
Abstract
Purpose
Erlotinib marginally improves survival when administered continuously with gemcitabine to patients with advanced pancreatic cancer; however, preclinical data suggest that there is antagonism between chemotherapy and epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors when these are delivered concurrently. We tested a pharmacodynamic separation approach for erlotinib plus gemcitabine and interrogated EGFR signaling intermediates as potential surrogates for the efficacy of this strategy.
Methods
Patients with measurable, previously untreated locally advanced unresectable or metastatic pancreatic cancer were treated with gemcitabine 1000 mg/m2 as an intravenous infusion over 30-min on days 1, 8, 15 and erlotinib 150 mg/day on days 2–5, 9–12, 16–26 of each 28-day cycle. The primary endpoint was progression-free survival (PFS); secondary endpoints included RECIST objective response rate (ORR) and safety. The study was terminated after thirty patients due to funding considerations.
Results
The median PFS was 2.07 months (95 % CI; 1.87–5.50 months) and the ORR was 11 %. No unexpected safety signals were seen: the most common grade 3 or higher adverse events were neutropenia (23 %), lymphopenia (23 %), and fatigue (13 %). Patients with mutant plasma Kirsten rat sarcoma virus (KRAS) had significantly lower median PFS (1.8 vs. 4.6 months, p = 0.014) and overall survival (3.0 vs. 10.5 months, p = 0.003) than those without detected plasma KRAS mutations.
Conclusions
Although pharmacodynamically separated erlotinib and gemcitabine were feasible and tolerable in patients with advanced pancreatic cancer, no signal for increased efficacy was seen in this molecularly unselected cohort. Detection of a KRAS mutation in circulating cell-free DNA was a strong predictor of survival.
Similar content being viewed by others


Introduction
Pancreatic cancer is among the most lethal of human malignancies and is the 4th leading cause of cancer death in the United States [1]. Until the very recent advent of the FOLFIRINOX and gemcitabine–abraxane chemotherapy combinations [2, 3], over a decade of clinical investigation in advanced pancreatic cancer had failed to significantly alter the therapeutic landscape beyond gemcitabine monotherapy. The epidermal growth factor receptor (EGFR) signaling pathway is frequently activated in advanced pancreatic cancer. While targeting EGFR kinase signaling with erlotinib alone has minimal activity [4], combining erlotinib with gemcitabine results in a statistically significant survival improvement in patients with advanced pancreatic cancer [5]. However, given the additional toxicities and the small magnitude of the effect, new EGFR targeting strategies or selection biomarkers are needed to optimize the use of EGFR inhibitors in patients with advanced pancreatic cancer.
Due to effects on the cell cycle, work by our group and others suggests that there is antagonism between chemotherapy and EGFR tyrosine kinase inhibitors (TKIs) when administered concurrently [6, 7]. In preclinical models, the administration of EGFR TKIs induces cytostasis due to G1 arrest, which antagonizes subsequent cell cycle phase-dependent activity of chemotherapy. Due to this hypothesized negative interaction of EGFR TKIs and chemotherapy, we proposed that temporal pharmacodynamic separation of the administration of erlotinib with gemcitabine would maximize the therapeutic potential of this combination.
Pancreatic adenocarcinomas frequently harbor activating mutations in the Kirsten rat sarcoma virus (KRAS) oncogene [8, 9]. As has been demonstrated in colorectal cancer, KRAS mutations may confer intrinsic resistance to EGFR inhibitors via oncogenic bypass, whereby constitutive activation of downstream signaling abrogates the role of the upstream kinase on tumor cell proliferation and survival [10]. In this trial, we tested a pharmacodynamic separation schedule for erlotinib and gemcitabine in patients with advanced pancreatic cancer. Additionally, we collected blood and tumor samples to interrogate components of EGFR signaling as potential biomarkers for the efficacy of this strategy.
Patients and methods
Patients
Adult patients with measurable, previously untreated locally advanced unresectable or metastatic pancreatic cancer with adequate organ function and Zubrod performance status 0–2 were eligible for this single arm phase II trial. Prior adjuvant chemotherapy was allowed provided that it was not gemcitabine and that it had been completed more than 6 months before study entry. No prior erlotinib was allowed. Submission of available tumor tissue was strongly encouraged but not required as an entry criterion. Further inclusion criteria included adequate organ function defined as leukocytes ≥3000/mm3, neutrophils ≥1500/mm3, platelets ≥100,000/mm3, bilirubin ≤1.5 times the upper limit of normal (ULN), AST and ALT ≤3 times ULN (≤5 × ULN for patients with liver metastases), and creatinine ≤1.5 times ULN or creatinine clearance ≥50 mL/min/1.73 m2 as measured by 24-h urine collection.
Exclusion criteria included treatment with other investigational agents, known brain metastases, and second primary malignancy within the previous 5 years apart from in-situ carcinoma or adequately treated non-melanomatous carcinoma of the skin. Because of the potential adverse treatment effects, pregnant women and patients with immune deficiency were excluded. Further exclusion criteria included uncontrolled intercurrent illness and history of allergic reactions attributed to compounds of similar chemical or biological composition to erlotinib or gemcitabine.
The study protocol was reviewed and approved by the institutional review boards of the University of California, Davis and the University of Southern California. All patients gave written informed consent before treatment.
Study procedures
Prior to registration, patients underwent history and physical examination including measurement of height, weight, performance status, vital status, and pregnancy test for women of reproductive potential. Tumor measurement was required within 4 weeks of study entry and was repeated every 8 weeks. Response was assessed using the Response Evaluation Criteria in Solid Tumors (RECIST) 1.0 criteria. History and physical examination were repeated every 4 weeks. Toxicity was monitored continuously throughout the trial using the Common Terminology Criteria for Adverse Events (CTCAE) version 3.0.
Treatment plan
Gemcitabine 1000 mg/m2 was administered as an intravenous infusion over 30 min on days 1, 8, and 15 of each 28-day cycle. Sequential reduced dose levels of 750 and 500 mg/m2 were given for severe gemcitabine-associated toxicities. Open label erlotinib was administered at a dose of 150 mg per day on days 2–5, 9–12, and 16–26 of each 28-day cycle. This schedule was designed to allow significant depletion of circulating erlotinib (≥2 half-lives) prior to administration of gemcitabine. Erlotinib was resumed 24 h after gemcitabine administration. The erlotinib dose could be reduced to 100 mg and then 75 mg per day on the same schedule for erlotinib-associated toxicities. Erlotinib-associated rash was managed symptomatically. If the erlotinib dose was reduced due to rash, the dose could be raised again as tolerated when the skin toxicity had improved by at least one grade level.
An absolute neutrophil count ≥1500/mm3 and platelet count ≥100,000/mm3 was required to start a cycle. Interval treatment (day 8 and 15) could proceed with an absolute neutrophil count ≥1000/mm3 and platelet count ≥75,000/mm3.
Correlative studies
Archival tumor specimens including paraffin-embedded tissue blocks containing formalin-fixed tumor or needle aspirate slides obtained prior to therapy were requested from all patients. Peripheral blood specimens (in EDTA) were obtained for correlative studies prior to each treatment cycle.
DNA was extracted from both peripheral blood and tumor specimens using the chemagen system (PerkinElmer) and the DNA concentration was quantified by NanoDrop (Thermo Scientific). The Scorpion amplification-refractory mutation system (ARMS) polymerase chain reaction (PCR) was utilized to detect KRAS mutations in codons 12 and 13 from both tumor and cell-free DNA (Qiagen). Additionally, plasma concentrations of the EGFR ligands amphiregulin (R&D Systems) and epigregulin (MyBioSource) were measured using enzyme-linked immunosorbent assays (ELISAs).
Statistical analysis
The primary endpoint of the trial was progression-free survival (PFS), defined from the day of registration to the first observation of disease progression or death due to any cause. We sought to improve the median PFS of approximately 3.75 months in a prior investigation with this drug combination to 5.25 months with pharmacodynamic separation. At the planned sample size of 70 patients, this study had 80 % power at 5 % significance to detect an increase in PFS from 3.75 to 5.25 months. PFS and overall survival (OS) were analyzed using the Kaplan–Meier method. Treatment response by RECIST criteria was examined as a secondary endpoint. Survival outcomes in biomarker subgroups were analyzed as exploratory endpoints. Amphiregulin results were dichotomized at the lower limit of the standard curve for the assay. Epiregulin and DNA concentration results were dichotomized at the median. Log-rank tests were used to compare survival distributions between biomarker groups with two-sided p values less than 0.05 considered statistically significant. The results of biomarker studies were not adjusted for multiple testing and are considered hypothesis generating.
Results
Baseline characteristics
Between November 2009 and July 2012, a total of 30 patients with locally advanced or metastatic pancreatic cancer were enrolled. The study was stopped after funding was terminated. The majority of patients had metastatic disease at the time of study entry and had good performance status (0 or 1 in 70 % of patients). Baseline characteristics for the entire cohort are summarized in Table 1.
Efficacy
The primary endpoint, median PFS, was 2.07 months (95 % CI; 1.87–5.50 months). The median OS of this treatment cohort was 5.67 months (95 % CI; 2.83–11.87 months). The median number of treatment cycles completed was 2 and ranged from 0 to 8. Two patients withdrew consent in the first cycle and were not evaluable for response. Responses are summarized in Table 2. The overall RECIST objective response rate was 11 %. There were no complete responses. Disease control (i.e. partial response or stable disease) was achieved in 46 % of patients.
Toxicity
Treatment-associated toxicity was observed in nearly all patients, but was similar to that described with gemcitabine and continuous erlotinib [5]. The most common events were hematologic and were primarily low grade (Table 3). Common non-hematological adverse events included rash (53 %), nausea (33 %), diarrhea (33 %), and fatigue (33 %). Grade 3 or higher adverse events occurred in 63 % of patients. The most common grade 3 or higher hematological adverse events were neutropenia and lymphopenia (23 % each); the most common severe non-hematological events were fatigue (13 %), rash, diarrhea, and nausea (10 % each).
Biomarkers
Archival tumor specimens were requested from each patient entering the trial. Tumor specimens were submitted for 20 patients (67 %). Of those, sufficient viable tumor for KRAS mutation testing was obtained from 9 patients (30 % of the entire study population). Pre-treatment plasma samples were available for 27 of 30 patients; these patients constituted the biomarker testing subgroup. OS outcomes were similar in the biomarker group to those in the overall study (Table 4). Tissue KRAS mutations were detected in 7 (78 %) of the 9 patients with sufficient available tumor. Plasma KRAS mutations were detected in the cell-free DNA from 10 (37 %) of 27 patients, including 3 (43 %) of those with known tumor KRAS mutations. Both patients without KRAS mutations detected in the tumor also did not have a mutation detected in the plasma. In the 3 patients with locally advanced pancreatic cancer with available plasma specimens, none had KRAS mutation detected in cell-free DNA. The most common plasma KRAS mutation detected was G12D (15 % of patients) followed by G12V and G12R (11 % of patients each).
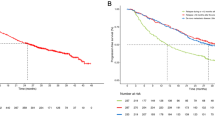
Patients with detectable mutant plasma KRAS had significantly lower median PFS (1.8 vs. 4.6 months, p = 0.014) and OS (3.0 vs. 10.5 months, p = 0.003) compared with those without KRAS mutations detected in the plasma (Table 4; Fig. 1). This effect was not explained by plasma DNA concentration, as DNA levels ≥ median did not predict shorter survival than low plasma DNA levels (9.0 vs. 5.0 months, p = 0.525). All three patients with partial response to treatment did not have a detected KRAS mutation in the plasma. Plasma amphiregulin and epiregulin did not correlate with PFS or OS.

a Progression-free survival of patients treated with pharmacodynamically separated gemcitabine and erlotinib by presence or absence of a detectable mutation in KRAS in pre-treatment cell-free DNA. b Overall survival of patients treated with pharmacodynamically separated gemcitabine and erlotinib by presence or absence of a detectable mutation in KRAS in pre-treatment cell-free DNA
Discussion
To our knowledge, this is the only clinical trial exploring the potential for a pharmacodynamic interaction between an EGFR TKI and cytotoxic therapy in advanced pancreatic cancer. This study was aborted before reaching its enrollment goal; nonetheless, we did not observe a promising signal for improved outcomes with pharmacodynamically separated erlotinib and gemcitabine in unselected patients. On the other hand, the presence of a KRAS mutation in pre-treatment cell-free plasma DNA detected using automated DNA isolation and high-sensitivity ARMS PCR was a strong predictor of survival for patients treated with this regimen.
Our results are in contrast to preliminary data from a similar phamacodynamic separation strategy of EGFR inhibitors in non-small cell lung cancer using docetaxel or pemetrexed [11, 12]. Possible reasons for this discrepancy include differences in cell cycle interactions with gemcitabine compared with other agents, differences in the cell cycle effects of erlotinib on pancreatic cancer versus non-small cell lung cancer, or failure of the schedule employed in this study to allow adequate pharmacodynamic separation. Alternatively, intrinsic resistance to erlotinib may be pervasive in patients with advanced pancreas cancer such that this study had insufficient power to detect any sequence-dependent effects.
A key finding from this study is the potential use of plasma KRAS mutation detection as a prognostic biomarker in advanced pancreatic cancer. Previous work has not confirmed tumor KRAS mutation as predictive of intrinsic resistance to this combination in pancreatic cancer [13]. Our analysis of KRAS mutations was limited by the small numbers of patients with concurrent tumor and plasma specimens. However, our ability to detect KRAS mutations in the circulating cell-free DNA in only a fraction of those patients with known tumor KRAS mutations is similar to the results from previous studies despite the higher sensitivity of the techniques for DNA extraction and KRAS mutation detection used in this study [14]. The detection of a KRAS mutation in this study was not a surrogate for cell-free DNA concentration, as there was no statistical difference in outcome between those with higher versus lower plasma DNA levels.
Few studies have addressed the prognostic significance of KRAS mutation detection in cell-free DNA; nevertheless, the results thus far are promising. With the use of restriction length polymorphism PCR, Castells and colleagues detected mutant KRAS in the cell-free DNA from 12 (27 %) of 44 patients with pancreatic ductal carcinoma. Survival rates at 6 and 12 months were significantly lower in patients with detected KRAS mutations compared with those without a detectable KRAS mutation in cell-free DNA (17 vs. 41 % survival at 6 months, and 0 vs. 24 % at 12 months) [15]. In a more recent study, Chen and colleagues were able to detect plasma KRAS mutations in 30 (33 %) of 91 pancreatic cancer patients. Median survival was only 3.9 months in those with mutant plasma KRAS, compared with 10.2 months in those without detected circulating KRAS gene mutations [16].
Our findings lend credence to the observation that detection of KRAS mutations in cell-free DNA predicts shorter survival in pancreatic cancer. Previous studies have not controlled for variability of therapeutic regimens in the analysis of outcome with respect to plasma KRAS. As all patients in this study were treated with the same regimen, the survival disadvantage in those with plasma KRAS mutations cannot be attributed to differences in therapy. We speculate that the release of KRAS-mutated DNA from the tumor into the plasma at levels detectable using ARMS PCR may be an indicator of more aggressive tumor biology; however, the mechanisms for generation of cell-free DNA from tumors are incompletely understood [17]. Given the absence of a randomized control arm in this study, further research is needed to determine if circulating mutant KRAS is primarily a prognostic indicator or if it has any role as a predictive biomarker for the efficacy of gemcitabine and erlotinib.
This study was aborted prior to reaching its planned accrual goal, limiting our ability to generalize the results of the study. Nonetheless, it is unlikely that we would have observed a substantially more promising result from pharmacodynamic separation with further accrual. Archival specimens were specifically requested as part of the protocol design but not required. Even so, specimens were submitted in only 67 % of patients and sufficient tumor for KRAS mutation testing was only available in 30 % of patients. These results highlight the challenges of biomarker studies in this patient population and the need for dedicated procurement procedures as recently proposed by an expert panel [18].
In conclusion, we observed no indication that pharmacodynamic separation of gemcitabine and erlotinib would improve the PFS of advanced pancreatic cancer patients. Different strategies are needed to optimize the use of EGFR inhibitors in advanced pancreatic cancer. The detection of a KRAS codon 12 mutation in cell-free DNA using a sensitive assay occurs in a fraction of patients with known intra-tumor KRAS mutation and is strongly associated with worse progression-free and overall survival in this population. If confirmed in larger studies, this biomarker could be used to identify a subgroup in need of alternative treatment options.
References
Siegel R, Naishadham D, Jemal A (2013) Cancer statistics, 2013. CA Cancer J Clin 63(1):11–30. doi:10.3322/caac.21166
Conroy T, Desseigne F, Ychou M et al (2011) FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med 364(19):1817–1825. doi:10.1056/NEJMoa1011923
Von Hoff DD, Ervin T, Arena FP et al (2013) Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med 369(18):1691–1703. doi:10.1056/NEJMoa1304369
Tang P, Gill S, Au HJ et al (2009) Phase II trial of erlotinib in advanced pancreatic cancer (PC). J Clin Oncol 27(15S):4609
Moore MJ, Goldstein D, Hamm J et al (2007) Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol 25(15):1960–1966. doi:10.1200/JCO.2006.07.9525
Davies AM, Ho C, Lara PN Jr et al (2006) Pharmacodynamic separation of epidermal growth factor receptor tyrosine kinase inhibitors and chemotherapy in non-small-cell lung cancer. Clin Lung Cancer 7(6):385–388. doi:10.3816/CLC.2006.n.021
Mahaffey CM, Davies AM, Lara PN Jr et al (2007) Schedule-dependent apoptosis in K-ras mutant non-small-cell lung cancer cell lines treated with docetaxel and erlotinib: rationale for pharmacodynamic separation. Clin Lung Cancer 8(9):548–553
Grunewald K, Lyons J, Frohlich A et al (1989) High frequency of Ki-ras codon 12 mutations in pancreatic adenocarcinomas. Int J Cancer 43(6):1037–1041
Smit VT, Boot AJ, Smits AM et al (1988) KRAS codon 12 mutations occur very frequently in pancreatic adenocarcinomas. Nucl Acids Res 16(16):7773–7782
Bardelli A, Siena S (2010) Molecular mechanisms of resistance to cetuximab and panitumumab in colorectal cancer. J Clin Oncol 28(7):1254–1261. doi:10.1200/JCO.2009.24.6116
Sangha R, Davies AM, Lara PN Jr et al (2011) Intercalated erlotinib-docetaxel dosing schedules designed to achieve pharmacodynamic separation: results of a phase I/II trial. J Thorac Oncol 6(12):2112–2119. doi:10.1097/JTO.0b013e31822ae061
Davies AM, Ho C, Beckett L et al (2009) Intermittent erlotinib in combination with pemetrexed: phase I schedules designed to achieve pharmacodynamic separation. J Thorac Oncol 4(7):862–868. doi:10.1097/JTO.0b013e3181a94b08
da Cunha SG, Dhani N, Tu D et al (2010) Molecular predictors of outcome in a phase 3 study of gemcitabine and erlotinib therapy in patients with advanced pancreatic cancer: National Cancer Institute of Canada Clinical Trials Group Study PA.3. Cancer 116(24):5599–5607. doi:10.1002/cncr.25393
Marchese R, Muleti A, Pasqualetti P et al (2006) Low correspondence between K-ras mutations in pancreatic cancer tissue and detection of K-ras mutations in circulating DNA. Pancreas 32(2):171–177. doi:10.1097/01.mpa.0000202938.63084.e3
Castells A, Puig P, Mora J et al (1999) K-ras mutations in DNA extracted from the plasma of patients with pancreatic carcinoma: diagnostic utility and prognostic significance. J Clin Oncol 17(2):578–584
Chen H, Tu H, Meng ZQ et al (2010) K-ras mutational status predicts poor prognosis in unresectable pancreatic cancer. Eur J Surg Oncol 36(7):657–662. doi:10.1016/j.ejso.2010.05.014
Schwarzenbach H, Hoon DS, Pantel K (2011) Cell-free nucleic acids as biomarkers in cancer patients. Nat Rev Cancer 11(6):426–437. doi:10.1038/nrc3066
Tempero MA, Klimstra D, Berlin J et al (2013) Changing the way we do business: recommendations to accelerate biomarker development in pancreatic cancer. Clin Cancer Res 19(3):538–540. doi:10.1158/1078-0432.CCR-12-2745
Acknowledgments
Research support was provided by Genentech BioOncology and the UC Davis Cancer Center Support Grant, P30CA093373-06. The research reported was also supported by the National Cancer Institute of the National Institutes of Health under Award Number K12CA138464. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Study Identifier: NCT00810719. Presented in part at the 2013 ASCO Annual Meeting (Abstract e15073).
Conflict of interest
Thomas J. Semrad has received honoraria as a consultant to Genentech and has received research grants from Genentech; Afsaneh Barzi has no conflict of interest; Heinz-Josef Lenz has received honoraria from Genentech and has received research grants from Genentech; Irene M. Hutchins has no conflict of interest; Edward J. Kim has no conflict of interest; I-Yeh Gong has no conflict of interest; Michael Tanaka has no conflict of interest; Laurel Beckett has no conflict of interest; William Holland has no conflict of interest; Rebekah A. Burich has no conflict of interest; Leslie Snyder-Solis has no conflict of interest; Philip Mack has no conflict of interest; Primo N. Lara, Jr. serves as a consultant to Genentech and has received research grants from Genentech including for the conduct of this study.
Author information
Authors and Affiliations
Corresponding author
About this article

Cite this article
Semrad, T., Barzi, A., Lenz, HJ. et al. Pharmacodynamic separation of gemcitabine and erlotinib in locally advanced or metastatic pancreatic cancer: therapeutic and biomarker results. Int J Clin Oncol 20, 518–524 (2015). https://doi.org/10.1007/s10147-014-0730-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10147-014-0730-2