 39 Lavoué et al. (2008b); 40 Yamanoue et al. (2008); 41 Azuma et al. (2008); 42 Setiamarga et al. (2008); 43 Lavoué et al. (2008a); 44 Yamanoue et al. (2009b); 45 Kawahara et al. (2009); 46 Yamanoue et al. (2009a); 47 Johnson et al. (2009); 48 Inoue et al. (2009); 49 Mayden et al. (2009); 50 Yagishita et al. (2009); 51 Poulsen et al. (2009); 52 Setiamarga et al. (2009); 53 Takada et al. (2010); 54 Miya et al. (2010); 55 Doosey et al. (2010); 56 Hirayama et al. (2010); 57 Inoue et al. (2010c); 58 Lavoué et al. (2010); 59 Tang et al. (2010); 60 Yang et al. (2010); 61 Inoue et al. (2010b); 62 Satoh et al. (2010); 63 Yamanoue et al. (2011); 64 Saitoh et al. (2011); 65 Lavoué et al. (2011); 66 Nakatani et al. (2011); 67 Tang et al. (2011); 68 Johnson et al. (2012); 69 Aschliman et al. (2012); 70 Lavoué et al. (2012b); 71 Lavoué et al. (2012a); 72 Yang et al. (2012a); 73 Yang et al. (2012b); 74 Imoto et al. (2013); 75 Lavoué et al. (2013); 76 Poulsen et al. (2013); 77 Tang et al. (2013); 78 Miya et al. (2013); 79 Campbell et al. (2013b); 80 Iwasaki et al. (2013); 81 Song et al. (2014); 82 Lavoué et al. (2014); 83 Campbell et al. (2014)
Taxonomic coverage of those studies is extensive (Fig. 1), encompassing three of the four major fish lineages from chondrichthyans (e.g., Inoue et al. 2010b; Aschliman et al. 2012) to various actinopterygians (e.g., Inoue et al. 2003a; Ishiguro et al. 2003; Miya et al. 2003; Saitoh et al. 2003) and even to sarcopterygians (coelacanths, Inoue et al. 2005). Notably, our research group has never performed de novo sequencing for mitogenomes from agnathans; however, Inoue et al. (2010b) used two agnathans as outgroups in their study of holocephalan evolutionary history. Thus, the temporal dimension of our studies is well over 450 million years, considering that a common ancestor of those four vertebrate lineages emerged in the Paleozoic (Benton et al. 2009). In terms of the spatial dimensions, our phylogenetic studies cover the global habitat diversity, including freshwaters on major continental landmasses (e.g., Otophysi, see Saitoh et al. 2003; Nakatani et al. 2011), coral reefs in tropical and subtropical waters (e.g., Labridae, see Mabuchi et al. 2004, 2007), open oceans (e.g., Pelagia, Miya et al. 2013), deep-sea midwaters (e.g., Myctophiformes, see Poulsen et al. 2013), demersal waters (e.g., Macrouridae, see Satoh et al. 2006), and combinations across those habitats (e.g., diadromous Anguillidae, see Minegishi et al. 2005).
As of 30 September 2014, those 83 mitogenomic papers have been cited 5,303 times in total, with the most frequently cited paper (Miya et al. 2003) reaching 543 citations, followed by 14 papers with ≥100 citations (Google Scholar: http://scholar.google.co.jp; Table 1). A number of the mitogenomic papers have been cited in standard textbooks in ichthyology, such as “Fishes of the World” (Nelson 2006) and “The Diversity of Fishes” (Helfman et al. 2009), and even in textbooks of other disciplines, such as “Vertebrate Palaeontology” (Benton 2009) and “Evolution” (Futuyma 2013). In the 4th edition of “Fishes of the World,” Nelson (2006) mentioned our research activities, stating “… Some of the new, exciting molecular work is being done in the laboratories of … Masaki Miya in Chiba, Japan; and Mutsumi Nishidain Tokyo, Japan. These workers and their colleagues as well as many others are finding support for new clades that must be taken seriously, especially in those areas where agreement is found when different approaches are taken.” In addition to those citations in the textbooks, our article demonstrating a sister group relationship between pike and salmon (Ishiguro et al. 2003) was featured in a review article of Trends in Ecology & Evolution for discussing the evolutionary origin of anadromous migrations of salmons (Ramsden et al. 2003); our recent discovery of “three deep-fish families into one” (Johnson et al. 2009) was highlighted in a “News & Views” section in Nature (Howlett 2009); and our discovery of deep-ocean origin of the freshwater eels was highlighted in a “Feature” section in Current Biology (Williams 2010). Other than those scientific impacts, some of our recent papers have attracted considerable attention from various media outlets, such as popular science magazines, newspapers, radio, and television [for a list of the media coverage of our studies, see the website of one of the authors (MM) lab: https://sites.google.com/site/masakimiyalab/media-coverage].
This review summarizes the achievements and novel perspectives that our mitogenomic project has brought to molecular phylogenetics and evolution of fishes. Although the review is necessarily a highly personal retrospect on our own studies, we attempted to describe those achievements and perspectives in an objective manner with several examples of criticisms from other studies. Finally, we briefly comment on the potential directions of future mitogenomics of fishes in the nuclear genomic age.
Phylogenetic background
Before reviewing the mitogenomics of fishes, we summarize a phylogenetic background prior to the inception of our project following Miya and Nishida (2000b). We use phylogenetic history of teleosts as an example because it is well documented and the teleosts cover a major portion of fish diversity, comprising over 26,840 extant species (about 96 % of all extant fishes) placed in 40 orders, 448 families, and 4,278 genera (Nelson 2006).
Earlier classifications of the teleosts (e.g., Berg 1940) simply reflected the recognition of a series of evolutionary “grades,” starting from a morphologically primitive group (e.g., Isospondyli), through intermediate steps, toward the most advanced forms (e.g., Percomorpha, see Nelson 1989). Following the publication of the seminal work by Greenwood et al. (1966) and the advent of cladistic theory (Hennig 1966; Wiley 1981), those evolutionary grades have been replaced by the monophyletic groups (“clades”), which are explicitly recognized through searches for the shared derived characters (“synapomorphies”). One early example of an important synapomorphy for a group of fishes was the recognition of the possession of leptocephalus larvae among members of the Elopomorpha (Greenwood et al. 1966). The influence of cladistics on fish systematics has been profound, with numerous comparative anatomical studies having been conducted in attempts to circumscribe monophyletic groups for both the basal (Rosen 1973, 1974, 1985; Fink and Weitzman 1982; Lauder and Liem 1983; Fink 1984; Begle 1991, 1992; Johnson 1992; Johnson and Patterson 1996; Lecointre and Nelson 1996) and the derived (Lauder and Liem 1983; Stiassny 1986; Stiassny and Moore 1992; Johnson 1993; Johnson and Patterson 1993) groups of teleosts based on nested sets of synapomorphies used to resolve their relationships. Despite the efforts of these early phylogeneticists, there remained much controversy over higher-level relationships among the major teleostean lineages. This controversy was especially evident in the transitions of the classification systems adopted in the first through fourth editions of “Fishes of the World” (Nelson 1976, 1984, 1994, 2006).
As independent lines of evidence, molecular characters were expected to be decisive in resolving persistent controversies in the teleostean phylogeny (Nelson 1989), although they had not fulfilled their promise before the late 1990s (Stepien and Kocher 1997). Indeed, an early study using partial amino acid sequences from three mitochondrial protein-coding genes (Normark 1991) suggested an unorthodox tree (e.g., nonmonophyletic teleosts) that Patterson et al. (1993) criticized as “goofy” from the morphologist’s point of view. Later, Bernardi et al. (1993) and Rubin and Dores (1995) analyzed amino acid sequences of growth hormone on the basis of a different data set, including 25 and 24 teleosts, respectively, comparing them with those from two outgroups, and reported that the resulting maximum parsimony tree agreed well with the morphology-based tree. Lê et al. (1993) analyzed nuclear 28S rRNA gene sequences from 31 gnathostomes (including 18 teleosts) and found two highly supported nodes within the teleosts (Osteoglossomorpha + Elopomorpha and Clupeomorpha + Ostariophysi) that were incongruent with the morphology-based tree (Nelson 1989). Using entire mitochondrial cytochrome b (cyt b) gene sequences from 31 fishes (including 30 teleosts), Lydeard and Roe (1997) reported that the resulting maximum parsimony trees were largely congruent with the morphology-based tree, although some incongruities (e.g., paraphyletic Neoteleostei) were observed. However, with the exception of the sister relationship between Clupeomorpha and Ostariophysi (see Lê et al. 1993), no novel molecular phylogenetic hypothesis has had a significant impact on those studies dealing with higher-level relationships among major teleostean lineages in the late 1990s (Johnson and Patterson 1996; Lecointre and Nelson 1996). Furthermore, some workers demonstrated the limits of partial mitochondrial rRNA (Ortí and Meyer 1997) and cyt b (Meyer 1994) genes in resolving higher-level relationships of teleosts.
It appears that adequate resolution of higher-level relationships in any organism requires longer DNA sequence reads, because historical signals are directional (= additive) by definition and expected to overcome nondirectional (= random) noise as more data are collected (Miya and Nishida 2000b). If so, what types of genes or genomes should we first explore for their usefulness in reconstructing higher-level relationships of teleosts? We chose the mitogenome, because it has many advantages as a phylogenetic marker such as maternal inheritance and haploidy, and the resulting short coalescence time (Avise 1994; Moore 1995; Curole and Kocher 1999; Springer et al. 1999). In mammals, for instance, the transition from an apparently unsolvable to a solvable problem came about mainly from the availability of complete mitochondrial DNA (mtDNA) sequences (Penny et al. 1999). There was, however, no simple and easy approach for sequencing entire mitogenomes in the late 1990s and we need to develop the novel method by ourselves.
Development of the novel sequencing method for fish mitogenomes
In an earlier stage of molecular phylogenetic studies of fishes, we published three papers on the deep-sea stomiiform fish genus Cyclothone (see Miya and Nishida 1996, 1997) and Sternoptyx (see Miya and Nishida 1998) based on partial sequences from the mitochondrial ribosomal genes (12S and 16S rRNAs). Concurrently, we attempted to expand those three studies to obtain the entire stomiiform phylogeny by sampling more molecular characters from additional taxa distributed across five families and 53 genera with about 400 species (Nelson 2006). Thus, our explorations of molecular phylogenetics and evolution of fishes had a very limited scope in an initial stage, only involving a single group of deep-sea fishes (Stomiiformes).
For the character sampling, we chose the mitochondrial cyt b gene sequences and attempted to amplify those sequences (approximately 1,150 bp) from seven gonostomatid species of Sigmops and Gonostoma (see Miya and Nishida 2000a) using two primers designed on the two flanking transfer RNA genes (tRNAGlu and tRNAThr). The PCR product from one species (Sigmops gracile), however, was unexpectedly small (approximately, 250 bp), and subsequent direct sequencing revealed that, instead of the cyt b, tRNAPro was found between these two tRNA genes (Fig. 2). Further PCR and sequencing experiments demonstrated that the cluster of these three tRNAs (tRNAGlu, tRNAPro, tRNAThr) was adjacent to the 3′ end of the cyt b gene, suggesting that tRNA rearrangements relative to the latter have occurred in S. gracile (see Miya and Nishida 1999). Such gene rearrangements had not been previously reported for any vertebrates at that time (see Macey 1997) and prompted us to sequence the whole mitogenome for this species (Miya and Nishida 1999).

Schematic representation of the mitogenome from Sigmops gracile (see Miya and Nishida 1999). The entire mitogenome is amplified using two long PCRs (gray lines with arrowheads 16S rRNA–cyt b and cyt b–16S rRNA gene regions) and the two long PCR products are used as templates for subsequent short PCRs using 30 pairs of fish-versatile primers that amplify contiguous, overlapping segments of the entire mitogenome (denoted by broken lines inside the gene map)
For sequencing the whole mitogenome (ca. 16,500 bp), purification of the intact mtDNA is an essential step for avoiding erroneous sequencings of mitochondrial pseudogenes in the nuclear genome (Dowling et al. 1996). We found that the use of a long PCR technique (Cheng et al. 1994) was an easy and effective method for the genomic purification from such small deep-sea fish (<120 mm in standard length) rather than the traditional method, such as physical isolation of mitochondria from the fish tissue (Miya and Nishida 1999). After a period of trial and error, we finally obtained excellent amplifications of the entire mitogenome from Sigmops gracile with two long PCR reactions using two sets of species-specific primers on the 16S rRNA and cyt b genes (Fig. 2) in late 1997. Those two long PCR products (approximately, 12 kb and 6 kb in length) were overlapped, being considered as the purified whole mitogenome and used as templates for subsequent DNA sequencings.
For massive sequencings from across various fish taxa, traditional methods, such as digestion with a restriction enzyme and subsequent cloning, were labor-intensive and time-consuming. Accordingly, we designed 30 sets of fish-versatile PCR primers with reference to the aligned whole mitogenome sequences from seven fish species available at that time (loach, Tzeng et al. 1992; carp, Chang et al. 1994; trout, Zardoya et al. 1995; cod, Johansen and Bakke 1996; bichir, Noack et al. 1996; lungfish, Zardoya and Meyer 1996; coelacanth, Zardoya and Meyer 1997), which cover the entire mitogenome in a series of partially overlapping fragments. Fortunately, those newly designed PCR primers worked very well for the species (Sigmops gracile) and successfully amplified contiguous, overlapping segments of the entire genome using the two long PCR products as templates (Fig. 2). We then assembled the whole mitogenome from direct sequencings for those 30 short PCR products.
This simple and quick approach (a combination of long and short PCRs with fish-versatile primers) has revolutionized the mitogenomics of fishes, enabling a rapid development in this field. With this novel technique, our research group has published a series of five papers describing whole mitogenomes from commercially or zoologically important fish species (Japanese sardine, Inoue et al. 2000b; Japanese eel, Inoue et al. 2000a; Japanese anchovy, Inoue et al. 2001b; ayu, Ishiguro et al. 2001; Mangrove rivulus, Lee et al. 2001) and is now assembling ≈1,340 whole mitogenome sequences as of 30 September 2014 (including unpublished ones). We abandoned this publication strategy at an earlier stage of the project, because our major interests were not in the descriptions of individual fish mitogenomes, but in their utility as phylogenetic marker sets for resolving higher-level relationships of fishes. Interestingly, Sorenson et al. (1999) concurrently developed a similar PCR-based approach to bird mitogenomes, and their research group has published a number of phylogenetic studies of birds (e.g., Mindell et al. 1999). We also applied this approach to invertebrates, such as decapods (Yamauchi et al. 2004a), copepods (Machida et al. 2002), insects (Yamauchi et al. 2004b), and lancelets (Nohara et al. 2005).
Use of mitogenomic data in molecular phylogenetics of fishes
It appears that adequate resolution of higher-level relationships in any organism requires longer DNA sequence reads (Miya and Nishida 2000b). In this regard, our PCR-based approach for fish mitogenomes (Miya and Nishida 1999) has overcome the technical difficulties in obtaining a number of the longer sequences from a wide variety of taxa. However, the usefulness of the whole mitogenome sequences in molecular phylogenetics of fishes was unknown and we had to demonstrate the phylogenetic utility and limits of the mitogenomic data before further sequencing efforts with this PCR-based method.
Miya and Nishida (2000b) addressed this issue using the whole mitogenome sequences of eight teleosts (including three newly determined sequences), whose relative phylogenetic positions are noncontroversial (Fig. 3). Maximum parsimony (MP) analyses of the nucleotide and amino acid sequences of 13 protein-coding genes from eight teleosts, plus two outgroups (a bichir and a shark), indicated that all of the individual protein-coding genes, with the exception of ND5, failed to recover the expected phylogeny, although unambiguously aligned sequences from 22 concatenated transfer RNA (tRNA) genes (stem regions only) recovered the expected phylogeny successfully with moderate statistical support (Miya and Nishida 2000b), consistent with a suggestion of Kumazawa and Nishida (1993). Miya and Nishida (2000b) also observed that the combined data sets comprising nucleotide sequences from the several protein-coding genes with higher phylogenetic performance (no third codon positions) plus the 22 concatenated tRNA genes (stem regions only) best recovered the expected phylogeny with all internal branches being supported by bootstrap values ≥90 %.

Basal teleostean relationships (a) following Nelson (1994). Miya and Nishida (2000b) chose eight teleosts (closed and open circles) from the four major lineages, whose relative phylogenetic positions are noncontroversial. They performed maximum parsimony analysis with various weighting schemes to evaluate the phylogenetic utility of the mitogenomic data by comparing the recovery rate of the expected phylogenies, (b) and among different data sets. Redrawn from Miya and Nishida (2000b)
Based on comparisons of the recovery rates of the expected phylogeny (Fig. 3) and statistical support for the internal branches among individual and concatenated genes, Miya and Nishida (2000b) concluded that judicious choice of mitochondrial genes and appropriate data weighting (including or excluding transitional and transversional changes at the third codon positions), in conjunction with purposeful taxonomic sampling to bisect long branches (Hillis 1998), are prerequisites for resolving higher-level relationships in teleosts under the maximum parsimony optimality criterion. Kawaguchi et al. (2001) reconfirmed this conclusion with an additional whole mitogenome sequence from an aulopiform, Aulopus japonicus, which occupies an important phylogenetic position between the basal and higher euteleosts. However, in our subsequent studies, we have had to concatenate all genes in our phylogenetic analyses, because there is no a priori reason to include or exclude specific genes (except for the ND6 gene owing to its distinct heterogeneous base composition).
It should be noted that comparisons of phylogenetic performance of the mitogenomic data had to be performed under the maximum parsimony (MP) optimality criterion in Miya and Nishida (2000b). Before the advent of the Bayesian (BA) inference implemented in MrBayes (Ronquist and Huelsenbeck 2003) and subsequent developments of a fast maximum likelihood (ML) algorithm implemented in RAxML (Stamatakis 2006), the MP method was the only character-based approach applicable to large data sets in the early 2000s. Our research group employed those model-based phylogenetic inferences (BA and ML) for large data sets (including ≥25 species) from Inoue et al. (2003a, 2004), and Simmons and Miya (2004) have not performed MP analysis since then, because MP is generally more sensitive to long branch attraction issues than ML (e.g., Philippe et al. 2005).
Mitogenomic resolution of fish phylogenies
In the early 2000s, no novel molecular phylogenetic hypotheses have been considered significant in those studies addressing higher-level relationships among major teleostean lineages (Johnson and Patterson 1996; Lecointre and Nelson 1996) with the exception of the sister group relationship demonstrated for Clupeomorpha and Ostariophysi (see Lê et al. 1993). This situation was in part due to the lack of longer DNA sequences that were necessary for adequate resolution of higher-level relationships (Stepien and Kocher 1997; Miya and Nishida 2000b). The novel method for sequencing whole mitogenomes of fishes (Miya and Nishida 1999), together with explicit demonstration of phylogenetic utility of the mitogenomic data (Miya and Nishida 2000b), enabled us to address various controversial issues in fish phylogenetics.
Inoue et al. (2001c) was the first study from our research group to address these phylogenetic issues, reexamining the interrelationships of the five major, basal teleostean lineages (Osteoglossomorpha, Elopomorpha, Clupeomorpha, Ostariophysi, Protacanthopterygii) using the mitogenomic data. There were five alternative phylogenetic hypotheses on the basis of both morphological (Greenwood et al. 1966; Greenwood 1973; Patterson and Rosen 1977; Arratia 1997) and molecular analyses (Lê et al. 1993) (Fig. 4), and Inoue et al. (2001c) performed MP and ML analyses with the mitogenomic data from purposefully chosen eight teleosts and two outgroups. The resultant tree topologies from the two methods were congruent, although they differed from any of the previously proposed hypotheses, with Osteoglossomorpha being sister to other teleosts, followed by the divergences between Elopomorpha and Otophysi + Protacanthopterygii (Fig. 4). Furthermore, the mitogenomic data confidently rejected all of those proposed hypotheses with high statistical significance. Note that recent phylogenetic studies using multiple nuclear genes (Li et al. 2008; Near et al. 2012; Betancur et al. 2013a; Broughton et al. 2013; Chen et al. 2013; Faircloth et al. 2013) have consistently recovered Elopomorpha to be the sister group to other teleosts instead of Osteoglossomorpha; none of those studies, however, performed statistical comparisons (e.g., AU-test, Shimodaira 2002) among alternative phylogenetic hypotheses and thus the issue requires further clarification.

Unlike the depauperate basal teleosts that have only a few alternative phylogenetic hypotheses possible, higher teleosts are enormously species rich (>18,000 species, Nelson 2006) and their phylogenetic problems are so complex that G. Nelson (1989) even described them as the “(unresolved) bush at the top of the tree” with no consensus having been reached. As a first step toward resolution of higher teleostean phylogenetics, Miya et al. (2001) attempted to circumscribe a well-supported monophyletic group encompassing the “bush” (i.e., Percomorpha) and to determine the phylogenetic position of that clade relative to other major lineages using mitogenomic sequences from 48 species of teleosts (including 38 newly determined sequences).
In their MP tree, which was statistically indistinguishable from the ML tree, Miya et al. (2001) confirmed the monophyly of the more comprehensive clades, such as Euteleostei, Neoteleostei, and Eurypterygii with high bootstrap support (93–100 %), while interrelationships among more derived Myctophiformes, Ateleopodiformes, and Lampridiformes were ambiguous within the Ctenosquamata. These lineages together formed a sister clade to other higher teleosts in their MP tree. In the latter clade, the phylogenetically problematic Polymixiiformes, Paracanthopterygii, and Zeioidei formed a sister clade to other higher teleosts. Although taxonomic sampling was still sparse, Miya et al. (2001) successfully found a highly supported monophyletic group at the top of the tree (Fig. 5), which is the sister group to a clade comprising non-reciprocally monophyletic Beryciformes and Stephanoberyciformes. This study provided an important stepping stone toward more taxonomically densely sampled analyses.

Four mitogenomic studies encompassing the entire actinopterygian phylogenies
The year of 2003 represented a significant milestone for our research group. Following Miya et al. (2001), we published four phylogenetic papers that together encompassed whole actinopterygian diversity, beginning from the basal actinopterygians (Inoue et al. 2003a) to ostariophysans (Saitoh et al. 2003), basal euteleosts (Ishiguro et al. 2003), and the higher teleosts (Miya et al. 2003). Those four pioneering studies have invariably had profound impacts on comparative studies of fishes, being cited 1,063 times in total as of 30 September 2014 (Table 1). Interestingly, a French group led by Guillaume Lecointre in Paris concurrently began publishing significant contributions in molecular phylogenetics of fishes mainly based on nuclear genes (e.g., Zaragüeta-Bagils et al. 2002; Chen et al. 2003; Dettaï and Lecointre 2005; Li et al. 2009).
The basal actinopterygians comprise four major fish lineages (polypteriforms, acipenseriforms, lepisosteids, Amia) and have been collectively called “ancient fish.” Inoue et al. (2003a) analyzed their relationships in relation to teleosts using the mitogenomic data and compared the resulting trees to the various alternative hypotheses. Inoue et al. (2003a) resolved polypteriforms as the sister lineage to all other actinopterygians and found that the other three ancient fish groups (acipenseriforms, lepisosteids, Amia) together formed a clade, which was sister to teleosts (Fig. 6). Although the latter hypothesis was congruent with that advocated by Venkatesh et al. (2001) based on nuclear markers (patterns of insertion/deletion in the RAG1 gene), subsequent mitogenomic reanalysis based on the different alignments (Azuma et al. 2008; Setiamarga et al. 2009; Nakatani et al. 2011) supported the neopterygian clade (lepisosteids + Amia being sister to teleosts). These results are in agreement with the analyses of multiple nuclear DNA sequences (Li et al. 2008; Near et al. 2012; Betancur et al. 2013a; Broughton et al. 2013) and morphological characters (Patterson 1973; Grande 2010).

Ostariophysi is the second largest superorder within Teleostei, comprising five mostly freshwater orders: Gonorynchiformes, Cypriniformes, Characiformes, Gymnotiformes, and Siluriformes. Saitoh et al. (2003) analyzed their higher-level relationships using whole mitogenome sequences from 11 ostariophysans and nine non-ostariophysan teleosts used as outgroups. Saitoh et al. (2003) reconfirmed clupeiforms as the closest relatives of ostariophysans and provided convincing evidence supporting monophyly and a sister-group relationship of Cypriniformes and Characiphysi (Characiformes + Siluriformes + Gymnotiformes). Saitoh et al. (2003) further argued a Pangean origin of the Otophysi (Cypriniformes + Characiphysi) based on their geographic distributions on the major continents. The publication of this article has led to our participation in the international project “Cypriniformes Tree of Life (CToL)” initiated by Dr. Richard L Mayden at Saint Louis University (see below).
Higher-level relationships of the basal Euteleostei (= Protacanthopterygii) are so complex and controversial that at least nine alternative morphology-based hypotheses were proposed before 2000. Ishiguro et al. (2003) investigated their relationships using mitogenomic data from 34 purposefully chosen species that fully represented major basal euteleostean lineages (Argentinoidea, Alepocephaloidea, Osmeriformes, Esociformes, Salmoniformes). Surprisingly, the resulting tree confidently placed alepocephaloids within the Otocephala (Fig. 7), a sister group of the euteleosts. This unexpected result has been reconfirmed by mitogenomic studies with denser taxonomic sampling from alepocephaloids (Lavoué et al. 2008b; Poulsen et al. 2009; Broughton 2010) as well as those studies based on multiple nuclear genes (Near et al. 2012; Betancur et al. 2013a; Broughton et al. 2013; Chen et al. 2013). Also Ishiguro et al. (2003) convincingly demonstrated a sister-group relationship between Esociformes and Salmoniformes (Fig. 7) (see also Campbell et al. 2013b), which was featured in a review article in Trends in Ecology & Evolution (Ramsden et al. 2003) in a discussion on the evolutionary origin of the anadromous migrations of salmonids. Later, Ishiguro et al. (2005) confidently demonstrated that Sundasalanx (formerly osmeriform Sundasalangidae, see Siebert 1997) is not a member of Osmeriformes, but is a member of Clupeiformes on the mitogenomic phylogeny.

Basal euteleostean relationships recovered in the maximum parsimony analysis of Ishiguro et al. (2003). Numbers above internal branches indicate jackknife values obtained for 500 replicates. Note that Alepocephaloidea falls outside the Euteleostei, and Protacanthopterygii is recovered as the nonmonophyletic group. Redrawn from Ishiguro et al. (2003)
Following a successful circumscription of a well-supported monophyletic group at the top of the higher teleostean tree and explicit demonstration of the phylogenetic position of such a monophyletic group relative to other major lineages (Miya et al. 2001), Miya et al. (2003) further expanded the taxonomic sampling to 100 purposefully chosen species and performed unweighted and weighted MP analyses using mitogenomic data. Miya et al. (2003) revealed that all major, comprehensive groups above the ordinal level as currently defined in higher teleosts (with the exception of the Neoteleostei and several monotypic groups), such as the Eurypterygii, Ctenosquamata, Acanthomorpha, Paracanthopterygii, Acanthopterygii, and Percomorpha, appeared to be non-monophyletic in the resulting tree. Such incongruities largely resulted from differences in the placement and/or limits of the orders Ateleopodiformes, Lampridiformes, Polymixiiformes, Ophidiiformes, Lophiiformes, Beryciformes, Stephanoberyciformes, and Zeiformes, all of them being long-standing problematic taxa in systematic ichthyology. Of these, the resulting phylogenetic positions of the Ophidiiformes and Lophiiformes were totally unexpected, because, although they have consistently been considered relatively primitive groups within higher teleosts (Paracanthopterygii), they were confidently placed within a crown group of teleosts (Fig. 8). These unexpected results have generally been supported by recent molecular phylogenetic studies based on multiple nuclear genes (Li et al. 2008; Near et al. 2012, 2013; Wainwright et al. 2012; Betancur et al. 2013a; Broughton et al. 2013). Later, Miya et al. (2005) included two species of the Batrachoidiformes not sampled in Miya et al. (2003) and performed partitioned Bayesian analysis. Miya et al. (2005) found that batrachoidiforms are also members of the “top” of the tree (Percomorpha) and this finding was reconfirmed by those studies based on multiple nuclear genes (Li et al. 2008; Near et al. 2012; Betancur et al. 2013a; Broughton et al. 2013).

It should be noted that large data matrices used in Miya et al. (2003) have been useful for addressing some issues in molecular phylogenetics, such as “efficient resolution of the basal clades in a phylogenetic tree” (Simmons and Miya 2004) and “overconfidence of posterior probabilities in Bayesian inferences” (Simmons et al. 2004). In particular, the latter paper has attracted considerable attention from the molecular systematics community and cited as many as 258 times as of 30 September 2014 (Table 1).
Gene rearrangement as a phylogenetic marker
Most vertebrates share an identical mitochondrial gene order, and deviations from such structural property have been employed as a useful phylogenetic marker in various animals (Kumazawa and Nishida 1995; Boore 1999). Although our first discovery of the unique mitochondrial gene order in Sigmops gracile (see Miya and Nishida 1999) was not shared by any closely related gonostomatid species, subsequent findings of gene rearrangements in fishes have represented molecular synapomorphies to diagnose subsets of various fish taxa.
For example, Inoue et al. (2001a) found a novel gene order in a mitogenome from congrid eel (Conger myriaster), in which the ND6, tRNAGlu, and tRNAPro genes were translocated between the control region and tRNAPhe genes that are contiguously located at the 5′ end of the 12S rRNA gene in typical vertebrates. Based on the partial sequences of this region from 11 other anguilliform species representing 11 families, Inoue et al. (2001a) found that this unique gene order was shared by four other families belonging to the suborder Congroidei. Subsequently, Inoue et al. (2010c) published anguilliform phylogenies based on whole mitogenome sequences from all of the 19 families. Inoue et al. (2010c) confirmed that the unique gene order was shared by six families of the suborder Congroidei (Colocongridae, Congridae, Derichthyidae, Nettastomidae, Ophichthidae, Muraenesocidae) and those six families exclusively formed a monophyletic group supported by 99 % bootstrap support.
Inoue et al. (2003b) found identical, unusual large-scale gene rearrangements in the two deep-sea gulper eels (Eurypharynx pelecanoides and Saccopharynx lavenbergi) placed in two different families (Eurypharyngidae and Saccopharyngidae) within the suborder Saccopharyingoidei (Fig. 9) and were recovered as sister groups in the phylogenetic analysis. Mabuchi et al. (2004) found mitochondrial gene rearrangements in a cluster of tRNAIle (I), tRNAGln (Q), and tRNAMet (M) between the two protein coding genes (ND1 and ND2) in scarid fishes; typically vertebrates including closely related species of scarids (labrids and odacids) exhibit a gene order of IQM, while 10 scarid species representing seven of the 10 currently recognized genera exhibit IMQ followed by a pseudogene of tRNAMet. This finding may represent a molecular synapomorphy to diagnose the entire family (Scaridae), but an example from unique gene orders found in two of the four deep-sea macrourid subfamilies was counterintuitive in terms of external morphologies (Satoh et al. 2006). While Squalogadus modificatus (Macrouroidinae) is characterized by a round head with no noticeable snout, Trachyrincus murrayi (Trachyrincinae) has a rugged head with the most pointed snout among macrourids. Despite these morphological differences, the two subfamilies share a unique gene order among vertebrates, and partitioned Bayesian analysis strongly indicated monophyly of the two among the four currently recognized macrourid subfamilies (Fig. 10) (Satoh et al. 2006).

Unusual gene order of the mitogenomes from the two gulper eels (below) compared with that of the typical vertebrates (center) and its derivatives (around the circular genome). Four conservative blocks of genes are colored and note the remarkable difference of the order between that of the two gulper eels and typical vertebrates. Redrawn from Inoue et al. (2003b)

More recently, Poulsen et al. (2013) attempted to resolve major myctophiform phylogenies from both mitogenomic sequences and the unique mitochondrial gene order rearrangements. Poulsen et al. (2013) observed a total of eight unique gene orders within the myctophids and found that mitogenomic sequences and those unique gene orders are highly congruent concerning phylogenetic resolution on several myctophiform classifications based on osteology, larval ontogeny, and photophore patterns (Fig. 11). In particular, the enigmatic Notolychnus valdiviae was placed as the sister taxon to all other myctophids and exhibited an unusual second copy of the tRNA-Met gene—a gene order rearrangement reminiscent of that found in the tribe Diaphini, although their analyses show it to be independently derived.

Myctophiform relationships recovered in the Bayesian analysis of Poulsen et al. (2013). Numbers beside internal branches are the bootstrap values and posterior probabilities only if those values are less than 100 %. Ten distinct myctophid lineages are noted (A–J) with respect to the current tribal classification. Patterns of eight gene rearrangements shared by subsets (total = 5) or all of the myctophiforms (total = 3) are shown in the colored boxes. Redrawn from Poulsen et al. (2013)
Resolution of inter- and intraspecific relationships
Other than the resolution of higher-level relationships in fishes, we also applied mitogenomic data to address inter- and intraspecific relationships. For example, Minegishi et al. (2005) analyzed mitogenome sequences of all 18 species/subspecies of the freshwater eel genus Anguilla to infer their phylogenetic relationships and to evaluate hypotheses about the possible historical dispersal routes of this genus. The resultant tree clearly indicated a sister relationship between the Atlantic and Oceanian species, which now have distantly separated geographic distributions (Fig. 12) due to the collision of the Eurasian and African continents. Minegishi et al. (2005) argued that the previous hypotheses to estimate the historical dispersal route of anguillid eels into the Atlantic Ocean based on the current geographic distribution of species are unsupported by the mitogenomic analysis and should be reconsidered.

Mabuchi et al. (2006) determined a whole mitogenome sequence from the Lake Biwa wild strain of common carp (Cyprinus carpio) as a reference sequence for further comparisons with a number of already-published mtDNA sequences of multiple mitochondrial gene regions from various strains. This approach enabled more geographically comprehensive analyses including additional strains from Southeast Asia (Vietnam and Indonesia) to evaluate the origin of the Lake Biwa strain. Accordingly, Mabuchi et al. (2006) convincingly reconfirmed a previous finding of principal phylogenetic dichotomy between the “Lake Biwa wild” and “Eurasian” strains (Mabuchi et al. 2005) from more comprehensive data sets.
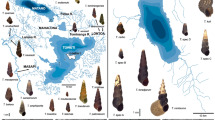
Carassius auratus is a primary freshwater fish with bisexual diploid and unisexual gynogenetic triploid lineages (Kobayasi et al. 1970), distributed widely in Eurasia. As the first step in clarifying the evolutionary entity of Carassius, Takada et al. (2010) determined nucleotide sequences for four mitochondrial genes [control region (CR) + ND4 + ND5 + cyt b; 4,669 bp] from 53 individuals of C. auratus and constructed a supermatrix with additional sequences from the 672 individuals. The supermatrix was subjected to phylogenetic analysis and the resulting trees revealed two lineages, one distributed mainly among the Japanese main islands and the other in various regions in and around the Eurasian continent, including Ryukyus and Taiwan (Fig. 13). The two lineages include seven sub-lineages with high regional specificity that are composed of endemic populations indigenous to each region. Triploids of C. auratus did not form a monophyletic group, but were clustered mostly with sympatric diploids (Fig. 13). Takada et al. (2010) argued that the lack of substantial genetic separation between triploids and diploids indicates that triploids are not composed of a single independent lineage.

Intraspecific relationships among Carassius auratus complex (upper right) recovered in the maximum likelihood analysis of Takada et al. (2010). Global geographical distributions of seven major lineages shown (lower left). Numbers above internal branches indicate neighbor-joining and ML bootstrap values and Bayesian posterior probabilities. Redrawn from Takada et al. (2010)
Hirayama et al. (2010) investigated intraspecific variations in the mitogenomic sequences from eight local populations and four inbred strains of medaka (Oryzias latipes). They found that the number of tandemly repeated 11 nucleotide units in the control region varied greatly among local populations. Also, they noted that the number of repeats was more variable in the northern Japanese group (10–34) than in the southern group (7–12), while two other species of Oryzias, inhabiting tropical regions, had no such repeats. A comprehensive comparison between the number of repeat units and meteorological data indicated that the number of repeats correlated to the index data of a cold environment and seasonal climatic change. Hirayama et al. (2010) argued that the repeated sequences in the CR might function in mitochondrial gene expression and that the number of tandem repeats is likely related to adaptation to a harsh habitat. Note that the northern group of medaka was recently described as a new species in Asai et al. (2011)
Participation in an international project (Cypriniformes Tree of Life)
In March 2004 Prof. Richard L. Mayden of Saint Louis University submitted a proposal entitled “Collaborative research: Systematics of Cypriniformes, Earth’s most diverse clade of freshwater fishes” to the AToL initiative (Assembling the Tree of Life), a large research effort sponsored by the National Science Foundation, USA. MM was nominated as one of the core participants along with many researchers from various countries. In response to this international initiative, MM submitted a separate grant proposal to the Japan Society for the Promotion of Science (JSPS) in November 2004, focusing on the mitogenomic resolution of the cypriniform phylogenies. Fortunately, those two proposals were successfully funded in 2005 and the Cypriniformes Tree of Life (CToL) project was officially initiated as an international project (for details, see http://bio.slu.edu/mayden/cypriniformes/home.html).
Before massive mitogenomic sequencings, Miya et al. (2006) designed a set of PCR primers that effectively amplified the mitochondrial ND4/ND5 gene region. The two genes exhibited good phylogenetic performance in a previous study (Miya and Nishida 2000b) and we had supposedly used them as markers for resolving phylogenetic issues of subsets of the Cypriniformes (e.g., within tribes, subfamilies, or families). These two genes showed better phylogenetic performance than the more commonly used cyt b gene (Miya et al. 2006) and they have been widely used in subsequent studies of the cypriniform phylogenies (see below).
One of the most important steps in resolving the higher-level relationships of a species-rich clade, such as that of the Cypriniformes, is to include a number of taxa that can bisect long branches (Miya and Nishida 2000b). With this in mind, Saitoh et al. (2006) carefully chose 53 cypriniforms and assembled whole mitogenome sequences from them along with six outgroups. The unambiguously aligned sequences were subjected to partitioned Bayesian analyses and the resultant phylogenies strongly supported monophyly of the Cypriniformes as well as that of the families Cyprinidae, Catostomidae, and a clade comprising Balitoridae + Cobitidae, with the two latter loach families being reciprocally paraphyletic (Fig. 14). Saitoh et al. (2006) also demonstrated that the RY-coding (Phillips and Penny 2003; Harrison et al. 2004), which takes only transversions into account, effectively removes the “noise” from the saturated third codon positions and counters the apparent lack of signal by retaining all available positions in the data set. Such use of the RY-coding in the third codon positions has been followed in subsequent mitogenomic analyses of fishes (see below).

Cypriniform relationships recovered in the Bayesian analysis of Saitoh et al. (2006). Note that both the loach families (Cobitidae and Balitoridae) are non-monophyletic. Numbers above internal branches indicate Bayesian posterior probabilities (shown as percentages). Redrawn from Saitoh et al. (2006)
Following the pioneering work of Saitoh et al. (2006), a number of papers have been published to resolve the relationships of the entire Cypriniformes (see Mayden et al. 2008; Mayden et al. 2009) and subsets of the order, such as Catostomidae (see Doosey et al. 2010), Danioninae (see Tang et al. 2010), Cyprinini (see Yang et al. 2010, 2012b), Gobioninae (see Tang et al. 2011), Labeonini (see Yang et al. 2012a), Oxygastrinae (see Tang et al. 2013), and Leuciscinae (see Imoto et al. 2013), and even to specific genera, such as Danio (see Mayden et al. 2007), Carassius (see Takada et al. 2010), Cyprinus (see Mabuchi et al. 2006), and taxonomically problematic genera, such as Discherodontus, Chagunius, and Hypselobarbus (see Yang et al. 2012b), using mitogenomic data (>2,000 bp).
Further resolution of non-euteleostean relationships
Publications of the four mitogenomic papers in 2003 (Inoue et al. 2003a; Ishiguro et al. 2003; Miya et al. 2003; Saitoh et al. 2003) provided useful frameworks for the taxonomic sampling in phylogenetic analysis in actinopterygians (e.g., choice of outgroups and limits of the targeted taxa). These frameworks in turn greatly facilitated further resolution of actinopterygian phylogenies at various taxonomic levels, leading to a number of unexpected discoveries that are in striking contrast to the textbook classification (Nelson 2006).
For basal teleosts below Euteleostei (excluding Cypriniformes), we resolved the lower- and higher-level relationships of Osteoglossomorpha, such as Notopteridae (see Inoue et al. 2009) and Mormyroidea (see Lavoué et al. 2012a); Elopomorpha (see Inoue et al. 2004; Chen et al. 2013), such as Anguilliformes (see Minegishi et al. 2005; Inoue et al. 2010c; Johnson et al. 2012) and Anguillidae (see Minegishi et al. 2005); and Otophysi (see Nakatani et al. 2011), such as Gonorynchiformes (see Lavoué et al. 2005, 2012b), Clupeiformes (see Lavoué et al. 2007, 2008a, 2010, 2013), Alepocephaliformes (see Lavoué et al. 2008b; Poulsen et al. 2009), and Gymnotiformes (see Lavoué et al. 2012a). Of those studies, an explicit demonstration of the deep-ocean origin of the freshwater eels (Inoue et al. 2010c), subsequent discovery of the “living fossil” eel from an undersea cave in Palau (Johnson et al. 2012), and surprisingly long morphological stasis found in the two allopatric lineages in the African butterflyfish (Lavoué et al. 2011) should be highlighted here.
Freshwater eels spend most of their lives in freshwater during their catadromous life cycle and migrate back to their specific breeding places in the ocean (Tsukamoto et al. 2002). However, the evolutionary origin of such an enigmatic behavior remained elusive because of the uncertain phylogenetic position of freshwater eels within the principally marine anguilliforms. Inoue et al. (2010c) performed phylogenetic analysis using mitogenomic sequences from all of the 19 currently recognized anguilliform families (including the four families of Saccopharyngiformes) and found that the freshwater eels occupied an apical position within the anguilliform phylogeny, forming a highly supported monophyletic group with various oceanic midwater eel species (Fig. 15). Moreover, reconstruction of the growth habitats on the resulting tree unequivocally indicated an origination of the freshwater eels from midwaters of the deep ocean (Fig. 15). This finding showed significant concordance with the recent collection of mature adults of the Japanese eel in the upper midwater of the Pacific (Chow et al. 2009) and Inoue et al. (2010c) argued that they have retained their evolutionary origin as a behavioral trait in their spawning areas.

A portion of the anguilliform relationships recovered in the maximum likelihood (ML) analysis of Inoue et al. (2010c). Numbers beside internal branches indicate bootstrap probabilities of ≥50 % based on 1000 replicates. Evolution of the adult growth habitats is reconstructed on the ML tree under an ML optimality criterion. A pie chart at each node indicates the likelihoods for these four character states. Redrawn from Inoue et al. (2010c)
Shortly after the publication of Inoue et al. (2010c), Johnson et al. (2012) reported the discovery of an enigmatic, small eel that exhibits an unusual suite of morphological characters, in a 35 m-deep fringing-reef cave in Palau (Fig. 16). Detailed morphological analysis explicitly placed this species as the most basal lineage (i.e., the sister group of extant anguilliforms). Phylogenetic analysis and divergence time estimation based on whole mitogenome sequences from various actinopterygians, including representatives of all eel families, demonstrated that this fish represents one of the most basal, independent lineages of the true eels, with a long evolutionary history comparable to that of the entire Anguilliformes (approximately 200 million years). Such a long, independent evolutionary history dating back to the early Mesozoic and retention of primitive morphological features warrant recognition of this species as a “living fossil” of the true eels and Johnson et al. (2012) described it as Protanguilla palau, a new genus and species of the new family Protanguillidae.

Phylogenetic position of the “living fossil” eel Protanguilla palau recovered in the time-calibrated phylogenies of Johnson et al. (2012). The tree topology is one of the three best scoring ML trees that is consistent with the morphology-based hypothesis. Note that the best scoring ML tree supports a sister-group relationship between Protanguilla and synaphobranchids (Johnson et al. 2012). Inlet photo courtesy of Jiro Sakaue. Redrawn from Johnson et al. (2012)
Finally, Lavoué et al. (2011) studied the African freshwater butterflyfish (Pantodon buchholzi), whose distinctive morphology earns it recognition as a monotypic family. Phylogenetic analysis using mitogenomic sequences showed that the two allopatric populations from the Congo and Niger basins formed a monophyletic group (Fig. 17), but they differed by 15.2 % in their coding sequences with no morphological divergences as shown by 15 morphometric measurements compared with those of other osteoglossomorphs. The mitogenomic divergence time between these populations was estimated to be greater than 50 million years (Fig. 17), and the deep genetic divergence was confirmed by nuclear sequence data. Thus, morphological stasis in these two allopatric lineages of Pantodon offers a living vertebrate model for investigating phenotypic stability over millions of generations in tropical Africa, which has experienced major climatic oscillations resulting in repeated cycles of forest expansion and fragmentation (Lavoué et al. 2011).

Further resolution of euteleostean relationships below percomorphs
For euteleostean relationships below percomorphs, we resolved the lower- and higher-level relationships of Esociformes + Salmoniformes (see Campbell et al. 2013b), Myctophiformes (see Poulsen et al. 2013), Stylephoriformes (see Miya et al. 2007), and Cetomimidae and associated families (Johnson et al. 2009).
Of those studies, two should be highlighted here because of their remarkable impact on systematic ichthyology. One study includes the rare, monotypic deep-sea fish family Stylephoridae, which has long been considered a member of the order Lampridiformes. Miya et al. (2007) demonstrated that the family was not a lampridiform, but that it was a sister to Gadiformes based on mitogenomic and nuclear data sets (Fig. 18). This result was totally unexpected, because it had been demonstrably placed within the Lampridiformes based on four morphological synapomorphies and it was deeply nested within the order in the cladogram (Olney et al. 1993). Subsequent studies based on multiple molecular markers corroborated this unusual phylogenetic position with confidence (Near et al. 2012; Betancur et al. 2013a; Grande et al. 2013) and a recent morphological study found that myological characters in the caudal fin of both gadiforms and stylephoriforms show uniquely derived states (Borden et al. 2013).

Another example also includes rare deep-sea fishes, which have been long misclassified into three different families. In one of the four mitogenomic studies published in 2003, Miya et al. (2003) found that the mitogenomic sequence from a mirapinnid specimen (Parataeniophorus sp. cf. gulosus) was almost identical with that of a whalefish (Cetostoma regani), differing in only seven among 16,508 bp sequenced. Paxton and Johnson (2005) questioned these results because of the striking morphological differences between these two families and absence of a voucher specimen for the mirapinnid (totally lost during DNA extraction), even stating “Our conclusion is that it is virtually impossible anatomically for Parataeniophorus gulosus to transform into Cetostoma regani” (Paxton and Johnson 2005). Availability of excellent new Gulf of Mexico megalomycterid specimens with closing-net data led Johnson et al. (2009) to re-examine the issue and they finally showed that fishes currently assigned to three families with greatly differing morphologies, Mirapinnidae, Megalomycteridae and Cetomimidae (Fig. 19), are larvae, males and females, respectively, of a single family Cetomimidae, based on morphology and mitogenomic sequences. These striking results were featured in a “News & Views” section in Nature (Howlett 2009), which was entitled “Three into one will go.”

Misclassified three deep-sea fish families proven to be a single family Cetomimidae in Johnson et al. (2009): a Eutaeniophorus festivus postlarva (Mirapinnidae), photo courtesy of Donald Hughes; b Ataxolepis apus adult male, 58 mm in standard length (Megalomycteridae), photo courtesy of G.D. Johnson; c Gyrinomimus sp., juvenile female (Cetomimidae), photo courtesy of Bruce Robison
Further resolution of percomorph relationships
We have successfully circumscribed the limits of percomorphs in a series of papers in the early 2000s (Miya et al. 2001, 2003, 2005) and those studies have hinted at previously unrecognized close relationships between some groups of percomorphs. For example, Gasterosteus aculeatus (Gasterosteiformes) and Helicolenus hilgendorfi (Scorpaeniformes) were recovered as a strongly supported monophyletic group with a bootstrap value of 100 % in Miya et al. (2001) (Fig. 5). The novel clade was also recovered in Miya et al. (2003) with additional groups, such as zoarcoids (Perciformes); however, it did not contain a gasterosteiform (Indostomus) and scorpaeniforms (Dactyloptena spp.).
In addition to this unexpected clade, one of the most striking findings in Miya et al. (2003) was a close relationship between carangids and pleuronectiforms, which had never been suggested but was supported by a jackknife value of 100 % (Fig. 8d). The placement is currently well established and the clade has been subsequently expanded to include those perciform taxa with diverse morphologies, such as Toxotidae, Carangidae, Centropomidae, Latidae, Xiphiidae, Istiophoridae, Polynemidae, Echeneidae, Coryphaenidae, Rachycentridae, Sphyraenidae, and Menidae (see Chen et al. 2003; Smith and Wheeler 2006; Smith and Craig 2007; Little et al. 2010; Near et al. 2012; Wainwright et al. 2012; Betancur et al. 2013a, b; Miya et al. 2013).
A close affinity of lophiiforms, caproids, and tetraodontiforms represents another example of the striking findings in Miya et al. (2003) (Fig. 8c). It was reconfirmed by Holcroft (2004) and Yamanoue et al. (2007) and later supported by those studies based on multiple nuclear genes (Dettaï and Lecointre 2005; Li et al. 2009; Near et al. 2012; Wainwright et al. 2012; Betancur et al. 2013a) as well as morphology (Chanet et al. 2013), leading to a series of mitogenomic studies of tetraodontiforms (Yamanoue et al. 2004, 2006, 2007, 2008, 2009a, 2009b, 2011) and lophiiforms (Miya et al. 2010).
Miya et al. (2003) also found a strongly supported clade (jackknife value = 87 %), comprising perciforms (4 spp.), mugiliforms (2 spp.), and atherinomorphs (6 spp.), within the percomorphs. In particular, four small, benthic perciforms (two blennids and two gobiesocids) were nested in the clade, which was totally unexpected from the previous studies (Fig. 8d). Subsequently, the clade has been reconfirmed with broader taxonomic sampling by Smith and Craig (2007) and Setiamarga et al. (2008) and both of them noticed that all members of the clade (excluding mugilids) spawn demersal eggs with filaments, later named Ovarentaria by Wainwright et al. (2012) with additional taxa in reference to the unique reproductive mode.
The partial resolution of the percomorph “bush” in three studies (Miya et al. 2001, 2003, 2005) facilitated further resolution of the complex relationships of higher teleosts. For example, Mabuchi et al. (2007) explicitly demonstrated that the Labridae and the remaining three labroid families (Cichlidae, Pomacentridae, Embiotocidae) have diverged early within the percomorphs, and the monophyly of the suborder Labroidei was confidently rejected by statistical tests. The resultant phylogenies indicated that the complex pharyngeal jaw apparatus (PJA) shared by those four labroid families evolved independently at least twice, once in Labridae and once in the common ancestor of the remaining three families (Fig. 20), supporting the idea that the evolution of the specialized PJA provided these lineages with the morphological potential for their spectacular trophic radiations from the aspect of historical repeatability (Mabuchi et al. 2007).

Diversity in the skulls of the Cichlidae (a–h) and Labridae (i–p), and diagrammatic representation of the principal components of the specifically modified pharyngeal jaw apparatus (PJA) of cichlids. Redrawn from Mabuchi et al. (2007)
Yamanoue et al. (2007), on the other hand, re-examined the phylogenetic position of the tetraodontiforms and demonstrated that they occupy the apical position in the percomorph tree together with lophiiforms and caproids. Yamanoue et al. (2008) further resolved tetraodontiform phylogenies and found the basal split into the two previously unrecognized clades, which involves ecological diversification into shallow and deep-water habitats (Fig. 21). This novel hypothesis has been at odds with both morphological and nuclear DNA-based analyses (see Yamanoue et al. 2008), but recent phylogenetic analyses based on 22 loci convincingly supported their evolutionary hypothesis on the early ecological diversification (Santini et al. 2013). Yamanoue and his colleagues further investigated subsets of tetraodontiform relationships, such as Molidae (see Yamanoue et al. 2004) and Balistoidea (see Yamanoue et al. 2009b), and revealed intriguing evolutionary events, such as explosive speciation in Takifugu (Fig. 22, Yamanoue et al. 2009a) and multiple invasions into freshwater by tetraodontids (Fig. 23, Yamanoue et al. 2011).

Tetraodontiform relationships recovered in the Bayesian analysis of Yamanoue et al. (2008). Numbers near internal branches indicate Bayesian posterior probabilities. Solid, open, and double circles, and triangles indicate that main habitats of a family are deep waters, coastal waters, open sea, and brackish and freshwater, respectively. Redrawn from Yamanoue et al. (2008)

Timescale for the divergence of species of Takifugu recovered in the ML analysis of Yamanoue et al. (2009a). Note that the divergence of Takifugu has occurred in a time window of 2.4 million years, which is comparable to the explosive speciation of the cichlids in Lake Malawi. Redrawn from Yamanoue et al. (2009a)

Kawahara et al. (2008) investigated another problematic issue concerning interrelationships of 11 gasterosteiform families within the larger context of percomorph relationships. Although members of this group commonly exhibit unique appearances, many of which are derived from various forms of dermal plate armor (Nelson 2006), seven synapomorphies were proposed for their monophyly (Johnson and Patterson 1993). The relationships derived from mitogenomic phylogenetic analyses indicated explicitly that previously recognized members of gasterosteiforms had diverged early within the percomorphs into three different clades with the following subgroups: Syngnathoidei, Gasterosteoidei (minus Indostomidae), and Indostomidae (see Kawahara et al. 2008). Kawahara et al. (2008) further found that Syngnathoidei forms a monophyletic group if it includes Dactylopteroidei, and that Indostomidae is nested within the Synbranchiformes, rendering the latter group paraphyletic. Subsequently, Kawahara et al. (2009) resolved gasterosteid relationships based on whole mitochondrial genomes (14,807 bp) and 11 single-copy nuclear genes (8,703 bp) from all the currently recognized genera. They proposed a novel hypothesis of their relationships (Fig. 24). Their results clearly indicated that morphological and behavioral similarities between Spinachia spinachia and two aulorhynchids (Auloryhnchus japonicus and Auloryhnchus flavidus) result from the independent adaptation to similar marine habitats (Kawahara et al. 2009). Notably, the resulting two trees from the whole mitogenomes and combined 11 nuclear genes exhibited complete topological congruence, yet no single nuclear gene was able to recover that topology (Kawahara et al. 2009).

Stickleback relationships recovered in the maximum likelihood (ML) analyses of Kawahara et al. (2009) using mitogenomes and 11 nuclear genes. Topologies of the resulting two trees are identical and numbers beside internal branches are bootstrap values from the two datasets. Boxes on the right side of the tree indicate the habitat of the gasterosteoid species. Redrawn from Kawahara et al. (2009)
Based on the results from Kawahara et al. (2008) on the relationships of the 11 gasterosteiform families, together with those from previous molecular phylogenetic studies, Song et al. (2014) attempted to circumscribe a novel clade mainly comprising Syngnathoidei using the mitogenomic data. They sampled a wide variety of percomorphs that have been suggested to have close affinities with syngnathoids and performed Bayesian and maximum likelihood analyses. The resulting trees revealed a highly supported monophyletic group comprising seven families in Syngnathoidei (Gasterosteiformes), Dactylopteridae (Scorpaeniformes), Mullidae in Percoidei and two families in Callionymoidei (Perciformes) (Fig. 25). No previous molecular phylogenetic studies have sampled all of those families (see table 2 in Song et al. 2014), and this previously unrecognized clade found across four different percomorph suborders with diverse morphologies (Fig. 25) represents another example of unique patterns of diversification in percomorphs with no trace of morphological synapomorphies.

Patterns of facial nerves, such as those of the ramus lateralis accessorius (RLA), have been considered as one of the candidate characters to delimit a monophyletic group within the percomorphs (Freihofer 1963). Six families of the suborder Percoidei (Arripidae, Dichistiidae, Kyphosidae, Terapontidae, Kuhliidae, Oplegnathidae) and suborder Stromateoidei (including six families) demonstrably share the unique pattern 10 of RLA (Fig. 26). Yagishita et al. (2009) investigated its evolutionary origin using mitogenomic data and found that there were at least two independent origins of the unique facial nerve pattern: one in the common ancestor of Kyphosidae, Terapontidae, Kuhliidae, and Oplegnathidae and another in the common ancestor of the percoids, Arripidae and Stromateoidei (Fig. 26), showing that the morphological character complex can evolve independently in different lineages within the percomorphs.

The order Lophiiformes, commonly known as the anglerfishes, contains a diverse array of marine fishes, comprising 321 living species placed in 68 genera, 18 families and five suborders, and ranging from benthic shallow-water dwellers to highly modified deep-sea midwater species (Pietsch 2009). Miya et al. (2010) assembled whole mitogenome sequences from 39 lophiiforms, representing 17 of the 18 currently recognized families, and analyzed their evolutionary history based on the time-calibrated phylogenies, paying special attention to that of the deep-sea ceratioids. The resultant trees revealed previously unappreciated phylogenetic relationships among the lophiiform suborders and ceratioid families and Miya et al. (2010) concluded that the acquisition of novel features, such as male dwarfism (Fig. 27), bioluminescent lures, and unique reproductive modes allowed the deep-sea ceratioids to diversify rapidly in a largely unexploited, food-poor bathypelagic zone (200–2,000 m depth) relative to the other lophiiforms occurring in shallow coastal areas.

Evolution of the male sexual parasitism in the ceratioid anglerfishes recovered in the maximum likelihood reconstruction of Miya et al. (2010). Four discrete character states were assigned to each terminal and the ancestral character states were reconstructed on the ML tree. Redraws from Miya et al. (2010)
One of the most striking findings in our mitogenomic studies of percomorphs was the discovery of an enigmatic clade comprising 15 (or possibly 16) pelagic fish families (Miya et al. 2013). Previous molecular phylogenetic studies have sporadically recovered a portion of this clade based on the three to seven families mostly with strong statistical support (91–100 %, see table 3 in Miya et al. 2013). Miya et al. (2013) found that the 15 families (Fig. 28) share a common ancestry based on bioinformatic analyses using partial mitochondrial and nuclear gene sequences from all percomorphs deposited in GenBank (10,733 sequences) and subsequent mitogenomic analysis based on 57 species from those targeted 15 families and 67 outgroup taxa. Morphological heterogeneity among these 15 families is so extraordinary that they have been placed in six different perciform suborders; however, members of the 15 families are either coastal or oceanic pelagic in their ecology with diverse modes of life, suggesting that they represent a previously undetected adaptive radiation in the pelagic realm. The time-calibrated phylogenies implied that those 15 families originated from a deep-ocean ancestor and began to radiate after the end-Cretaceous when large predatory epipelagic fishes were selective victims of the Cretaceous–Paleogene mass extinction (Friedman 2009). Miya et al. (2013) named this clade of open-ocean fishes “Pelagia” in reference to the common habitat preference that links the 15 families.

While writing this review, two mitogenomic papers on the percomorph relationships have been accepted for publication. The first one is that of Lavoué et al. (2014), who addressed phylogenetic issues of an enigmatic family Percichthyidae within the context of the Centrarchiformes. The latter taxon is a newly (but informally) recognized order based on the multiple nuclear genes (Near et al. 2012) and Lavoué et al. (2014) found that the mitogenomic data support the monophyly of the Percichthyidae only when the catadromous genus Percalates is excluded from the family. The second one is that of Campbell et al. (2014), who addressed an issue on phylogenetic affinities of the flatfish (Pleuronectiformes) based on the mitogenomic data. Campbell et al. (2014) found that the monophyly of the more comprehensive clade called Carangimorpha is strongly supported, but that of the flatfish is not, which is congruent with the recent molecular studies based on multiple nuclear genes (Betancur et al. 2013b; Campbell et al. 2013a).
Finally, it should be noted that our mitogenomic studies have consistently recovered eight major clades within the Percomorpha with the exception of Ophidiiformes and Batrachoidiformes despite differences in taxonomic coverage (Miya et al. 2003, 2005; Mabuchi et al. 2007; Yamanoue et al. 2007; Kawahara et al. 2008; Yagishita et al. 2009; Song et al. 2014). Kawahara et al. (2008) labeled those eight clades A to H and these clades are labeled (or named) in the latest two studies based on multiple nuclear genes as shown in Table 2. Interrelationships among those clades, however, are not congruent among those studies and should be clarified in future studies.
Divergence time estimation
The advent of Bayesian inference in phylogenetic analyses has provided a powerful framework for integrating different sources of information (molecules and fossils) to estimate divergence times on molecular phylogenies (Inoue et al. 2010a). Inoue et al. (2005) was the first study in our research group to employ this approach, estimating the divergence time between Indonesian and Comoran coelacanths based on the whole mitogenome sequences. Using the sarcopterygian–actinopterygian split as a calibration point [450 million years ago (Mya)], their estimate fell in the range of 30–40 Mya between the two species (Fig. 29). The estimated age was in agreement with the hypothesis that their diversification is linked to the collision of India with Eurasia (50 Mya) and the subsequent siltation caused by the formation of major rivers that resulted in disjunction of the ancestral coelacanth habitat (Springer 1999). Inoue et al. (2005) argued that this geological event allowed the ancestral coelacanth populations on either side of Indian subcontinents to diverge, resulting in the speciation into two species.

Subsequent studies using the whole mitogenomes (Peng et al. 2006; Yamanoue et al. 2006) provided similar node ages for the major actinopterygian divergences based on the same calibration points as those used in Inoue et al. (2005). Hurley et al. (2007), on the other hand, re-investigated the timing of the evolutionary origins of basal actinopterygians using morphological, and nuclear and mitochondrial DNA data with additional fossil evidence. Their results indicated that the node age of the crown-group Neopterygii, including the teleosts, Lepisosteus and Amia, was at least 40 million years (Myr) older than the existing fossil-based estimate and that the nuclear estimates were always younger than the mitochondrial ones. Also their estimates were generally much younger than the previous molecular estimates based on the whole mitogenomes (Inoue et al. 2005; Peng et al. 2006; Yamanoue et al. 2006), because the three most basal nodes were constrained with both minimum and maximum ages with narrow, younger confidence intervals based on the revised fossil records. Accordingly, we employed those revised fossil records from Hurley et al. (2007) as the more reliable time constraints in subsequent studies and provided much younger node ages across the trees (Azuma et al. 2008; Inoue et al. 2009, 2010b; Setiamarga et al. 2009; Miya et al. 2010; Nakatani et al. 2011; Saitoh et al. 2011; Johnson et al. 2012).
One of the most critical problems in divergence time estimation in fishes is the lack of adequate fossil records that can be used as the time constraints at multiple nodes, especially those of percomorphs. Azuma et al. (2008) explored an alternative source of time constraints in the higher teleostean phylogenies by evaluating a biogeographic hypothesis concerning freshwater fishes from the family Cichlidae (Perciformes). Azuma et al. (2008) conducted phylogenetic analyses and divergence time estimations using mitogenomic sequences from 10 cichlids and 44 outgroups. The resulting timetree indicated that the phylogenetic relationships and timing of diversifications in continental cichlids were much more congruent with Gondwanaland origin and Cretaceous vicariant divergences than with Cenozoic transmarine dispersal between major continents (Azuma et al. 2008) (Fig. 30). On the basis of such a remarkable congruence between the cichlid phylogenies and geological events, Azuma et al. (2008) proposed adding the biogeographic assumption of cichlid divergences by continental fragmentation as effective time constraints in dating teleostean divergence times. Their proposal, however, has been criticized by Friedman et al. (2013) who provided independent palaeontological and relaxed-molecular-clock estimates for the time of cichlid origin that collectively reject the antiquity of the group required by the Gondwanan vicariance scenario. It appears that congruence between paleontological and molecular time scales for cichlid evolution of Friedman et al. (2013) is better than that of Azuma et al. (2008), but the results from the former study demand what can only be considered a series of highly unlikely trans-oceanic dispersal events.

Timetree of the 10 continental cichlids recovered in the Bayesian relaxed-molecular clock analysis of Azuma et al. (2008). Horizontal bars indicate 95 % credible intervals of the divergence time estimates. Cichlid distributions are indicated by blue (India), green (Madagascar), orange (South America) and red (Africa) circles, which correspond to those of paleogeographic maps. Redrawn from the original data in Azuma et al. (2008)
In general, our mitogenomic estimations of the evolutionary history of actinopterygians (see below) are more consistent with older vicariance scenarios than with the conservative and literal ages of the fossil record (Inoue et al. 2005, 2009; Setiamarga et al. 2009; Nakatani et al. 2011; Saitoh et al. 2011). The resulting “ghost range” is extremely large, sometimes reaching >100 million years (Myr) without fossil evidence. Nakatani et al. (2011) argued that this formidable ghost range partially reflects a genuine difference between the estimated ages of stem group origin (molecular divergence time) and crown group morphological diversification (fossil divergence time) (Brown et al. 2008) and the ghost range may be filled with future discoveries of older fossils that can be used as more reasonable time constraints as well as with the development of more realistic models that accurately capture the divergence rates of molecular sequences.
Historical biogeography
The disjunct distributions of the osteoglossomorph family Notopteridae across Africa, India, and Southeast Asia constitute a long-standing issue of freshwater fish biogeography (Fig. 31). Inoue et al. (2009) investigated historical biogeography of the notopterids based on whole mitogenome sequences and the resultant time-calibrated phylogenies suggested that the Asian notopterids diverged from the African notopterids in Gondwanaland and migrated into Eurasia on the Indian subcontinent from the Cretaceous to the Tertiary. However, Inoue et al. (2009) could not exclude an alternative explanation that the African and Asian notopterids diverged in Pangaea before complete separation into Laurasia and Gondwanaland, to which these two lineages were later confined, respectively.

Geographic distributions of the African and Asian members of the family Notopteridae. Redrawn from Inoue et al. (2009)
The southern and northern Japanese populations of the medaka (Oyzias spp., currently recognized as two distinct species; see Asai et al. 2011) provide useful tools to gain insights into the comparative genomics and speciation of vertebrates. These fishes can breed to produce healthy and fertile offspring despite their highly divergent genetic backgrounds compared to those of human–chimpanzee (Ishikawa 2000). On the basis of Bayesian relaxed molecular-clock analyses of whole mitogenome sequences from 72 ray-finned fishes (including 14 medakas), Setiamarga et al. (2009) demonstrated that the divergence time between the two populations (18 Myr; Fig. 32) is about four times older than that of the previous study (Takehana et al. 2003). This remarkably older estimate, however, can be reconciled with the vicariant events of the Japanese archipelago, and the resulting rates of molecular evolution are found to be almost identical between the medaka and hominid lineages (Setiamarga et al. 2009).

Divergence times of the medaka fishes recovered in the Bayesian relaxed-molecular clock analysis of Setiamarga et al. (2009). Horizontal bars indicate 95 % credible intervals of the divergence time estimates. a–c Plate tectonic process of the Japanese archipelago and (d) the present distribution range of the Southern and Northern populations. Redrawn from Setiamarga et al. (2009)
Clupeoidei includes 397 extant species classified into five families and their distribution follows two general patterns of species richness, the longitudinal and latitudinal gradients. To test historical hypotheses explaining the formation of these two gradients, Lavoué et al. (2013) examined the early biogeography of the Clupeoidei by reconstructing the evolution of their habitat preferences (Fig. 33) along with their ancestral range distributions on a time-calibrated mitogenomic phylogeny. The resulting timetree and the ancestral range reconstruction suggested that the probable region of origin and diversification of the Clupeoidei was the tropical marine precursor to present-day Indo-West Pacific region during the Cretaceous, favoring the hypotheses of “region of origin” (Briggs 2007) and “tropical conservatism” (Mittelbach et al. 2007) to explain the origins of the longitudinal and latitudinal gradients of clupeoid species richness, respectively.

Geographic distribution patterns of the Clupeoidei. a Distribution and species diversity of Clupeoidei. Number of species per grid cell (4 by 4 degree latitude–longitude resolution) is represented by cool (low diversity) to warm (high diversity) colors. b The 12 biogeographical units used in the ancestral ranges reconstruction analysis; each unit was delimited by land masses, vast expanses of open ocean and water temperature. Redrawn from Lavoué et al. (2013)
Freshwater fishes can expand their distributions only through connection of freshwater habitats owing to their inability to acclimate to the sea (Saitoh et al. 2011). Saitoh et al. (2011) analyzed phylogenetic relationships and divergence times of the Cypriniformes, one of the largest teleost freshwater fish clade. Based on the resulting timetree, they performed historical biogeography analysis and found the lower Mesozoic of South Asia as the time and place of basal divergence of cypriniforms. Concurrently, Nakatani et al. (2011) investigated the evolutionary history of the more comprehensive clade called Otophysi, which includes the Cypriniformes and Characiphysi (Gymnotiformes, Characiformes, Siluriformes), together accounting for 43 % of all freshwater fish species. Nakatani et al. (2011) performed phylogenetic analyses and divergence time estimations and the resulting timetree suggested that survival of the ancestral lineages through the two consecutive mass extinctions on Pangaea, and subsequent radiations during the Jurassic through early Cretaceous, shaped the modern familial diversity of otophysans (Fig. 34).

Timetree of the otophysans recovered in the Bayesian relaxed-molecular clock analysis of Nakatani et al. (2011). Horizontal bars indicate 95 % credible intervals of the divergence time estimates. ML reconstruction of ancestral habitats is indicated on selected nodes with pie charts showing the likelihoods for two character states (blue, freshwater; light green, saltwater). All marine species are indicated by asterisks. Dagger symbols indicate the three big mass extinction events after 300 million years ago. Paleocoastline maps are shown below the timetree with moist zones schematically illustrated in green. Redrawn from Nakatani et al. (2011)
Evolutionary origin
One of the most remarkable examples of convergent evolution among vertebrates is illustrated by the independent origins of an active electric sense in weakly electric fishes from South America and Africa, the Gymnotiformes and Mormyroidea, respectively (Lavoué et al. 2012a) (Fig. 35). Lavoué et al. (2012a) examined the timing of the origins of the Gymnotiformes and the Mormyroidea using whole mitogenome sequences and estimated similar ages for the independent origins of the Mormyroidea and Gymnotiformes, which slightly postdate, or just predate, the final separation of Africa and South America by continental drift. Lavoué et al. (2012a) argued that such a convergent evolution with similar age enhances the comparative value of the weakly electric fish system for investigating pathways to evolutionary novelty, as well as the influences of key innovations in communication on the process of species radiation.

Morphological convergences between African and South American electric fishes. Mormyroid African electric fishes (left column) are facing gymnotiform South American electric fishes (right column) with similar aspects of morphology. Electric organ discharge waveform shown for every species (each trace 5 ms in total duration with head-positivity plotted upward). a Mormyrops zanclirostris ; b Sternarchorhynchus oxyrhynchus; c Mormyrus proboscirostris; d Rhamphichthys sp.; e Mormyrops anguilloides; f Gymnotus sp.; g Petrocephalus sullivani; h Eigenmannia sp. Reproduced from Lavoué et al. (2012a)
The chondrichthyans, comprising holocephalans (chimaeras) and elasmobranchs (shark, skates, rays), are the oldest living group of jawed vertebrates that diverged from the common ancestor of bony vertebrates in the early Silurian about 420 Mya (Benton 2009). The modern holocephalans—a previously successful and diverse group currently represented by only 39 extant species—and their relationship with elasmobranchs and other jawed vertebrates has been poorly documented largely owing to a lack of well-preserved fossil materials after the end-Permian about 250 Mya (Didier 2004). Inoue et al. (2001b) investigated their evolutionary history based on whole mitogenome sequences from eight representatives from all three families, finding a single origin of the modern holocephalans and a sister-group relationship with elasmobranchs. The mitogenomic tree recovered the most basal callorhinchids within the chimaeriforms, which is the sister group to a clade comprising the remaining two families (rhinochimaerids and chimaerids). The timetree suggests that the holocephalans originated in the Silurian about 420 Mya, having survived the end-Permian (250 Mya) mass extinction and undergoing familial diversifications during the late Jurassic to early Cretaceous (170–120 Mya) (Fig. 36). This postulated evolutionary scenario agrees well with that based on the paleontological observations (Grogan and Lund 2004).

Aschliman et al. (2012), on the other hand, investigated evolutionary history of the batoids (skates and rays)—one of the three major lineages of chondrichthyans—using mitogenomes, nuclear genes, and fossils, sampling densely across taxa. Aschliman et al. (2012) found widespread body plan convergence in batoids; for example, a depressed, rounded pectoral disk supported to the snout tip by fin radials, common to skates and stingrays, is indicated to have been derived independently by each group, while the long, spiny rostrum of sawfishes similarly appears to be convergent with that of sawsharks, which are not batoids. The major extant batoid lineages are inferred to have arisen relatively rapidly from the Late Triassic into the Jurassic, with long stems followed by subsequent radiations in each group around the Cretaceous/Paleogene boundary.
Comparative mitogenomics
Accumulation by our research group of the whole mitogenome sequences from a diverse array of fishes allowed us to compare the structural properties of the mitogenomes and we found that mitochondrial gene arrangement has been highly conserved among the major lineages of fishes (including tetrapods) for more than 500 Myr with a few exceptions (see Inoue et al. 2003b). It remained unclear whether high conservation of gene order is a consequence of some constraints, or whether it results only from shared common ancestry. To address this question, Satoh et al. (2010) analyzed codon usage and tRNA gene arrangements of the vertebrate mitogenomes to examine possible constraints on the gene order of vertebrate mitogenomes. They found that the tRNA genes specifying the hydrophobic residues were positioned close to the control region (CR), where the transcription efficiency is estimated to be relatively high. Using 47 vertebrate mitogenome sequences representing jawless fishes to mammals, Satoh et al. (2010) further found a correlation between codon usage and tRNA gene positions. They suggested the existence of translational constraint acting on the vertebrate gene arrangement of the mitogenome and concluded that such translational constraints, together with the deamination-related constraint, may have contributed to long-term maintenance of gene order.
Developments of the database and annotation pipeline
In 2004 we launched a publicly accessible, specialized nucleotide sequence database for fish mitogenomes named MitoFish (http://mitofish.aori.u-tokyo.ac.jp). Since then, MitoFish has received an average of more than 30,000 unique hits annually, and currently deposits 1,489 whole, plus 315,214 partial, mitogenome sequences from fishes as of 2 August 2014 (Fig. 37). Although older versions of MitoFish had a few simple functions, such as a homology search, it was recently updated by Iwasaki et al. (2013) to provide the web site with an automatic annotation pipeline called MitoAnnotator. The latter function is particularly important, because a severe bottleneck occurs during mitogenomic annotation, which is time consuming and involves difficulties in annotating sequences with degenerating tRNA structures, divergent start/stop codons of the coding elements, and the overlapping of adjacent elements (Iwasaki et al. 2013). Actually, MitoAnnotator automatically annotates a fish mitogenome with a high degree of accuracy in approximately five minutes (Iwasaki et al. 2013). It should be noted that as many as 38 papers have already used this novel annotation system as a useful tool since the publication of Iwasaki et al. (2013), less than a year ago.

MitoFish home page. A vertical menu bar on the right-hand side allows users to access the main functions of MitoFish including MitoAnnotator. The fish species/taxonomy search and sequence similarity searches can also be performed directly from the home page
Education
We have jointly supervised portions of M.S. or Ph.D. studies of nine graduate students in Japan who earned those degrees with the mitogenomics of fishes at MN’s home institutes (Fukui Prefectural University and Ocean Research Institute, The University of Tokyo), where we have regularly held monthly discussion meetings with them up to MN’s move to University of Ryukyus (April 2013) after his retirement from UT (March 2012). Among many graduate students whom we advised, J.G. Inoue, N.B. Ishiguro, A. Kawaguchi, T.P. Satoh, Y. Minegishi, Y. Yamanoue, R. Kawahara, D.H.E. Setiamarga, and M. Nakatani have greatly contributed to the development of the mitogenomics of fishes (see the reference section below).
In addition, we have hosted a number of overseas graduate students who wanted to apply the mitogenomic data to their own studies and have learned the DNA experimental methods and data analysis in our laboratories. Those oversea graduate students include N.C. Aschliman, M.A. Campbell, S.C. DeVaney, N.J. Lang, M.H. Doosey, J.J.D. Egge, and K.M. Laumann, all from the U.S., and J.Y. Poulsen from Denmark. The U.S. graduate students came to our labs through an EAPSI (East Asia and Pacific Summer Institute for U.S. Graduate Students) program jointly sponsored by JSPS (Japan Society for the Promotion of Science) and NSF (National Science Foundation), which not only provides them with great educational opportunities, but also will help them initiate professional relationships to enable collaborations with us. Some of them have published their own papers based on the mitogenomic data taken during their stay (Doosey et al. 2010; Aschliman et al. 2012; Campbell et al. 2013b) and those papers helped decide the future directions of their independent studies.
Concluding remarks
We have summarized the major achievements and novel perspectives that our project has brought to molecular phylogenetics and evolution of fishes from 1999 to 2014. During this period, two major international projects, which are targeted to address large-scale resolution of organismal phylogenies and taxonomy, have been launched: 1) Assembling the Tree of Life (AToL; currently replaced by Genealogy of Life “GoLife”), a phylogeny-oriented initiative (Cracraft 2004) sponsored by U.S. National Science Foundation (NSF); and 2) Barcode of Life (BOL), a molecular taxonomy-oriented project (Hebert et al. 2003) mainly sponsored by Genome Canada. As we stated earlier in this review, we have played significant roles in one of the AToL projects “Cypriniformes Tree of Life” (CToL) using the mitogenomic data and actually published a number of papers on cypriniform relationships. For the latter project, we were appointed as members of a regional working group (Northeast Asia) in one of the initiatives (FISH-BOL), although we were unable to apply the mitogenomic data to taxonomy and species identification. This is largely because we lacked various resources deemed necessary for development of this burgeoning field in biodiversity research while performing the phylogenetic studies at the same time.
Concerning phylogenetic issues addressed by the former project (AToL), our pioneering work based on mitogenomic data has provided a number of novel phylogenetic hypotheses across multiple taxonomic levels, most of which have been verified by subsequent studies based on multiple nuclear genes with intensive taxonomic sampling (e.g., see Table 2). We acknowledge, however, that more definite resolution of the higher-level relationships of fishes should be expected to use nuclear genomes, where large amounts of phylogenetic information spanning hundreds or thousands of mega base pairs (Mbp) remain unexplored. This plethora of data is in contrast to the 16.5 kilo base pairs (Kbp) maximally available in mitogenomes. Also, Lavoué et al. (2014) recently observed that the phylogenetic signal contained in the mitogenomes is lower than that of the concatenated multiple nuclear genes in resolving the percomorph relationships, where most of the unresolved phylogenetic questions still persist across the “bush at the top” (Nelson 1989) or the “new bush at the top” (Betancur et al. 2013a).
Nevertheless, caution should be exercised when using nuclear genes in phylogenetic analysis, in particular, for those lineages undergoing polyploidization or whole genome duplication events, where paralogous comparisons of the amplified genes from different species are likely to mislead the phylogenetic analysis (Martin and Burg 2002). For example, mitogenomic analysis supported a sister-group relationship between Coregoninae and Thymallinae among three salmoniform subfamilies (Li et al. 2010; Campbell et al. 2013b), while those studies based on multiple nuclear genes (Near et al. 2012) and combined partial mitochondrial and nuclear genes (Crête-Lafrenière et al. 2012) supported a sister-group relationship between Coregoninae and Salmoninae. With strict assessment of orthology, however, Macqueen and Johnston (2014) convincingly demonstrated strong support for the former sister-group relationship (Coregoninae and Thymallinae) based on the 36 nuclear genes, which is congruent with the results from mitogenomic analyses (Li et al. 2010; Campbell et al. 2013b) and reanalysis of the 13 mitochondrial protein-coding genes by themselves (Macqueen and Johnston 2014). Although it remains unclear that the different subfamilial relationships in salmoniforms resulted from paralogous comparisons of nuclear genes, such polyploidization is commonly found in fishes of the Cypriniformes and broadly observed across fish lineages from the Lepidosireniformes (lungfish) to the Perciformes (perches) (Comber and Smith 2004), which are likely to compound the phylogenetic analysis. Considering the supposed whole genome duplication event in a common ancestor of the extant teleosts (Sato and Nishida 2010), putatively orthologous genes from different species should be inspected more carefully before and after phylogenetic analysis.
The utility of mitogenome sequences in fish phylogenetics is not over in this nuclear genomics age (Li et al. 2008; Near et al. 2012, 2013 Wainwright et al. 2012; Betancur et al. 2013a; Broughton et al. 2013). The mitogenomic data will continue to contribute to the resolution of the higher-level relationships of fishes, such as those shown in the novel findings from recent studies (Miya et al. 2013; Song et al. 2014), and they will still be useful molecular markers as independent lines of evidence. We envisage that more promising use of the mitogenomic data would be at the interface between species and populations. Whole mitogenome sequences (ca. 16,500 bp) should be able to provide more informative data than the partial sequences widely used in the fish barcode (655 bp from the COI gene, Ward et al. 2005). For example, COI sequences are unable to unequivocally distinguish the eight currently recognized species of tunas (Thunnus spp.) (Ward et al. 2005), but whole mitogenome sequences are demonstrably able to do so (Miya et al. 2013). In insects, for example, Timmermans et al. (2010) even claim that the mitogenomes would be alternative molecular markers to delimit species boundaries by using next-generation sequencing (NGS) technologies, which produced 21 nearly complete and 7 partial sets of protein-coding mitochondrial genes from beetles in a single run. Mitogenome sequencing also revealed shallow evolutionary history of fishes, as seen in complete sampling from all the 18 species/subspecies of the freshwater eels (Minegishi et al. 2005), which eventually led to the discovery of a new species in conjunction with morphological analysis (Watanabe et al. 2009). More recently, NGS technologies made it possible to quickly and economically generate whole mitogenome sequences (Morin et al. 2010), and Jacobsen et al. (2012) successfully demonstrated recent divergences between morphologically and ecologically distinct European whitefish using 106 mitogenome sequences from the Coregonus species complex. Intensive taxonomic sampling from the interface between species and populations together with the massive character sampling from mitogenome sequences using NGS technologies would enable simultaneous attempts to delimit species boundaries (plus the resulting discoveries of cryptic species) and to reconstruct their evolutionary relationships at much finer resolution, eventually unraveling the fish part of the Tree of Life in a bottom-up manner with more accurate estimations of species diversity.
References
Arratia G (1997) Basal teleosts and teleostean phylogeny. Palaeo-Ichthyology 7:5–168
Asai T, Senou H, Hosoya K (2011) Oryzias sakaizumii, a new ricefish from northern Japan (Teleostei: Adrianichthyidae). Ichthyol Explor Freshwat 22:289–299
Aschliman NC, Nishida M, Miya M, Inoue JG, Rosana KM, Naylor GJ (2012) Body plan convergence in the evolution of skates and rays (Chondrichthyes: Batoidea). Mol Phylogenet Evol 63:28–42
Avise JC (1994) Molecular markers, natural history and evolution. Chapman & Hall, New York
Azuma Y, Kumazawa Y, Miya M, Mabuchi K, Nishida M (2008) Mitogenomic evaluation of the historical biogeography of cichlids toward reliable dating of teleostean divergences. BMC Evol Biol 8:215. doi:10.1186/1471-2148-8-215
Begle DP (1991) Relationships of the osmeroid fishes and the use of reductive characters in phylogenetic analysis. Syst Zool 40:33–53
Begle DP (1992) Monophyly and relationships of the argentinoid fishes. Copeia 1992:350–366
Benton MJ (2009) Vertebrate palaeontology, third ed. Blackwell, Malden, MA
Benton MJ, Donoghue PCJ, Asher RJ (2009) Calibrating and constraining molecular clocks. In: Hedges SB, Kumar S (eds) Timetree of life. Oxford University Press, Oxford, UK, pp 35–86
Berg LS (1940) Classification of fishes both recent and fossil. Trav Inst Zool Acad Sci USSR 5:87–517
Bernardi G, D’Onofrio G, Bernardi G (1993) Molecular phylogeny of bony fishes, based on the amino acid sequence of the growth hormone. J Mol Evol 37:644–649
Betancur-R. R, Broughton RE, Wiley EO, Carpenter K, López JA, Li C, Holcroft NI, Arcila D, Sanciangco M, Cureton II JC, Zhang F, Buser T, Campbell MA, Ballesteros JA, Roa-Varon A, Willis S, Borden WC, Rowley T, Reneau PC, Hough DJ, Lu G, Grande T, Arratia G, Ortí G (2013a) The tree of life and a new classification of bony fishes. PLoS Currents Tree of Life. doi:10.1371/currents.tol.53ba26640df0ccaee75bb165c8c26288
Betancur-R. R, Li C, Munroe TA, Ballesteros JA, Ortí G (2013b) Addressing gene tree discordance and non-stationarity to resolve a multi-locus phylogeny of the flatfishes (Teleostei: Pleuronectiformes). Syst Biol 62:763–785
Boore JL (1999) Animal mitochondrial genomes. Nucleic Acids Res 27:1767–1780
Borden WC, Grande T, Smith WL (2013) Comparative osteology and myology of the caudal fin in the Paracanthopterygii (Teleostei: Acanthomorpha). In: Arratia G, Schultze H-P, Wilson MVH (eds) Mesozoic fishes 5. Verlag Dr. Friedrich Pfeil, München, pp 419–455
Briggs JC (2007) Marine longitudinal biodiversity: causes and conservation. Divers Distrib 13:544–555
Broughton RE (2010) Phylogeny of teleosts based on mitochondrial genome sequences. In: Nelson JS, Shultze H-S, Wilson MVH (eds) Origin and phylogenetic interrelationships of teleosts. Verlag Dr. Friedrich Pfeil, München, pp 61–76
Broughton RE, Betancur-R R, Li C, Arratia G, Ortí G (2013) Multi-locus phylogenetic analysis reveals the pattern and tempo of bony fish evolution. PLoS Currents Tree of Life. doi:10.1371/currents.tol.2ca8041495ffafd0c92756e75247483e
Brown JW, Rest JS, García-Moreno J, Sorenson MD, Mindell DP (2008) Strong mitochondrial DNA support for a Cretaceous origin of modern avian lineages. BMC Biol 6:6. doi:10.1186/1741-7007-6-6
Campbell MA, Chen W-J, López JA (2013a) Are flatfishes (Pleuronectiformes) monophyletic? Mol Phylogenet Evol 69:664–673
Campbell MA, López JA, Sado T, Miya M (2013b) Pike and salmon as sister taxa: detailed intraclade resolution and divergence time estimation of Esociformes + Salmoniformes based on whole mitochondrial genome sequences. Gene 530:57–65
Campbell MA, López JA, Satoh TP, Chen W-J, Miya M (2014) Mitochondrial genomic investigation of flatfish monophyly. Gene 551:176–182
Chanet B, Guintard C, Betti E, Gallut C, Dettaï A, Lecointre G (2013) Evidence for a close phylogenetic relationship between the teleost orders Tetraodontiformes and Lophiiformes based on an analysis of soft anatomy. Cybium 37:179–198
Chang YS, Huang FL, Lo TB (1994) The complete nucleotide sequence and gene organization of carp (Cyprinus carpio) mitochondrial genome. J Mol Evol 38:138–155
Chen J-N, López JA, Lavoué S, Miya M, Chen W-J (2013) Phylogeny of the Elopomorpha (Teleostei): evidence from six nuclear and mitochondrial markers. Mol Phylogenet Evol 70:152–161
Chen W-J, Bonillo C, Lecointre G (2003) Repeatability of clades as a criterion of reliability: a case study for molecular phylogeny of Acanthomorpha (Teleostei) with larger number of taxa. Mol Phylogenet Evol 26:262–288
Cheng S, Chang SY, Gravitt P, Respess R (1994) Long PCR. Nature 369:684–685
Chow S, Kurogi H, Mochioka N, Kaji S, Okazaki M, Tsukamoto K (2009) Discovery of mature freshwater eels in the open ocean. Fish Sci 75:257–259
Comber SC, Smith C (2004) Polyploidy in fishes: patterns and processes. Biol J Linn Soc 82:431–442
Cracraft J (2004) Assembling the Tree of Life: where we stand at the beginning of the 21st century. In: Cracraft J, Donoghue MJ (eds) Assembling the Tree of Life. Oxford University Press, NY, pp 553–559
Crête-Lafrenière A, Weir LK, Bernatchez L (2012) Framing the Salmonidae family phylogenetic portrait: a more complete picture from increased taxon sampling. PLoS ONE 7:e46662. doi:10.1371/journal.pone.0046662
Curole J, Kocher T (1999) Mitogenomics: digging deeper with complete mitochondrial genomes. Trends Ecol Evol 14:394–398
Dettaï A, Lecointre G (2005) Further support for the clades obtained by multiple molecular phylogenies in the acanthomorph bush. C R Biol 328:674–689
Didier DA (2004) Phylogeny and classification of extant Holocephali. In: Carrier JC, Musick JA, Heithanus MR (eds) Biology of sharks and their relatives. CRC Press, London, pp 115–135
Doosey MH, Bart HLJ, Saitoh K, Miya M (2010) Phylogenetic relationships of catostomid fishes (Actinopterygii: Cypriniformes) based on mitochondrial ND4/ND5 gene sequences. Mol Phylogenet Evol 54:1028–1034
Dowling T, Moritz C, Palmer J, Rieseberg L (1996) Nucleic acids III. Analysis of fragments and restriction sites. In: Hillis D, Moritz C, Mable B (eds) Molecular systematics. Sinauer Associates, Sunderland, MA, pp 249–320
Faircloth BC, Sorenson L, Santini F, Alfaro ME (2013) A phylogenomic perspective on the radiation of ray-finned fishes based upon targeted sequencing of ultraconserved elements (UCEs). PLoS ONE 8:e65923. doi:10.1371/journal.pone.0065923
Fink WL (1984) Basal euteleosts: relationships. In: Moser HG, Richards WJ, Cohen DM, Fahay MP, Kendall Jr AW, Richardson SL (eds) Ontogeny and systematics of fishes. Am. Soc. Ichthyol. Herpetol. Spec. Publ. 1, Lawrence, KS, pp 202–206
Fink WL, Weitzman SH (1982) Relationships of stomiiform fishes (Teleostei), with a description of Diplophos. Bull Mus Comp Zool 150:31–93
Freihofer WC (1963) Patterns of the ramus lateralis accessorius and their systematic significance in teleostean fishes. Stanford Ichthyol Bull 8:80–189
Friedman M (2009) Ecomorphological selectivity among marine teleost fishes during the end-Cretaceous extinction. Proc Natl Acad Sci USA 106:5218–5223
Friedman M, Keck BP, Dornburg A, Eytan RI, Martin CH, Hulsey CD, Wainwright PC, Near TJ (2013) Molecular and fossil evidence place the origin of cichlid fishes long after Gondwanan rifting. Proc R Soc B 280:20131733
Futuyma DJ (2013) Evolution. Sinauer Associates, Sunderland, MA
Grande L (2010) An empirical synthetic pattern study of gars (Lepisosteiformes) and closely related species, based mostly on skeletal anatomy: The resurrection of Holostei. Am. Soc. Ichthyol. Herpetol. Spec. Publ. 6, Lawrence, KS
Grande T, Borden WC, Smith WL (2013) Limits and relationships of Paracanthopterygii: A molecular framework for evaluating past morphological hypotheses. In: Arratia G, Schultze H-P, Wilson MVH (eds) Mesozoic fishes 5. Verlag Dr. Friedrich Pfeil, München, pp 385–418
Greenwood PHJ (1973) Interrelationships of osteoglossomorphs. In: Greenwood PHJ, Miles RS, Patterson C (eds) Interrelationships of fishes. Academic Press, London, pp 307–332
Greenwood PHJ, Rosen DE, Weitzman SH, Myers GS (1966) Phyletic studies of teleostean fishes, with a provisional classification of living forms. Bull Am Mus Nat Hist 131:339–456
Grogan D, Lund R (2004) The origin and relationships of early Chondrichthyes. In: Carrier JC, Musick JA, Heithanus MR (eds) Biology of sharks and their relatives. CRC Press, London, pp 3–31
Harrison GL, McLenachan PA, Phillips MJ, Slack KE, Cooper A, Penny D (2004) Four new avian mitochondrial genomes help get to basic evolutionary questions in the late Cretaceous. Mol Biol Evol 21:974–983
Hebert PD, Cywinska A, Ball SL (2003) Biological identifications through DNA barcodes. Proc R Soc B 270:313–321
Helfman GS, Collette BB, Facey DE, Bowen BW (2009) The diversity of fishes. Wiley-Blackwell, Oxford
Hennig W (1966) Phylogenetic systematics. University of Illinois Press, Urbana, IL
Hillis DM (1998) Taxonomic sampling, phylogenetic accuracy, and investigator bias. Syst Biol 47:3–8
Hirayama M, Mukai T, Miya M, Murata Y, Sekiya Y, Yamashita T, Nishida M, Watabe S, Oda S, Mitani H (2010) Intraspecific variation in the mitochondrial genome among local populations of Medaka Oryzias latipes. Gene 457:13–24
Holcroft N (2004) A molecular test of alternative hypotheses of tetraodontiform (Acanthomorpha: Tetraodontiformes) sister group relationships using data from the RAG1 gene. Mol Phylogenet Evol 32:749–760
Howlett R (2009) Taxonomy: Three into one will go. Nature 457:973
Hurley I, Mueller R, Dunn K, Schmidt E, Friedman M, Ho R, Prince V, Yang Z, Thomas M, Coates M (2007) A new time-scale for ray-finned fish evolution. Proc R Soc B 274:489–498
Imoto JM, Saitoh K, Sasaki T, Yonezawa T, Adachi J, Kartavtsev YP, Miya M, Nishida M, Hanzawa N (2013) Phylogeny and biogeography of highly diverged freshwater fish species (Leuciscinae, Cyprinidae, Teleostei) inferred from mitochondrial genome analysis. Gene 514:112–124
Inoue JG, Miya M, Tsukamoto K, Nishida M (2000a) Complete mitochondrial DNA sequence of the Japanese eel, Anguilla japonica. Fish Sci 67:118–125
Inoue JG, Miya M, Tsukamoto K, Nishida M (2000b) Complete mitochondrial DNA sequence of the Japanese sardine, Sardinops melanostictus. Fish Sci 66:924–932
Inoue JG, Miya M, Tsukamoto K, Nishida M (2001a) Complete mitochondrial DNA sequence of Conger myriaster (Teleostei: Anguilliformes): novel gene order for vertebrate mitochondrial genomes and the phylogenetic implications for anguilliform families. J Mol Evol 52:311–320
Inoue JG, Miya M, Tsukamoto K, Nishida M (2001b) Complete mitochondrial DNA sequence of the Japanese anchovy, Engraulis japonicus. Fish Sci 67:828–835
Inoue JG, Miya M, Tsukamoto K, Nishida M (2001c) A mitogenomic perspective on the basal teleostean phylogeny: resolving higher-level relationships with longer DNA sequences. Mol Phylogenet Evol 20:275–285
Inoue JG, Miya M, Tsukamoto K, Nishida M (2003a) Basal actinopterygian relationships: a mitogenomic perspective on the phylogeny of the “ancient fish”. Mol Phylogenet Evol 26:110–120
Inoue JG, Miya M, Tsukamoto K, Nishida M (2003b) Evolution of the deep-sea gulper eel mitochondrial genomes: large-scale gene rearrangements originated within the eels. Mol Biol Evol 20:1917–1924
Inoue JG, Miya M, Tsukamoto K, Nishida M (2004) Mitogenomic evidence for the monophyly of elopomorph fishes (Teleostei) and the evolutionary origin of the leptocephalus larva. Mol Phylogenet Evol 32:274–286
Inoue JG, Miya M, Venkatesh B, Nishida M (2005) The mitochondrial genome of Indonesian coelacanth Latimeria menadoensis (Sarcopterygii: Coelacanthiformes) and divergence time estimation between the two coelacanths. Gene 349:227–235
Inoue JG, Kumazawa Y, Miya M, Nishida M (2009) The historical biogeography of the freshwater knifefishes using mitogenomic approaches: a Mesozoic origin of the Asian notopterids (Actinopterygii: Osteoglossomorpha). Mol Phylogenet Evol 51:486–499
Inoue J, Donoghue PCJ, Yang Z (2010a) The impact of the representation of fossil calibrations on Bayesian estimation of species divergence times. Syst Biol 59:74–89
Inoue JG, Miya M, Lam K, Tay B-H, Danks JA, Bell J, Walker TI, Venkatesh B (2010b) Evolutionary origin and phylogeny of the modern holocephalans (Chondrichthyes: Chimaeriformes): a mitogenomic perspective. Mol Biol Evol 27:2576–2586
Inoue JG, Miya M, Miller MJ, Sado T, Hanel R, Hatooka K, Aoyama J, Minegishi Y, Nishida M, Tsukamoto K (2010c) Deep-ocean origin of the freshwater eels. Biol Lett 6:363–366
Ishiguro NB, Miya M, Nishida M (2001) Complete mitochondrial DNA sequence of ayu Plecoglossus altivelis. Fish Sci 67:474–481
Ishiguro NB, Miya M, Nishida M (2003) Basal euteleostean relationships: a mitogenomic perspective on the phylogenetic reality of the “Protacanthopterygii”. Mol Phylogenet Evol 27:476–488
Ishiguro NB, Miya M, Inoue JG, Nishida M (2005) Sundasalanx (Sundasalangidae) is a progenetic clupeiform, not a closely-related group of salangids (Osmeriformes): mitogenomic evidence. J Fish Biol 67:561–569
Ishikawa Y (2000) Medakafish as a model system for vertebrate developmental genetics. Bioessays 22:487–495
Iwasaki W, Fukunaga T, Isagozawa R, Yamada K, Maeda Y, Satoh TP, Sado T, Mabuchi K, Takeshima H, Miya M, Nishida M (2013) MitoFish and MitoAnnotator: a mitochondrial genome database of fish with an accurate and automatic annotation pipeline. Mol Biol Evol 30:2531–2540
Jacobsen MW, Hansen MM, Orlando L, Bekkevold D, Bernatchez L, Willerslev E, Gilbert MTP (2012) Mitogenome sequencing reveals shallow evolutionary histories and recent divergence time between morphologically and ecologically distinct European whitefish (Coregonus spp.). Mol Ecol 21:2727–2742
Johansen S, Bakke I (1996) The comlete mitochondrial DNA sequence of Atlantic cod (Gadus morhua): relevance to taxonomic studies among codfishes. Mol Mar Biol Biotechnol 5:203–214
Johnson GD (1992) Monophyly of the euteleostean clades—Neoteleostei, Eurypterygii, and Ctenosquamata. Copeia 1992:8–25
Johnson GD (1993) Percomorph phylogeny: progress and problems. Bull Mar Sci 52:3–28
Johnson GD, Patterson C (1993) Percomorph phylogeny: a survey of acanthomorphs and a new proposal. Bull Mar Sci 52:554–626
Johnson GD, Patterson C (1996) Relationships of lower euteleostean fishes. In: Stiassny MLJ, Parenti LR, Johnson GD (eds) Interrelationships of fishes. Academic Press, San Diego, CA, pp 251–332
Johnson GD, Paxton JR, Sutton TT, Satoh TP, Sado T, Nishida M, Miya M (2009) Deep-sea mystery solved: astonishing larval transformations and extreme sexual dimorphism unite three fish families. Biol Lett 5:232–239
Johnson GD, Ida H, Skaue J, Sado T, Asahida T, Miya M (2012) A ‘living fossil’ eel (Anguilliformes: Protanguillidae, fam. nov.) from an undersea cave in Palau. Proc R Soc B 279:934–943
Kawaguchi A, Miya M, Nishida M (2001) Complete mitochondrial DNA sequence of Aulopus japonicus (Teleostei: Aulopiformes), a basal Eurypterygii: longer DNA sequences and the higher-level relationships. Ichthyol Res 48:213–223
Kawahara R, Miya M, Mabuchi K, Lavoué S, Inoue JG, Satoh TP, Kawaguchi A, Nishida M (2008) Interrelationships of the 11 gasterosteiform families (sticklebacks, pipefishes, and their relatives): a new perspective based on whole mitogenome sequences from 75 higher teleosts. Mol Phylogenet Evol 46:224–236
Kawahara R, Miya M, Mabuchi K, Near T, Nishida M (2009) Stickleback phylogenies resolved: evidence from mitochondrial genomes and 11 nuclear genes. Mol Phylogenet Evol 50:401–404
Kobayasi H, Kawashima Y, Takeuchi N (1970) Comparative chromosome studies in the genus Carassius, especially with a finding of polyploidy in the ginbuna (C. auratus langsdorfii). Jpn J Ichthyol 17:153–160
Kumazawa Y, Nishida M (1993) Sequence evolution of mitochondrial tRNA genes and deep-branch animal phylogenetics. J Mol Evol 37:380–398
Kumazawa Y, Nishida M (1995) Variations in mitochondrial tRNA gene organization of reptiles as phylogenetic markers. Mol Biol Evol 12:759–772
Lauder GV, Liem KF (1983) The evolution and interrelationships of the actinopterygian fishes. Bull Mus Comp Zool 150:95–197
Lavoué S, Miya M, Inoue JG, Saitoh K, Ishiguro NB, Nishida M (2005) Molecular systematics of the gonorynchiform fishes (Teleostei) based on whole mitogenome sequences: implications for higher-level relationships within the Otocephala. Mol Phylogenet Evol 37:165–177
Lavoué S, Miya M, Saitoh K, Ishiguro N, Nishida M (2007) Phylogenetic relationships among anchovies, sardines, herrings and their relatives (Clupeiformes), inferred from whole mitogenome sequences. Mol Phylogenet Evol 43:1096–1105
Lavoué S, Miya M, Kawaguchi A, Yoshino T, Nishida M (2008a) The phylogenetic position of an undescribed paedomorphic clupeiform taxon: mitogenomic evidence. Ichthyol Res 55:328–334
Lavoué S, Miya M, Poulsen JY, Møller PR, Nishida M (2008b) Monophyly, phylogenetic position and inter-familial relationships of the Alepocephaliformes (Teleostei) based on whole mitogenome sequences. Mol Phylogenet Evol 47:1111–1121
Lavoué S, Miya M, Nishida M (2010) Mitochondrial phylogenomics of anchovies (family Engraulidae) and recurrent origins of pronounced miniaturization in the order Clupeiformes. Mol Phylogenet Evol 56:480–485
Lavoué S, Miya M, Arnegard ME, McIntyre PB, Mamonekene V, Nishida M (2011) Remarkable morphological stasis in an extant vertebrate despite tens of millions of years of divergence. Proc R Soc B 278:1003–1008
Lavoué S, Miya M, Arnegard ME, Sullivan JP, Hopkins CD, Nishida M (2012a) Comparable ages for the independent origins of electrogenesis in African and South American weakly electric fishes. PLoS ONE 7:e36287. doi:10.1371/journal.pone.0036287
Lavoué S, Miya M, Moritz T, Nishida M (2012b) A molecular timescale for the evolution of the African freshwater fish family Kneriidae (Teleostei: Gonorynchiformes). Ichthyol Res 52:1–9
Lavoué S, Miya M, Musikasinthorn P, Chen W-J, Nishida M (2013) Mitogenomic evidence for an Indo-west Pacific origin of the Clupeoidei (Teleostei: Clupeiformes). PLoS ONE 8:e56485. doi:10.1371/journal.pone.0056485
Lavoué S, Nakayama K, Jerry DR, Yamanoue Y, Yagishita N, Suzuki N, Nishida M, Miya M (2014) Mitogenomic phylogeny of the Percichthyidae and Centrarchiformes (Percomorphaceae): comparison with recent nuclear gene-based studies and simultaneous analysis. Gene 549:46–57
Lê H, Lecointre G, Perasso R (1993) A 28S rRNA-based phylogeny of the gnathostomes: first steps in the analysis of conflict and congruence with morphologically based cadograms. Mol Phylogenet Evol 2:31–51
Lecointre G, Nelson GJ (1996) Clupeomorpha, sister-group of Ostariophysi. In: Stiassny MLJ, Parenti LR, Johnson GD (eds) Interrelationships of Fishes. Academic Press, San Diego, CA, pp 193–207
Lee JS, Miya M, Lee YS, Kim CG, Park EH, Aoki Y, Nishida M (2001) The complete DNA sequence of the mitochondrial genome of the self-fertilizing fish Rivulus marmoratus (Cyprinodontiformes, Rivulidae) and the first description of duplication of a control region in fish. Gene 280:1–7
Li B, Dettaï A, Cruaud C, Couloux A, Desoutter-Meniger M, Lecointre G (2009) RNF213, a new nuclear marker for acanthomorph phylogeny. Mol Phylogenet Evol 50:345–363
Li C, Lu G, Ortí G (2008) Optimal data partitioning and a test case for ray-finned fishes (Actinopterygii) based on ten nuclear loci. Syst Biol 57:519–539
Li J, Xia R, McDowall RM, López JA, Lei G, Fu C (2010) Phylogenetic position of the enigmatic Lepidogalaxias salamandroides with comment on the orders of lower euteleostean fishes. Mol Phylogenet Evol 57:932–936
Little AG, Lougheed SC, Moyes CD (2010) Evolutionary affinity of billfishes (Xiphiidae and Istiophoridae) and flatfishes (Plueronectiformes): Independent and trans-subordinal origins of endothermy in teleost fishes. Mol Phylogenet Evol 56:897–904
Lydeard C, Roe KJ (1997) The phylogenetic utility of the mitochondrial cytochrome b gene for inferring relationships among actinopterygian fishes. In: Kocher TD, Stepien CA (eds) Molecular systematics of fishes. Academic Press, San Diego, CA, pp 285–311
Mabuchi K, Miya M, Satoh TP, Westneat MW, Nishida M (2004) Gene rearrangements and evolution of tRNA pseudogenes in the mitochondrial genome of the parrotfish (Teleostei: Perciformes: Scaridae). J Mol Evol 59:287–297
Mabuchi K, Senou H, Suzuki T, Nishida M (2005) Discovery of an ancient lineage of Cyprinus carpio from Lake Biwa, central Japan, based on mtDNA sequence data, with reference to possible multiple origins of koi. J Fish Biol 66:1516–1528
Mabuchi K, Miya M, Senou H, Suzuki T, Nishida M (2006) Complete mitochondrial DNA sequence of the Lake Biwa wild strain of common carp (Cyprinus carpio L.): further evidence for an ancient origin. Aquaculture 257:68–77
Mabuchi K, Miya M, Azuma Y, Nishida M (2007) Independent evolution of the specialized pharyngeal jaw apparatus in cichlid and labrid fishes. BMC Evol Biol 7:10. doi:10.1186/1471-2148-7-10
Macey J (1997) Two novel gene orders and the role of light-strand replication in rearrangement of the vertebrate mitochondrial genome. Mol Biol Evol 14:91–104
Machida RJ, Miya MU, Nishida M, Nishida S (2002) Complete mitochondrial DNA sequence of Tigriopus japonicus (Crustacea: Copepoda). Mar Biotechnol 4:406–417
Macqueen DJ, Johnston IA (2014) A well-constrained estimate for the timing of the salmonid whole genome duplication reveals major decoupling from species diversification. Proc R Soc B 281:20132881. doi:10.1098/rspb.2013.2881
Martin AP, Burg TM (2002) Perils of paralogy: using HSP70 genes for inferring organismal phylogenies. Syst Biol 51:570–587
Mayden RL, Tang KL, Conway KW, Freyhof J, Chamberlain S, Haskins M, Schneider L, Sudkamp M, Wood RM, Agnew M, Bufalino A, Sulaiman Z, Miya M, Saitoh K, He S (2007) Phylogenetic relationships of Danio within the order Cypriniformes: a framework for comparative and evolutionary studies of a model species. J Exp Zool B Mol Dev Evol 308B:642–654
Mayden RL, Tang KL, Wood RM, Chen W-J, Agnew MK, Conway KW, Yang L, Simons AM, Bart HL, Harris PM, Li J, Wang X, Saitoh K, He S, Liu H, Chen Y, Nishida M, Miya M (2008) Inferring the Tree of Life of the order Cypriniformes, the earth’s most diverse clade of freshwater fishes: implications of varied taxon and character sampling. J Systemat Evol 46:424–438
Mayden RL, Chen WJ, Bart HL, Doosey MH, Simons AM, Tang KL, Wood RM, Agnew MK, Yang L, Hirt MV, Clements MD, Saitoh K, Miya M, Nishida M (2009) Reconstructing the phylogenetic relationships of the earth’s most diverse clade of freshwater fishes —order Cypriniformes (Actinopterygii: Ostariophysi): A case study using multiple nuclear loci and the mitochondrial genome. Mol Phylogenet Evol 51:500–514
Meyer A (1994) Shortcomings of the cytochrome b gene as a molecular marker. Trends Ecol Evol 9:278–280
Mindell D, Sorenson M, Dimcheff D, Hasegawa M (1999) Interordinal relationships of birds and other reptiles based on whole mitochondrial genomes. Syst Biol 48:138–152
Minegishi Y, Aoyama J, Inoue JG, Miya M, Nishida M, Tsukamoto K (2005) Molecular phylogeny and evolution of the freshwater eels genus Anguilla based on the whole mitochondrial genome sequences. Mol Phylogenet Evol 34:134–146
Mittelbach GG, Schemske DW, Cornell HV, Allen AP, Brown JM, Bush MB, Harrison SP, Hurlbert AH, Knowlton N, Lessios HA (2007) Evolution and the latitudinal diversity gradient: speciation, extinction and biogeography. Ecol Lett 10:315–331
Miya M, Nishida M (1996) Molecular phylogenetic perspective on the evolution of the deep-sea fish genus Cyclothone (Stomiiformes: Gonostomatidae). Ichthyol Res 43:375–398
Miya M, Nishida M (1997) Speciation in the open ocean. Nature 389:803–804
Miya M, Nishida M (1998) Molecular phylogeny and evolution of the deep-sea fish genus Sternoptyx. Mol Phylogenet Evol 10:11–22
Miya M, Nishida M (1999) Organization of the mitochondrial genome of a deep-sea fish, Gonostoma gracile (Teleostei: Stomiiformes): first example of transfer RNA gene rearrangements in bony fishes. Mar Biotechnol 1:416–426
Miya M, Nishida M (2000a) Molecular systematics of the deep-sea fish genus Gonostoma (Stomiiformes: Gonostomatidae): two paraphyletic clades and resurrection of Sigmops. Copeia 2000:378–389
Miya M, Nishida M (2000b) Use of mitogenomic information in teleostean molecular phylogenetics: a tree-based exploration under the maximum-parsimony optimality criterion. Mol Phylogenet Evol 17:437–455
Miya M, Kawaguchi A, Nishida M (2001) Mitogenomic exploration of higher teleostean phylogenies: a case study for moderate-scale evolutionary genomics with 38 newly determined complete mitochondrial DNA sequences. Mol Biol Evol 18:1993–2009
Miya M, Takeshima H, Endo H, Ishiguro NB, Inoue JG, Mukai T, Satoh TP, Yamaguchi M, Kawaguchi A, Mabuchi K, Shirai SM, Nishida M (2003) Major patterns of higher teleostean phylogenies: a new perspective based on 100 complete mitochondrial DNA sequences. Mol Phylogenet Evol 26:121–138
Miya M, Satoh TP, Nishida M (2005) The phylogenetic position of toadfishes (order Batrachoidiformes) in the higher ray-finned fish as inferred from partitioned Bayesian analysis of 102 whole mitochondrial genome sequences Biol J Linn Soc 85:289–306
Miya M, Saitoh K, Wood R, Nishida M, Mayden RL (2006) New primers for amplifying and sequencing the mitochondrial ND4/ND5 gene region of the Cypriniformes (Actinopterygii: Ostariophysi). Ichthyol Res 53:75–81
Miya M, Holcroft NI, Satoh TP, Yamaguchi M, Nishida M, Wiley EO (2007) Mitochondrial genome and a nuclear gene indicate a novel phylogenetic position of deep-sea tube-eye fish (Stylephoridae). Ichthyol Res 54:323–332
Miya M, Pietsch TW, Orr JW, Arnold RJ, Satoh TP, Shedlock AM, Ho HC, Shimazaki M, Yabe M, Nishida M (2010) Evolutionary history of anglerfishes (Teleostei: Lophiiformes): a mitogenomic perspective. BMC Evol Biol 10:58
Miya M, Friedman M, Satoh TP, Takeshima H, Sado T, Iwasaki W, Yamanoue Y, Nakatani M, Mabuchi K, Inoue JG, Poulsen JY, Fukunaga T, Nishida M (2013) Evolutionary origin of the Scombridae (tunas and mackerels): members of a Paleogene adaptive radiation with 14 other pelagic fish families. PLoS ONE 8:e73535. doi:10.1371/journal.pone.0073535
Moore WS (1995) Inferring phylogenies from mtDNA variation: mitochondrial-gene trees versus nuclear-gene trees. Evolution 49:718–726
Morin PA, Archer FI, Foote AD, Vilstrup J, Allen EE, Wade P, Durban J, Parsons K, Pitman R, Li L (2010) Complete mitochondrial genome phylogeographic analysis of killer whales (Orcinus orca) indicates multiple species. Genome Res 20:908–916
Nakatani M, Miya M, Mabuchi K, Saitoh K, Nishida M (2011) Evolutionary history of Otophysi (Teleostei), a major clade of the modern freshwater fishes: Pangaean origin and Mesozoic radiation. BMC Evol Biol 11:177. doi:10.1186/1471-2148-11-177
Near TJ, Eytan RI, Dornburg A, Kuhn KL, Moore JA, Davis MP, Wainwright PC, Friedman M, Smith WL (2012) Resolution of ray-finned fish phylogeny and timing of diversification. Proc Natl Acad Sci USA 109:13698–13703
Near TJ, Dornburg A, Eytan RI, Keck BP, Smith WL, Kuhn KL, Moore JA, Price SA, Burbrink FT, Friedman M, Wainwright PC (2013) Phylogeny and tempo of diversification in the superradiation of spiny-rayed fishes. Proc Natl Acad Sci USA 110:12738–12743
Nelson GJ (1989) Phylogeny of major fish groups. In: Fernholm B, Bremer K, Jörnvall H (eds) The hierarchy of life. Excerpta Medica, Amsterdam, pp 325–336
Nelson JS (1976) Fishes of the world. John Wiley & Sons, NY
Nelson JS (1984) Fishes of the world. John Wiley & Sons, NY
Nelson JS (1994) Fishes of the world. John Wiley & Sons, NY
Nelson JS (2006) Fishes of the world. John Wiley & Sons, Hoboken, NJ
Noack K, Zardoya R, Meyer A (1996) The complete mitochondrial DNA sequence of the bichir (Polypterus ornatipinnis), a basal ray-finned fish: ancient establishment of the consensus vertebrate gene order. Genetics 144:1165–1180
Nohara M, Nishida M, Miya M, Nishikawa T (2005) Evolution of the mitochondrial genome in Cephalochordata as inferred from complete nucleotide sequences from two Epigonichthys species. J Mol Evol 60:526–537
Normark B (1991) Phylogenetic relationships of neopterygian fishes, inferred from mitochondrial DNA sequences. Mol Biol Evol 8:819–834
Olney JE, Johnson GD, Baldwin CC (1993) Phylogeny of lampridiform fishes. Bull Mar Sci 52:137–169
Ortí G, Meyer A (1997) The radiation of characiform fishes and the limits of resolution of mitochondrial ribosomal DNA sequences. Syst Biol 46:75-100
Patterson C (1973) Interrelationships of holosteans. In: Greenwood PHJ, Miles RS, Patterson C (eds) Interrelationships of fishes. Academic Press, London, pp 233–305
Patterson C, Rosen DE (1977) Review of ichthyodectiform and other Mesozoic teleost fishes and the theory and practice of classifying fossils. Bull Am Mus Nat Hist 158:81–172
Patterson C, Williams DM, Humphries CJ (1993) Congruence between molecular and morphological phylogenies. Annu Rev Ecol Syst 24:153–188
Paxton JR, Johnson GD (2005) Cetomimidae: Whalefishes; Mirapinnidae: tapetails & hairyfish; Megalomycteridae: bignose fishes. In: Richards WJ (ed) Early stages of Atlantic fishes. CRC Press, Boca Raton, FL, pp 1089–1102
Peng Z, He S, Wang J, Wang W, Diogo R (2006) Mitochondrial molecular clocks and the origin of the major otocephalan clades (Pisces: Teleostei): a new insight. Gene 370:113–124
Penny D, Hasegawa M, Waddell P, Hendy M (1999) Mammalian evolution: timing and implications from using the LogDeterminant transform for proteins of differing amino acid composition. Syst Biol 48:76–93
Philippe H, Zhou Y, Brinkmann H, Rodrigue N, Delsuc F (2005) Heterotachy and long-branch attraction in phylogenetics. BMC Evol Biol 5:50. doi:10.1186/1471-2148-5-50
Phillips M, Penny D (2003) The root of the mammalian tree inferred from whole mitochondrial genomes. Mol Phylogenet Evol 28:171–185
Pietsch TW (2009) Oceanic anglerfishes: extraordinary diversity in the deep sea. University of California Press, Berkeley, CA
Poulsen JY, Møller PDR, Lavoué S, Knudsen SW, Nishida M, Miya M (2009) Higher and lower-level relationships of the deep-sea fish order Alepocephaliformes (Teleostei: Otocephala) inferred from whole mitogenome sequences. Biol J Linn Soc 98:923–936
Poulsen JY, Byrkjedal I, Willassen E, Rees D, Takeshima H, Satoh TP, Shinohara G, Nishida M, Miya M (2013) Mitogenomic sequences and evidence from unique gene rearrangements corroborate evolutionary relationships of Myctophiformes (Neoteleostei). BMC Evol Biol 13:111. doi:10.1186/1471-2148-13-111
Ramsden SD, Brinkmann H, Hawryshyn CW, Taylor JS (2003) Mitogenomics and the sister of Salmonidae. Trends Ecol Evol 18:607–610
Ronquist F, Huelsenbeck JP (2003) MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19:1572–1574
Rosen DE (1973) Interrelationships of higher euteleostean fishes. In: Greenwood PHJ, Miles RS, Patterson C (eds) Interrelationships of fishes. Academic Press, London, pp 397–513
Rosen DE (1974) Phylogeny and zoogeography of salmoniform fishes and relationships of Lepidogalaxias salamandroides. Bull Am Mus Nat Hist 153:263–326
Rosen DE (1985) An essay on euteleostean classification. Am Mus Novit 2782:1–45
Rubin DA, Dores RM (1995) Obtaining a more resolute teleost growth hormone phylogeny by the introduction of gaps in sequence alignment. Mol Phylogenet Evol 4:129–138
Saitoh K, Miya M, Inoue JG, Ishiguro NB, Nishida M (2003) Mitochondrial genomics of ostariophysan fishes: perspectives on phylogeny and biogeography. J Mol Evol 56:464–472
Saitoh K, Sado T, Mayden RL, Hanzawa N, Nakamura K, Nishida M, Miya M (2006) Mitogenomic evolution and interrelationships of the Cypriniformes (Actinopterygii: Ostariophysi): the first evidence toward resolution of higher-level relationships of the World’s largest freshwater fish clade based on 59 whole mitogenome sequences. J Mol Evol 63:826–841
Saitoh K, Sado T, Doosey MH, Bart HLJ, Inoue JG, Nishida M, Mayden RL, Nishida M, Miya M (2011) Evidence from mitochondrial genomics supports the lower Mesozoic of South Asia as the time and place of basal divergence of cypriniform fishes (Actinopterygii: Ostariophysi). Zool J Linn Soc 161:633–662
Santini F, Sorenson L, Alfaro ME (2013) A new phylogeny of tetraodontiform fishes (Tetraodontiformes, Acanthomorpha) based on 22 loci. Mol Phylogenet Evol 69:177–187
Sato Y, Nishida M (2010) Teleost fish with specific genome duplication as unique models of vertebrate evolution. Environ Biol Fishes 88:169–188
Satoh TP, Miya M, Endo H, Nishida M (2006) Round and pointed-head grenadier fishes (Actinopterygii: Gadiformes) represent a single sister group: evidence from the complete mitochondrial genome sequences. Mol Phylogenet Evol 40:129–138
Satoh TP, Sato Y, Masuyama N, Miya M, Nishida M (2010) Transfer RNA gene arrangement and codon usage in vertebrate mitochondrial genomes: a new insight into gene order conservation. BMC Genomics 11:479. doi:10.1186/1471-2164-11-479
Setiamarga DHE, Miya M, Yamanoue Y, Mabuchi K, Satoh TP, Inoue JG, Nishida M (2008) Interrelationships of Atherinomorpha (medakas, flyingfishes, killifishes, silversides, and their relatives): the first evidence based on whole mitogenome sequences. Mol Phylogenet Evol 49:598–605
Setiamarga DHE, Miya M, Inoue JG, Ishiguro NB, Mabuchi K, Nishida M (2009) Divergence time of the two regional medaka populations in Japan as a new time scale for comparative genomics of vertebrates. Biol Lett 5:81–86
Shimodaira H (2002) An approximately unbiased test of phylogenetic tree selection. Syst Biol 51:492–508
Siebert DJ (1997) Notes on the anatomy and and relationships of Sundasalanx Roberts (Teleostei, Clupeidae), with descriptions of four new species from Borneo. Bull Nat Hist Mus (Zool Ser) 63:13–26
Simmons MP, Miya M (2004) Efficiently resolving the basal clades of a phylogenetic tree using Bayesian and parsimony approaches: a case study using mitogenomic data from 100 higher teleost fishes. Mol Phylogenet Evol 31:351–362
Simmons MP, Pickett KM, Miya M (2004) How meaningful are Bayesian support values? Mol Biol Evol 21:188–199
Smith WL, Craig MT (2007) Casting the percomorph net widely: the importance of broad taxonomic sampling in the search for the placement of serranid and percid fishes. Copeia 2007:35–55
Smith WL, Wheeler WC (2006) Venom evolution widespread in fishes: a phylogenetic road map for the bioprospecting of piscine venoms. J Hered 97:206–217
Song HY, Mabuchi K, Satoh TP, Moore JA, Yamanoue Y, Miya M, Nishida M (2014) Mitogenomic circumscription of a novel percomorph fish clade mainly comprising “Syngnathoidei” (Teleostei). Gene 542:146–155
Sorenson MD, Ast JC, Dimcheff DE, Yuri T, Mindell DP (1999) Primers for a PCR-based approach to mitochondrial genome sequencing in birds and other vertebrates. Mol Phylogenet Evol 12:105–114
Springer M, Amrine H, Burk A, Stanhope M (1999) Additional support for Afrotheria and Paenungulata, the performance of mitochondrial versus nuclear genes, and the impact of data partitions with heterogeneous base composition. Syst Biol 48:65–75
Springer V (1999) Are the Indonesian and western Indian Ocean coelacanths conspecific: a prediction. Environ Biol Fishes 54:453–456
Stamatakis A (2006) RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22:2688–2690
Stepien CA, Kocher TD (1997) Molecules and morphology in studies of fish evolution. In: Kocher TD, Stepien CA (eds) Molecular systematics of fishes. Academic Press, San Diego, CA, pp 1–11
Stiassny MLJ (1986) The limits and relationships of the acanthomorph teleosts. J Zool 1986:411–460
Stiassny MLJ, Moore JA (1992) A review of the pelvic girdle of acanthomorph fishes, with comments on hypotheses of acanthomorph interrelationships. Zool J Linn Soc 104:209–242
Takada M, Tachihara K, Kon T, Yamamoto G, Iguchi K, Miya M, Nishida M (2010) Biogeography and evolution of the Carassius auratus-complex in East Asia. BMC Evol Biol 10:7. doi:10.1186/1471-2148-10-7
Takehana Y, Nagai N, Matsuda M, Tsuchiya K, Sakaizumi M (2003) Geographic variation and diversity of the cytochrome b gene in Japanese wild populations of medaka, Oryzias latipes. Zool Sci 20:1279–1291
Tang KL, Agnew MK, Chen W-J, Vincent Hirt M, Raley ME, Sado T, Schneider LM, Yang L, Bart HL, He S, Liu H, Miya M, Saitoh K, Simons AM, Wood R, Mayden RL (2011) Phylogeny of the gudgeons (Teleostei: Cyprinidae: Gobioninae). Mol Phylogenet Evol 61:103–124
Tang KL, Conway KW, Agnew MK, Chen W-J, Hirt MV, Sado T, Schneider LM, Freyhof J, Swartz E, Vidthayanon C, Bart HL, Miya M, Saitoh K, Simons AM, Wood RM, Mayden RL (2010) Systematics of the subfamily Danioninae (Teleostei: Cypriniformes: Cyprinidae). Mol Phylogenet Evol 57:189–214
Tang KL, Agnew MK, Hirt MV, Lumbantobing DN, Raley ME, Sado T, Teoh V-H, Yang L, Bart HL, Harris PM, He S, Miya M, Saitoh K, Simons AM, Wood RM, Mayden RL (2013) Limits and phylogenetic relationships of East Asian fishes in the subfamily Oxygastrinae (Teleostei: Cypriniformes: Cyprinidae). Zootaxa 3681:101–135
Timmermans MJ, Dodsworth S, Culverwell C, Bocak L, Ahrens D, Littlewood DT, Pons J, Vogler AP (2010) Why barcode? High-throughput multiplex sequencing of mitochondrial genomes for molecular systematics. Nucleic Acids Res 38:e197. doi:10.1093/nar/gkq807
Tsukamoto K, Aoyama J, Miller MJ (2002) Migration, speciation, and the evolution of diadromy in anguillid eels. Can J Fish Aquat Sci 59:1989–1998
Tzeng CS, Hui CF, Shen SC, Huang PC (1992) The complete nucleotide sequence of the Crossostoma lacustre mitochondrial genome: conservation and variations among vertebrates. Nucleic Acids Res 20:4853–4858
Venkatesh B, Erdmann M, Brenner S (2001) Molecular synapomorphies resolve evolutionary relationships of extant jawed vertebrates. Proc Natl Acad Sci USA 98:11382–11387
Wainwright PC, Smith WL, Price SA, Tang KL, Sparks JS, Ferry LA, Kuhn KL, Eytan RI, Near TJ (2012) The evolution of pharyngognathy: a phylogenetic and functional appraisal of the pharyngeal jaw key innovation in labroid fishes and beyond. Syst Biol 61:1001–1027
Ward RD, Zemlak TS, Innes BH, Last PR, Hebert PDN (2005) DNA barcoding Australia’s fish species. Philos Trans R Soc Lond, Ser B: Biol Sci 360:1847–1857
Watanabe S, Aoyama J, Tsukamoto K (2009) A new species of freshwater eel Anguilla luzonensis (Teleostei: Anguillidae) from Luzon Island of the Philippines. Fish Sci 75:387–392
Wiley EO (1981) Phylogenetics: the theory and practice of phylogenetic systematics. Wiley & Sons, NY
Williams N (2010) Deep secrets. Curr Biol 20:R43–R44
Yagishita N, Miya M, Yamanoue Y, Shirai SM, Nakayama K, Suzuki N, Satoh TP, Mabuchi K, Nishida M, Nakabo T (2009) Mitogenomic evaluation of the unique facial nerve pattern as a phylogenetic marker within the percifom fishes (Teleostei: Percomorpha). Mol Phylogenet Evol 53:258–266
Yamanoue Y, Miya M, Matsuura K, Katoh M, Sakai H, Nishida M (2004) Mitochondrial genomes and phylogeny of the ocean sunfishes (Tetraodontiformes: Molidae). Ichthyol Res 51:269–273
Yamanoue Y, Miya M, Inoue JG, Matsuura K, Nishida M (2006) The mitochondrial genome of spotted green pufferfish Tetraodon nigroviridis (Teleostei: Tetraodontiformes) and divergence time estimation among model organisms in fishes. Genes Genet Syst 81:29–39
Yamanoue Y, Miya M, Matsuura K, Yagishita N, Mabuchi K, Sakai H, Katoh M, Nishida M (2007) Phylogenetic position of tetraodontiform fishes within the higher teleosts: Bayesian inferences based on 44 whole mitochondrial genome sequences. Mol Phylogenet Evol 45:89–101
Yamanoue Y, Miya M, Matsuura K, Katoh M, Sakai H, Nishida M (2008) A new perspective on phylogeny and evolution of tetraodontiform fishes (Pisces: Acanthopterygii) based on whole mitochondrial genome sequences: basal ecological diversification? BMC Evol Biol 8:212. doi:10.1186/1471-2148-8-212
Yamanoue Y, Miya M, Matsuura K, Miyazawa S, Miyamoto N, Doi H, Takahashi H, Mabuchi K, Nishida M, Sakai H (2009a) Explosive speciation of Takifugu: another use of the fugu as a model system for evolutionary biology. Mol Biol Evol 26:623–629
Yamanoue Y, Miya M, Matsuura K, Sakai H, Katoh M, Nishida M (2009b) Unique patterns of pelvic fin evolution: a case study of balistoid fishes (Pisces: Tetraodontiformes) based on whole mitochondrial genome sequences. Mol Phylogenet Evol 50:179–189
Yamanoue Y, Miya M, Doi H, Mabuchi K, Sakai H, Nishida M (2011) Multiple invasions into freshwater by pufferfishes (Teleostei: Tetraodontidae): a mitogenomic perspective. PLoS ONE 6:e17410. doi:10.1371/journal.pone.0017410
Yamauchi MM, Miya MU, Machida RJ, Nishida M (2004a) PCR-based approach for sequencing mitochondrial genomes of decapod crustaceans, with a practical example from kuruma prawn (Marsupenaeus japonicus). Mar Biotechnol 6:419–429
Yamauchi MM, Miya MU, Nishida M (2004b) Use of a PCR-based approach for sequencing whole mitochondrial genomes of insects: two examples (cockroach and dragonfly) based on the method developed for decapod crustaceans. Insect Mol Biol 13:435–442
Yang L, Mayden RL, Sado T, He S, Saitoh K, Miya M (2010) Molecular phylogeny of the fishes traditionally referred to Cyprinini sensu stricto (Teleostei: Cypriniformes). Zool Scr 39:527–550
Yang L, Arunachalam M, Sado T, Levin BA, Golubtsov AS, Freyhof J, Friel JP, Chen W-J, Vincent Hirt M, Manickam R, Agnew MK, Simons AM, Saitoh K, Miya M, Mayden RL, He S (2012a) Molecular phylogeny of the cyprinid tribe Labeonini (Teleostei: Cypriniformes). Mol Phylogenet Evol 65:362–379
Yang L, Hirt V, Sado T, Arunachalam M, Manickam R, Tang KL, Simons AM, Wu H, Mayden R, Miya M (2012b) Phylogenetic placements of the barbin genera Discherodontus, Chagunius, and Hypselobarbus in the subfamily Cyprininae (Teleostei: Cypriniformes) and their relationships with other barbins. Zootaxa 3586:26–40
Zaragüeta-Bagils R, Lavoué S, Tillier A, Bonillo C, Lecointre G (2002) Assessment of otocephalan and protacanthopterygian concepts in the light of multiple molecular phylogenies. C R Biol 325:1191–1207
Zardoya R, Garrido-Pertierra A, Bautista JM (1995) The complete nucleotide sequence of the mitochondrial DNA genome of the rainbow trout, Oncorhynchus mykiss. J Mol Evol 41:942–951
Zardoya R, Meyer A (1996) The complete nucleotide sequence of the mitochondrial genome of the lungfish (Protopterus dolloi) supports its phylogenetic position as a close relative of land vertebrates. Genetics 142:1249–1263
Zardoya R, Meyer A (1997) The complete DNA sequence of the mitochondrial genome of a “living fossil,’’ the coelacanth (Latimeria chalumnae). Genetics 146:995–1010
Acknowledgments
We thank Keiichi Matsuura and other organizing committee members of the 9th Indo-Pacific Fish Conference (IPFC9) for providing us with an opportunity for reviewing our contributions to the molecular phylogenetics and evolution of fishes. This project would not have been possible without the collaborations with over 150 coauthors of the 83 papers as well as numerous people who kindly assisted our studies in various ways and are listed in the acknowledgment sections in the respective papers. We sincerely thank all of them who made this project successful. M.A. Campbell, J.G. Inoue, R. Kawahara-Miki, K.M. Laumann, S. Lavoué, K. Mabuchi, Y. Minegishi, J.Y. Poulsen, and Y. Yamanoue read the draft manuscript and gave us helpful comments. In particular, thoughtful inputs from M.A. Campbell greatly improved the manuscript. T. Sado kindly assisted redrawing of some of the older figures. Also, we thank P. Chakrabarty for providing us with useful comments and editing as a reviewer. Publication of this review was financially supported by IPFC9. Finally, we greatly appreciate continuing support from JSPS and other funding agencies.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Miya, M., Nishida, M. The mitogenomic contributions to molecular phylogenetics and evolution of fishes: a 15-year retrospect. Ichthyol Res 62, 29–71 (2015). https://doi.org/10.1007/s10228-014-0440-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10228-014-0440-9