
Abstract
Alternative splicing is a crucial mechanism of gene expression regulation that enormously increases the coding potential of our genome and represents an intermediate step between messenger RNA (mRNA) transcription and protein posttranslational modifications. Alternative splicing occupies a central position in the development and functions of the nervous system. Therefore, its deregulation frequently leads to several neurological human disorders. In the present review, we provide an updated overview on the impact of alternative splicing in Parkinson’s disease (PD), the second most common neurodegenerative disorder worldwide. We will describe the alternative splicing of major PD-linked genes by collecting the current evidences about this intricate and not carefully explored aspect. Assessing the role of this mechanism on PD pathobiology may represent a central step toward an improved understanding of this complex disease.
Similar content being viewed by others

Introduction
The flow of genetic information from DNA to RNA to protein has traditionally been considered the central dogma of molecular biology. Additional steps of regulation are currently well known, greatly expanding this simplistic framework and revealing the complex network that controls gene expression [1]. One of these steps is represented by alternative splicing (AS), whereby a single gene gives rise to multiple messenger RNA (mRNA) transcripts and protein isoforms with different functional properties [1]. It is estimated that 94 % of human protein-coding genes are alternatively spliced [2, 3], and the main site of alternative splicing events is the central nervous system [4, 5].
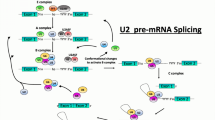
The alternative splicing process consists in the removal of the intronic regions from the RNA primary transcript and simultaneous assembly of the exonic regions in different combinations to form a mature mRNA, which is then polyadenilated, exported to the cytoplasm, and translated into protein. The accuracy and efficiency of pre-mRNA splicing process depend on a range of constitutive DNA sequence motifs: the donor and the acceptor splice sites, the lariat branch point, the polypyrimidine tract, and splicing enhancers and silencers (Fig. 1, panel a). These motifs are recognized by a large macromolecular splicing machinery (called the spliceosome), which models the pre-mRNA while RNA polymerase II synthesizes it in the nucleus. The splicing machinery includes five spliceosomal uridine-rich small nuclear ribonucleoproteins (snRNPs) (U1, U2, U4, U5, and U6) and several non-snRNP protein splicing factors such as the serine/arginine (SR)-rich protein family and hnRNP proteins [6, 7]. The splicing reaction relies on two transesterification steps that occur within the highly dynamic splicing machine. The stepwise molecular mechanisms of the splicing reaction are detailed in Fig. 1 (panel a).

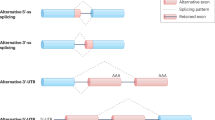
The alternative splicing mechanism. a Four main conserved DNA sequence motifs allow the splicing mechanism: the donor splice site GU (5′ SS), the acceptor splice site AG (3′ SS), the lariat branch point (A) located upstream of the acceptor site and the polypyrimidine tract (PPT) placed between the acceptor site and the branch point. The splicing machinery includes mainly five spliceosomal uridine-rich small nuclear ribonucleoproteins (snRNPs) (U1, U2, U4, U5, and U6) and further auxiliary RNA binding proteins. During the first step of spliceosome assembly, U1 snRNP base pairs with the 5′ splice site of the pre-mRNA (E complex), whereas U2 base pairs with the branch point (A complex). Then the tri-snRNP complex U4, U5, and U6 associates with the forming spliceosome (B complex), and both U1 and U4 are ejected. This allows U6 to replace U1 at the 5′ splice site (C complex) and leads to a U6–U2 interaction that gets close together the 5′ splice site and the branch point, allowing for a transesterification step. At the end, U5 brings near the two exons, joining them through a second transesterification reaction. b Five major alternative splicing events are currently known: exon skipping/inclusion, use of alternative 3′ splice site, use of alternative 5′ splice site, mutually exclusive exons, and intron retention. In blue, are represented the constitutive exons. Yellow and red represent the alternatively spliced exons. The splicing events rely on the interplay between the constitutive splicing motifs, the splicing regulatory sequences, the RNA secondary structures, the components of the spliceosome, and further auxiliary RNA-binding proteins. However, how the spliceosome decides which exons to include remains currently not clear
Alternative splicing works as an on–off switch in gene expression. It affects the expression levels, stability, half-life [via the nonsense-mediated mRNA decay (NMD)], and localization of the RNA messengers. It has also the potential to generate several protein isoforms with different biological properties, protein–protein interactions, subcellular localization, signaling pathway, or catalytic ability. During the last years, great efforts have been made to decipher the intricate alternative splicing code. Five major alternative splicing events (i.e., cassette exons, use of alternative acceptor and/or donor sites, intron retention, and mutually exclusive exons) have been described up to now and are detailed in Fig. 1 (panel b) [2, 8]. However, how the spliceosome recognizes alternative exons and decides which exons to include remains not fully understood. Undoubtedly, there is more diversity in splice transcript variants than in protein isoforms. Although this is still not clear, different variants encode the same protein, but probably translate it with different efficiencies [9].
The finely tuned splicing regulatory network can easily undergo alterations. An aberrant alternative splicing may arise from changes in regulatory sequences required for correct pre-mRNA processing (the so-called cis-acting mutations), as well as from mutations that affect components necessary for splicing regulation (trans-acting mutations). Cis- and trans-splicing aberrations represent direct causative agents of disease or more subtle contributions to the determinants of disease susceptibility or modulators of disease severity. An extensive range of neurological diseases has been already associated to both splicing defects, including Alzheimer’s disease, retinitis pigmentosa, spinal muscular atrophy, muscular dystrophy, neurofibromatosis, and fragile X-associated tremor/ataxia syndrome [10–12, 1, 13]. In this broad neurological disorder scenario, the relevance of alternative splicing in Parkinson’s disease (PD) is not still clear, and the splicing mechanisms that regulate PD-related genes remain mostly unknown.
Here, we provide an updated overview of the current knowledge about the impact of alternative splicing on Parkinson’s disease. Firstly, we will take into account the most common PD-related genes “one by one” by analyzing their alternative transcripts currently known and their involvement in this disease. Then, we will describe the few studies that have globally analyzed the changes of splice variant expression in PD patients through genome-wide RNA expression approaches. Finally, we will briefly describe the current evidences about the alternative splicing modulation in PD through noncoding RNAs [microRNA (miRNA) and long noncoding RNA (lcnRNA)].
Genetics of Parkinson’s disease
PD is the second most common neurodegenerative disorder worldwide, characterized by resting tremor, bradykinesia, stiffness of movement, and postural instability. These symptoms are derived from the progressive loss of neurons from the substantia nigra pars compacta, coupled with an accumulation of intraneuronal aggregates called Lewy bodies.
Despite significant progresses in the understanding of PD pathogenesis, the exact etiology of PD remains unknown. Over the past 15 years, an even more detailed knowledge of the genetic factors that contribute to PD has emerged through different research strategies [14, 15]. Linkage mapping analysis, genome-wide association studies (GWAS), and next-generation sequencing technologies are revealing an increasing number of locus and genes strongly linked to either autosomal dominant (SNCA-PARK1, LRRK2-PARK8, VPS35-PARK17, and GBA), or typical recessive (PARKIN-PARK2, PINK1-PARK6, and DJ1-PARK7) and atypical recessive (ATP13A2-PARK9, PLA2G6-PARK14, and FBXO7-PARK15) or X-linked (ATP6A2 and TAF1) forms of disease. For the sake of completeness, we mention here further monogenic loci, not confirmed genes, or risk factor genes (i.e., PARK3, UCHL1, PARK10, GIGYF2, PARK12, HTRA2, PARK16, EIF4G1, DNAJ, HLA-DR, GAK-DGKQ, SYNJ1, and GBAP1) [15–17]. Furthermore, a large-scale meta-analysis of genome-wide association data is revealing a wide range of additional loci having genome-wide significant association [18]. However, we will overlook their discussion because of the few data in the literature regarding their splicing regulation in pathological conditions.
In the next paragraphs, we will describe the alternative spliced mRNA variants of PD genes and the current scientific data demonstrating their involvement in PD pathogenesis. For a more complete picture, we have also added some further implicated genes (SRRM2, MAO-B, SNCAIP, MAPT, and GBA), indicated as other PD-related genes, which are not directly causative genes, but whose splicing regulation seems to be altered in PD states.
Autosomal dominant PD genes
SNCA
Alpha-synuclein, encoded by SNCA gene, is a small, natively unfolded presynaptic protein linked to PD [19]. Aggregates of alpha-synuclein protein represent the neuropathological hallmark lesions of PD and constitute the major components of Lewy bodies. Genetically, mutations in SNCA gene were the first to be associated with PD family inheritance. Missense mutations in coding regions (Ala53Thr, Ala30Pro, and Glu46Lys), single nucleotide substitution in 3′ untranslated region (3′ UTR), and dose-dependent genomic multiplications (duplications or triplications) of the gene cause both monogenic and sporadic forms of PD [20, 19, 21]. Some point mutations in splice donor sites have also been reported (IVS2 + 9A > C) [22].
SNCA gene maps to chromosome 4q22.1 and contains six exons spanning about 114 kb [21]. The set of mRNAs produced by SNCA gene includes the full-length transcript, commonly known as SNCA-140 from the amino acidic length of the encoded protein, and corresponds to SNCA-001, SNCA-002, SNCA-003, SNCA-006, and SNCA-008 mRNAs from Ensembl library (Table 1 and Fig. 2). Further additional splicing variants, known as SNCA-126, SNCA-112, and SNCA-98 and corresponding to (i) SNCA-004, SNCA-203, SNCA-201, (ii) SNCA-005, SNCA-202, and (iii) SNCA-010, respectively, are generated by in-frame excision of exons 3, 5, or both (Table 1 and Fig. 2). Two additional splice variants (SNCA-009 and SNCA-007) are generated from an inner transcription start and encode proteins of 115 and 97 amino acids, respectively (Table 1 and Fig. 2). SNCA-140, SNCA-126, and SNCA-112 are expressed in a broad spectrum of human tissues, while SNCA-98 seems to be a brain-specific splice variant with varying expression levels in different areas of fetal and adult brain [23].

Structures of the alternative splicing variants of human dominant PD genes. Structures of the described mRNA splicing variants are represented in the figure as reported in Ensembl library (http://www.ensembl.org/index.html). On the left, each variant is indicated with a number corresponding to that indicated in Table 1. LRRK2 gene is illustrated in 5′-3′ sense, while SNCA and VPS35 genes are illustrated in antisense corresponding to their 3′-5′ sense transcription
The expression profile of SNCA-140, SNCA-126, SNCA-112, and SNCA-98 splice variants is different in the various brain areas under normal and pathological states. Compared to healthy controls, in PD frontal cortex, all these four transcripts are overexpressed, with significant upregulation of SNCA-126 [24]. In PD substantia nigra, only the three shorter transcripts have been observed significantly overexpressed [25, 26], while higher SNCA-112 and SNCA-98 levels are also present in the cerebellum [25]. Different expression profiles of SNCA variants also occur in other forms of neurodegenerative disorders. Both SNCA-140 and SNCA-126 downregulation and SNCA-98 overexpression have been reported in dementia with Lewy bodies and Alzheimer’s disease, while SNCA-112 is upregulated in dementia with Lewy bodies and downregulated in Alzheimer’s disease [27, 28, 24].
Some interesting data emerge on SNCA-112 variant. An association between PD risk-associated single nucleotide polymorphisms (SNPs) within the 3′ region of SNCA gene and higher SNCA-112 ratio level has been observed in about 100 of frontal cortex samples. These data reveal the cis-regulatory effect of these mutations on splicing mechanism [29]. The expression of SNCA-112 is also abundantly induced by some parkinsonism mimetics (MPP+, rotenone) and related oxidants [30]. However, the reason for these effects remains unclear.
In addition to splice variants, specific RNA transcript isoforms of SNCA with an extended 3′ untranslated region have been described and appear selectively linked to pathological processes [31]. However, this review is focusing only on the mRNA splice variants; thus, their discussion will be omitted.
The 140 amino acid isoform is a small protein with a molecular weight of 14.5 kDa. It is composed of three distinct regions: (1) an amino terminus containing amphipathic helices conferring the propensity to bind membranes; (2) a central hydrophobic region, the so-called non-Ab component (NAC), which confers the b-sheet potential; and (3) an acid glutamatergic carboxyl terminus that is highly negatively charged and prone to be unstructured [19]. Structural changes in the shorter splicing isoforms can be predicted as a result of exon skipping events. SNCA-126-predicted isoform shows interruption of the N-terminal protein–membrane interaction domain [32]; SNCA-112 is significantly shorter in the unstructured C-terminal [32], while SNCA-98 isoform results in a truncated protein consisting almost only of the central region containing NAC [23]. Recently, a lower aggregation propensity of the shorter isoforms has been demonstrated in vitro [33]. In addition, morphology studies by using electron microscopy have shown straight fibrils for SNCA-140, shorter fibrils mostly arranged in parallel arrays for SNCA-126, and circular structures for SNCA-98 [33]. These data open new insights regarding the formation of Lewy bodies induced by alpha-synuclein.
Numerous functions of alpha-synuclein have been proposed, counting molecular chaperone, regulator of dopamine uptake and homeostasis, inhibitor of phospholipase D2, downregulator of p53 pathway [32], and promoter of the SNARE-complex assembling [34]. Unfortunately, nothing is known about the specific pathophysiological roles of each alpha-synuclein isoform and their relative posttranslational modifications (i.e., phosphorylations, nitration, sumoylation, oxidation, glycosylation, cleavage, and ubiquitination), which are known to play a key role in SNCA functions and regulation [32].
LRRK2
LRRK2 encodes for leucine-rich repeat kinase 2 (or dardarin), which is a large 2527 amino acid multidomain protein. The protein consists of multiple conserved well-defined domains including a small GTPase-like domain (Ras of complex proteins or ROC), a domain of unknown function termed the C-terminal of ROC (COR), a kinase domain, as well as several protein interaction domains [e.g., the leucine-rich repeat (LRR), the WD40 domain, the ankyrin repeat domain, and the armadillo repeat region]. The precise physiological function of LRRK2 is unknown. However, LRRK2 seems implicated in different cellular functions as neurite outgrowth, cytoskeletal maintenance, vesicle trafficking, and autophagic protein degradation [35].
The LRRK2 gene spans a genomic region of 144 kb, with 51 exons, and harbors the most common mutations linked to both autosomal dominant inherited late-onset and sporadic PD. The missense mutations known so far are spread over the whole LRRK2 gene and affect all functional domains. Some mutations have much higher frequencies than others, such as Gly2019Ser and mutations altering codon Arg1441, respectively, in the kinase and ROC domains. In addition, several unclear pathogenic mutations affecting splice sites have been observed (IVS19 + 5_8delGTAA, IVS25-8delT, IVS27-9C > T, IVS30-6C > T, IVS31 + 3A > G, IVS32 + 14G > A, IVS33 + 6 T > A, IVS37-9A > G, IVS38 + 7C > T, IVS46-14 T > A, and IVS46-8delT) [36, 22, 37–43].
In addition to the full-length transcript (LRRK2-004), further LRRK2 shorter transcripts are deposited in Ensembl library (Table 1 and Fig. 2). Despite the existence of these transcripts, there are currently no data analyzing the splicing profile of this gene in PD states. Recently, a gene expression and splicing analysis of the LRRK2 locus have been carried on [44]. Both exon array and RT-PCR methods confirm the existence of an isoform with spliced out exons 32–33 in the substantia nigra and an isoform with exon 32 alone spliced out in the occipital cortex, medulla, and cerebellum of healthy humans [44].
Further evidences on LRRK2 splicing have been observed by Giesert and collaborators [45], who have conducted a study in various brain regions and organs from adult mice. In this regard, it should be considered that LRRK2 is highly conserved in human and mouse and that several transgenic animal models have been created. Giesert et al. [45] have identified two LRRK2 splice variants: one with skipped exon 5, primarily expressed in astrocytes, and another truncated variant terminating with an alternative exon 42a barely detectable in the microglia but highly expressed in neurons and astrocytes. Protein-structure predictions reveal that the loss of exon 5 may generate a smaller protein with changed affinity of binding partners, while the alternative exon 42a may lead to changes of its enzymatic activity. In addition, the protein-interaction domain WD40 would also be absent in such truncation. Interestingly, the deletion of this domain in the Zebrafish LRRK2 ortholog (zLRRK2) causes parkinsonism-like phenotype including loss of dopaminergic neurons in diencephalon and locomotion defects [46]. Further studies will need to assess the involvement of LRRK2 alternative splice variants in PD.
VPS35
In 2011, two groups reported the identification of the same missense mutation (p.Asp620Asn) in the vacuolar protein sorting 35 (VPS35) gene as a novel cause of autosomal dominant PD [47, 48]. VPS35 was the first PD gene found by a direct whole exome sequencing in large families of Austrian and Swiss origins. An in-depth sequence analyses of all coding, noncoding, and exon–intron boundaries VPS35 genetic regions have been performed in a large well-characterized cohort of Lewy body disorders, including PD patients, PD with dementia, and dementia with Lewy bodies [49]. In addition to three novel missense mutations, silent and intronic variations, predicted to activate cryptic splice sites, have been observed in the patient’s group but not in controls. However, the pathogenicity of these mutations was not completely conclusive since these mutations were not supported by segregation analysis in family relatives [49].
Various spliced transcript variants of this gene are reported in Ensembl library (Table 1 and Fig. 2), but the majority of them are processed for degradation and do not encode proteins.
Autosomal recessive PD genes
Early-onset typical PD genes
PARK2
Mutations in PARK2 gene (also known as PARK2 parkin RBR E3 ubiquitin-protein ligase) are the most common cause (50 % of cases) of autosomal recessive juvenile parkinsonism (AR-JP), a form of early-onset parkinsonism characterized by good and prolonged response to levodopa and a benign, slow course. PARK2 mutations also explain ~15 % of the sporadic cases with onset before 45 [50, 51] and act as susceptibility alleles for late-onset forms of Parkinson’s disease (2 % of cases) [52]. Along with about 200 mutations currently identified in PARK2 coding region, several point mutations in splice acceptor or donor sites (introns 1, 6, 7, 10, 12, 13, and 16) have been identified in PD patients [53–57, 22, 58, 59].
PARK2 gene spans more than 1.38 Mb of genomic DNA in the long arm of chromosome 6 (6q25.2–q27) and contains 12 exons, which are alternatively spliced to produce at least 11 different splicing variants (Table 2 and Fig. 3) [59]. The full-length PARK2 transcript (PARK2-004) encodes a protein of 465 amino acids (parkin) [60, 61, 59] acting in numerous molecular pathways (protein turnover, stress response, mitochondrial homeostasis, mitophagy, mitochondrial DNA stability, metabolism, cell growth, and survival) [62]. Multiple parkin isoforms likely arising from PARK2 splicing variants have been observed in different brain areas through Western blot studies [9].

Structures of the alternative splicing variants of human recessive PD genes. Structures of the described mRNA splicing variants are represented in the figure as reported in Ensembl library (http://www.ensembl.org/index.html). On the left, each variant is indicated with a number corresponding to that indicated in Table 2. All transcripts are illustrated in 5′-3′ sense, except PARK2, ATP13A2, and PLA2G6 genes, which are illustrated in antisense corresponding to their 3′–5′ sense transcription
The extensive alternative splicing of PARK2 is differently regulated both at transcript and protein level in tissues and cells [63, 64, 24, 65–68]. Distinct expression patterns of PARK2 splice variants emerge in human brain regions [69] and leukocytes [68], in rat brain, in neuronal and glial cells [67], and in a wide variety of mouse tissues (brain, heart, lung, liver, skeletal muscle, kidney, and testis) [64]. At the protein level, PARK2 protein isoforms show a differential distribution in human leukocytes [70] and aged brain [71], as well as in different rat and mouse nervous system areas (cerebral cortex/diencephalons, hippocampus, cerebellum, brainstem, striatum, spinal cord, and substantia nigra), peripheral tissues (heart, liver, spleen, pancreas, and kidney), and developmental stages [72–76].
Emerging evidences support the importance of PARK2 splice variant expression changes in disease development. Differential expression of PARK2 transcripts have been identified in the frontal cortex of Parkinson’s disease, pure dementia with Lewy bodies, common Lewy body disease, and Alzheimer’s disease patients, compared to controls [65, 24]. Particularly, two PARK2 splicing variants are significantly overexpressed in PD [65]. Another study reports both an increase in the expression level of a parkin splice variant and a decrease of the wild type between PD patients and healthy controls [66]. The differential and disease-specific expression profiles of PARK2 alternative splice variants suggest a role for splicing deregulation in the development of neurodegenerative disorders.
PINK1
Homozygous or compound heterozygous loss-of-function mutations in PTEN-induced putative kinase 1 (PINK1) are the second most frequent cause of autosomal recessive early-onset parkinsonism. Mutation frequency varies geographically from 1 to 9 % depending on ethnic background [77]. The PINK1 mutation spectrum involves nonsense and missense mutations, insertions, or deletions, and whole gene or single/multiple exon copy number variants located across the entire gene [78].
PINK1 gene maps in the short arm of chromosome 1 (1p36.12), encompassing ~18 kb of genomic DNA. Its coding sequence is spread over eight exons. In addition to the full length (PINK1-001), two shorter variants exist but do not produce proteins (Table 2 and Fig. 3).
Some interesting findings emerge regarding the splicing regulation of exon 7. A 23-bp deletion disrupting the splice acceptor site of exon 7 has been detected in a sporadic parkinsonian patient, producing several aberrant mRNAs [79]. Moreover, whole exon 7 deletion and a novel U1-dependent 5′ splice-site mutation in exon 7 have been found in a large Spanish family with PD members [80].
The PINK1 protein is a putative serine/threonine kinase of 581 amino acids involved in mitochondrial response to cellular and oxidative stress [81]. It has been demonstrated that at least two isoforms are expressed in the human brain: a full-length protein of ~63 kDa and an N-terminally truncated isoform of 52 kDa [77, 82–84]. An additional isoform of approximately 45 kDa has been suggested, although it has not been extensively studied [85]. The 52-kDa isoform seems to originate by enzymatic cleavage of PARL [86]; however, the exact nature of the isoforms, the precise reason for the cleavage, and the functional roles of these three different isoforms require further studies.
DJ1
Mutations in the DJ1 (also known as PARK7) gene are the less common cause of autosomal recessive parkinsonism (~1 % of early-onset PD) [87, 88]. A large homozygous deletion and a missense mutation (L166P) in DJ-1 gene were first identified in both Italian and Dutch consanguineous families [89, 90]. Additional mutations have been collected in other PD families and include missense mutations in coding and promoter regions, frame shifts, copy number variations [91, 88], and splice site alterations [92, 93].
DJ-1 gene maps to chromosome 1 (1p36.23) and includes seven exons. Several spliced transcript variants have been identified encoding the same protein (Table 2 and Fig. 3). Two shorter transcripts (the first lacking exon 4 and the second starting in an inner transcription point) encode for smaller proteins (Table 2 and Fig. 3).
The product of DJ-1 gene is a highly conserved protein of 189 amino acids belonging to the peptidase C56 family [94]. It is a multifunctional protein, acting as a positive regulator of transcription, redox-sensitive chaperone, sensor for oxidative stress, and apparently protects neurons from ROS-induced apoptosis [95, 96]. In the human brain and peripheral blood, several DJ-1 isoforms exist and differ on their isoelectric point (pI) [97–100]. The relative abundance of these different DJ-1 isoforms appears to be altered in PD, and therefore, blood DJ-1 isoforms have been proposed as potential biomarkers for Parkinson’s disease [101]. The different pI of each variant are believed to result from posttranslational modifications that alter the intrinsic charge of the protein [101]. Interestingly, it has been demonstrated that one of the major binding partners of DJ-1 in dopaminergic neuronal cells is the splicing factor proline/glutamine-rich (SFPQ protein) [96, 102]. SFPQ, originally identified as a polypyrimidine tract-binding protein, is part of the spliceosome C complex and is required for in vitro splicing of pre-mRNA [96, 102]. DJ-1 binding to SFPQ modulates its transcriptional activity and, therefore, tunes its effect on splicing regulation. DJ-1 mutations could reverberate on its downstream targets, including the splicing factor SFPQ and altering the splicing control.
Juvenile atypical PD genes
ATP13A2
ATP13A2 mutations are associated with Kufor–Rakeb syndrome, a form of recessively levodopa-responsive inherited atypical parkinsonism [103]. It encodes a large protein belonging to the ATPase transmembrane transporters, and recently, it has been identified as a potent modifier of the toxicity induced by alpha-synuclein [104].
ATP13A2 is composed of 29 exons and lies on chromosome 1 covering about 26 kb of genomic DNA. One of the first identified disease-causing mutations was a guanine-to-adenine transition in the donor splice site of exon 13, leading to the skipping of exon 13 and resulting in a deletion of part of the third transmembrane domain [105].
According to data repositories, at least 15 alternatively spliced transcripts are expressed in humans (Table 2 and Fig. 3). The longest transcripts are ATP13A2-001, ATP13A2-002, and ATP13A2-005. Transcript variants ATP13A2-001 and ATP13A2-005 differ only in a nucleotide segment on exon 5, while transcript variant ATP13A2-002 lacks exons 22 and 28. The ATP13A2 mRNA is highly expressed in the brain, particularly in the substantia nigra of patients with classical late-onset PD [91]. However, nothing is known about the splicing expression profiles of this gene in PD and healthy subjects.
The products of these transcripts have been studied at the protein level. The isoform 1 encoded by ATP13A2-001 is a protein of 1180 amino acids with ten transmembrane domains. Isoform 2 encoded by ATP13A2-005 contains a five amino acid deletion near the N-terminus, while isoform 3, encoded by ATP13A2-002, contains two deletions, generating a highly diverged C-terminus [105]. Functional studies have shown that the isoform 1 is located in the lysosome membrane, whereas the isoform 3 protein is retained in the endoplasmic reticulum and rapidly degraded by the proteasome. In addition, both isoforms 1 and 3 are eliminated via the endoplasmic reticulum-associated degradation pathway [105].
PLA2G6
Recessive mutations in the phospholipase A2 group VI (PLA2G6) gene have been initially described as the cause of infantile neuroaxonal dystrophy and neurodegeneration associated with brain iron accumulation. Recently, this gene has also been associated with a particular parkinsonian phenotype, consisting of levodopa-responsive dystonia, pyramidal signs, and cognitive/psychiatric features, with onset in early adulthood [106]. Among PLA2G6 identified mutations, the c.1077G > A mutation at the last nucleotide of exon 7 (apparently a synonymous mutation) stands out as a cause of abnormal mRNA splicing. This single nucleotide substitution causes the activation of a cryptic splice site producing a 4-bp deleted transcript with altered frame shift in leukocytes [106].
PLA2G6 gene maps on chromosome 22 (q13.1), covering 70 kb of genomic DNA. Several transcript variants encoding multiple isoforms have been described up to now (Table 2 and Fig. 3). The longest PLA2G6 mRNA PLA2G6-001 includes 17 exonic regions and encodes the 85/88 kDa calcium-independent phospholipase known as A2 isoform a. The other two long transcripts (PLA2G6-002 and PLA2G6-201) differ in the start point, both lack of exon 9 and encode the same protein, called isoform b. The expression profile of this gene in healthy and disease states remains unknown.
FBXO7
Mutations in the F-box only protein 7 (FBXO7) gene cause parkinsonian pyramidal disease (PPD- or PARK15-associated parkinsonism), an autosomal recessive neurodegenerative disease with juvenile onset, severe levodopa-response, and additional pyramidal signs. Some pathogenic mutations have been identified (R378G, R498X, and T22M) including a compound heterozygous mutation (IVS7 + 1G/T) that removes the invariable splice donor of intron 7 and may disrupt FBXO7 messenger RNA splicing [107–109].
The FBXO7 gene, mapped on chromosome 22q12.3, contains nine exons spanning about 24.1 kb. It encodes a 522 amino acid protein consisting of several domains [108], which target proteins for ubiquitination [108]. Alternatively spliced transcript variants of this gene have been identified (Table 2 and Fig. 3) [107]. FBXO7-001 is the longest and more abundant transcript ubiquitously expressed [110], particularly in skin fibroblasts [111]. FBXO7-002 arises from an inner alternative exon 1, differs in the start codon, and produces a shorter isoform. Both these encoded protein isoforms have been detected in cells [111].
X-linked parkinsonism
X-linked dystonia parkinsonism (XDP) is an X-linked recessive adult-onset movement disorder characterized by both dystonia and parkinsonism. TATA-box binding protein-associated factor 1 (TAF1) gene, located in the disease locus Xq13.1, has been reported as the first related XPD gene, harboring disease-specific single-nucleotide changes and a small deletion within the multiple transcript panel [112]. This gene is part of a complex region of DNA (the TAF1/DYT3 multiple transcript systems), which encompasses the exonic regions of TAF1 gene and further additional downstream exons [112, 113]. This system includes multiple different transcription start sites and encodes multiple spliced transcripts and isoforms (Table 3 and Fig. 4) [112].

Structures of the alternative splicing variants of human X-linked PD genes. Structures of the described mRNA splicing variants are represented in the figure as reported in Ensembl library (http://www.ensembl.org/index.html). On the left, each variant is indicated with a number corresponding to that indicated in Table 3. All transcripts are illustrated in 5′-3′ sense
Recently, the ATP6AP2 gene (Table 3 and Fig. 4) has been proposed as a novel gene for X-linked parkinsonism with spasticity (XPDS) by exome sequencing analysis [114]. A silent mutation (p.S115S) in the ATP6AP2 gene has been identified in one affected individual, resulting in the aberrant splicing of ATP6AP2 mRNA and the overexpression of a minor splice isoform [114]. Noteworthy, the ATP6AP2 is an essential accessory component of the vacuolar ATPase required for lysosomal degradative functions and autophagy, a pathway frequently affected in PD.
Other PD-related genes
SNCAIP
Synphilin-1, encoded by SNCAIP gene, is a presynaptic protein containing several protein–protein interaction motifs, including ankyrin-like repeats, a coiled-coil domain, and an ATP/GTP-binding domain [115]. It interacts strongly with alpha-synuclein in neuronal tissue and may play a role in the formation of Lewy bodies during neurodegeneration. It is also implicated in parkinsonism as one of the parkin substrates. In addition, some studies have identified SNCAIP sequence variants in PD patients and have suggested it as a candidate PD gene [116, 117].
SNCAIP gene maps on chromosome 5 (5q23.2) and spans about 152 kb of genomic DNA. Although the database Gene reports SNCAIP composed of 11 exons, additional exonic regions emerge by aligning the sequence of the gene with each transcript. To date, at least 22 alternative spliced transcript variants have been identified (Table 4 and Fig. 5), but the most studied are synphilin-1 and 1A. Synphilin-1 (SNCAIP-001) is the full-length transcript, while synphilin-1A variant is a shorter form (SNCAIP-201). The latter lacks exons 4 and 5 and contains an extra exon located between exons 10 and 11. Synphilin-1A isoform is thought to be involved in the pathogenesis of PD and may play an important role in the formation of Lewy bodies [118–120]. Interestingly, synphilin-1A protein shows enhanced aggregation properties, which cause neuronal toxicity [118–120].

Structures of the alternative splicing variants of the other human PD-related genes. Structures of the described mRNA splicing variants are represented in the figure as reported in Ensembl library (http://www.ensembl.org/index.html). On the left, each variant is indicated with a number corresponding to that indicated in Table 4. All transcripts are illustrated in 5′-3′ sense, except MAO-B and GBA genes, which are illustrated in antisense corresponding to their 3′-5′ sense transcription
The mRNA expression levels of synphilin 1, 1A, and other two additional synphilin variants have been simultaneously investigated in the frontal cortex of PD patients. Their overall overexpression has been demonstrated when compared to healthy controls [24, 65].
MAPT
MAPT gene encodes the microtubule-associated protein tau, a protein involved in microtubule assembly and stability [121]. It is located on chromosome 17q21 and contains 15 exons. It gives rise to multiple splice transcripts (Table 4 and Fig. 5) which are differentially expressed in human tissues [11]. In the adult human central nervous system, MAPT splicing generates six tau isoforms composed of either three or four microtubule-binding repeat motifs in the C-terminal (3R- and 4R-tau).
A number of mutations within and around MAPT exon 10 disrupt exonic and intronic splicing elements as well as the formation of an RNA stem-loop structure at the 5′ splice site (which normally functions to restrict spliceosome assembly). This event results in an altered ratio of 3R/4R isoforms [10, 13]. The disruption of the balance between them results in hyperphosphorylation and aggregation of tau proteins into neurofibrillary tangles, causing the frontotemporal dementia with parkinsonism linked to chromosome 17 (FTDP-17) [10, 13]. These data support a direct relationship between aberrant alternative splicing of tau and neuropathology.
GBA
Mutations in β-glucocerebrosidase (GBA) gene cause Gaucher disease, a lysosomal storage disease characterized by an accumulation of glucocerebrosides. Some studies have identified GBA genetic variants as significant risk factors for the development of PD [122, 15].
GBA gene is located in a gene-rich region on chromosome 1q21. It spans 10.4 kb and contains 12 exons. Currently, there are four annotated alternative transcripts encoding proteins (GBA-001, GBA-002, GBA-015, and GBA-016; Table 4 and Fig. 5). Two of them originate from an alternative promoter located 2.6 kb upstream of the first ATG [123]. All transcripts share the same start codon, with the exception of GBA-012, whose open-reading frame starts upon exon 4 and produces a shorter protein isoform. Further transcripts are produced, but they do not encode proteins. The GBA splicing profile has not been studied still, and it is unknown if its alternative splicing is involved in PD.
MAO-B
MAO-B gene is located on chromosome X and includes 15 exons (Table 4 and Fig. 5). Although it is not a confirmed susceptibility gene [18], increased levels of monoamine oxidase B (MAO) mRNA and enzymatic activity have been reported in platelets from patients with both Parkinson’s and Alzheimer’s diseases [124]. Furthermore, it is well established that MAO-B inhibitors delay progression of both pathologies [125, 126].
Several DNA polymorphisms in the MAO-B gene have been described in populations with distinct ethnic backgrounds [124]. A SNP common in all ethnic groups and associated with two-fold risk of PD is the G/A dimorphism in intron 13 sequence [127–129]. This SNP does not change the coding sequence and does not affect the consensus acceptor and donor sites. However, it has been demonstrated the G/A dimorphism in intron 13 sequence creates a splicing enhancer that stimulates intron 13 removal and a spliceosomal complex assembly and alters splicing factors’ binding site efficiency [124].
SRRM2
Along with cis-acting elements, alternative splicing regulation relies on trans-splicing factors including the serine/arginine (SR) proteins. One of these proteins, the RNA splicing factor SRRM2 (or serine/arginine repetitive matrix 2), has been identified as the only gene that stood out as differentially expressed in multiple gene expression PD datasets [130].
SRRM2 gene generates two main alternative splicing transcripts different at their 3′ end (Table 4 and Fig. 5). The full-length isoform SRRM2-001 contains 15 exons, while the shorter isoform SRRM2-003 contains 11 exons and lacks exons 12–15. These two isoforms are differentially expressed in postmortem PD brain regions [130]. The shorter transcript was upregulated in the substantia nigra but unchanged in the amygdala of PD patients versus healthy controls. On the contrary, the longer transcript was downregulated in both substantia nigra and amygdala of PDs as compared to controls [130]. Furthermore, in the peripheral blood of patients with PD, SRRM2 short isoform is overexpressed, while the expression of longest isoform is reduced [130].
Genome-wide RNA expression analysis reveals global alternative splicing changes in PD
Although a “gene-by-gene” approach may simplify splicing analysis, global alternative splicing changes in PD have to be considered. The majority of the whole gene expression array studies in PD brain regions have unfortunately looked at a single transcript per gene, ignoring the multiple transcripts generated by alternative splicing [14]. Nonetheless, mRNA splicing has been identified as a mechanism significantly altered in cortical neurons of PD patients [131].
In order to investigate the splicing expression changes, some studies have used exon arrays. This kind of approach, enabling better monitoring and detection of the alternative splicing events, has allowed to observe significant changes in overall gene splicing in PD blood cells compared to healthy controls [130, 132]. Another exon array study has been conducted in blood of advanced PD patients prior to and following deep brain stimulation neurosurgery, a technique that efficiently improves the motor symptoms of PD [133]. This analysis has showed preliminary results suggesting brain electrical stimulation may correlate with significant profile changes in nonsense-mediated mRNA decay (an mRNA surveillance process that detects and selectively degrades splice transcripts harboring premature termination codons) in blood cell transcripts [133]. Potashkin et al. [134] have also used specific splice variant microarrays in PD patients in order to identify mRNAs splice transcripts as molecular biomarkers for an early PD diagnosis. Through this approach, they identified 13 splice variants with an altered expression in early-stage PD patients versus healthy controls [134, 135].
A recent technology to better study splicing defects is deep sequencing of RNA (RNAseq) [14]. The advantage of RNAseq is that it is theoretically feasible to measure both RNA expression levels and modifications such as splicing. In addition, RNAseq gives the possibility of discovering novel transcripts. Whole transcriptome RNAseq data have been obtained from blood leukocytes of PD patients’ predeep and postdeep brain stimulation treatment [136]. This approach has enabled to discover novel human exons and junctions in protein-coding RNA molecules, as well as a large range of differential splicing events pretreatment and posttreatment compared to healthy controls [136]. Although this is the first study using in-depth PD transcriptome sequencing, RNAseq represents a promising technique to better study PD alternative splicing.
The role of miRNA and lncRNA in PD alternative splicing modulation
A large number of alternative exon regions have been predicted as binding sites of microRNAs (miRNAs). The latter is a class of small noncoding RNA molecules, which mainly act as posttranscriptional modulators of multiple target genes by partial sequence complementarity. Through this mechanism, they may also influence splicing process.
The interplay between miRNA differential expression and alternative splicing modification in PD has been recently investigated [137]. Parallel changes in miRNA profiles and their spliced targets have been observed in PD leukocytes and PD-relevant brain regions (including the substantia nigra as well as the frontal lobe). This study was conducted through coupled analysis of small RNA sequencing data, splice junction arrays, and exon arrays [137].
Another novel fascinating class of RNAs with unknown functions is long noncoding RNAs (lncRNAs), defined as transcripts of over 200 nucleotides. The GENCODE noncoding RNA set collects all lncRNAs known so far, including several spliced transcript shorter than 200 bp. LncRNA profiling has been recently assessed in PD leukocytes predeep and postdeep brain stimulation via RNAseq [136]. This survey allowed to identify some lncRNAs overexpressed in PD and inversely decreased following deep brain stimulation [136]. Differentially expressed lncRNA includes the spliceosome component U1, supporting the hypothesis of disease-involved splicing modulations [136].
The identification of existing networks between noncoding mRNAs and alternative splicing modifications represents an important step forward the road to understanding the molecular basis of PD.
Conclusions
Alternative splicing is a highly harmonized process, based on a combination of DNA sequence motifs, intronic and exonic elements, regulatory factors, and temporal and spatial signaling pathways. Mutations that disrupt any of these critical features may alter the finely tuned splicing processes, upsetting the production or functions of the encoded proteins, and finally causing human diseases. Assessing the alternative splicing modulation of PD-related genes represents an important point to understand PD molecular etiology. Future studies, both with the standard or the new currently available large-scale techniques, will offer a complete data pool of the alternative splicing events in PD and will provide new possible insights in order to develop strategies for PD therapy and diagnosis.
References
Ward AJ, Cooper TA (2010) The pathobiology of splicing. J Pathol 220(2):152–163. doi:10.1002/path.2649
Yap K, Makeyev EV (2013) Regulation of gene expression in mammalian nervous system through alternative pre-mRNA splicing coupled with RNA quality control mechanisms. Mol Cell Neurosci 56:420–428. doi:10.1016/j.mcn.2013.01.003
Calarco JA, Zhen M, Blencowe BJ (2011) Networking in a global world: establishing functional connections between neural splicing regulators and their target transcripts. RNA 17(5):775–791. doi:10.1261/rna.2603911
Li Q, Lee JA, Black DL (2007) Neuronal regulation of alternative pre-mRNA splicing. Nat Rev Neurosci 8(11):819–831. doi:10.1038/nrn2237
Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ (2008) Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet 40(12):1413–1415. doi:10.1038/ng.259
Behzadnia N, Golas MM, Hartmuth K, Sander B, Kastner B, Deckert J, Dube P, Will CL, Urlaub H, Stark H, Luhrmann R (2007) Composition and three-dimensional EM structure of double affinity-purified, human prespliceosomal A complexes. EMBO J 26(6):1737–1748. doi:10.1038/sj.emboj.7601631
Shefer K, Sperling J, Sperling R (2014) The supraspliceosome—a multi-task machine for regulated pre-mRNA processing in the cell nucleus. Comput Struct Biotechnol J 11(19):113–122. doi:10.1016/j.csbj.2014.09.008
Gamazon ER, Stranger BE (2014) Genomics of alternative splicing: evolution, development and pathophysiology. Hum Genet 133(6):679–687. doi:10.1007/s00439-013-1411-3
Scuderi S, La Cognata V, Drago F, Cavallaro S, D’Agata V (2014) Alternative splicing generates different parkin protein isoforms: evidences in human, rat, and mouse brain. Biomed Res Int 2014:690796. doi:10.1155/2014/690796
Licatalosi DD, Darnell RB (2006) Splicing regulation in neurologic disease. Neuron 52(1):93–101. doi:10.1016/j.neuron.2006.09.017
Tazi J, Bakkour N, Stamm S (2009) Alternative splicing and disease. Biochim Biophys Acta 1792(1):14–26. doi:10.1016/j.bbadis.2008.09.017
Singh RK, Cooper TA (2012) Pre-mRNA splicing in disease and therapeutics. Trends Mol Med 18(8):472–482. doi:10.1016/j.molmed.2012.06.006
Cooper TA, Wan L, Dreyfuss G (2009) RNA and disease. Cell 136(4):777–793. doi:10.1016/j.cell.2009.02.011
Lewis PA, Cookson MR (2012) Gene expression in the Parkinson’s disease brain. Brain Res Bull 88(4):302–312. doi:10.1016/j.brainresbull.2011.11.016
Bonifati V (2014) Genetics of Parkinson’s disease—state of the art, 2013. Parkinsonism Relat Disord 20(1):S23–S28. doi:10.1016/S1353-8020(13)70009-9
Lubbe S, Morris HR (2014) Recent advances in Parkinson’s disease genetics. J Neurol 261(2):259–266. doi:10.1007/s00415-013-7003-2
Trinh J, Farrer M (2013) Advances in the genetics of Parkinson disease. Nat Rev Neurol 9(8):445–454. doi:10.1038/nrneurol.2013.132
Nalls MA, Pankratz N, Lill CM, Do CB, Hernandez DG, Saad M, DeStefano AL, Kara E, Bras J, Sharma M, Schulte C, Keller MF, Arepalli S, Letson C, Edsall C, Stefansson H, Liu X, Pliner H, Lee JH, Cheng R, International Parkinson’s Disease Genomics C, Parkinson’s Study Group Parkinson’s Research: The Organized GI, andMe, GenePd, NeuroGenetics Research C, Hussman Institute of Human G, Ashkenazi Jewish Dataset I, Cohorts for H, Aging Research in Genetic E, North American Brain Expression C, United Kingdom Brain Expression C, Greek Parkinson’s Disease C, Alzheimer Genetic Analysis G, Ikram MA, Ioannidis JP, Hadjigeorgiou GM, Bis JC, Martinez M, Perlmutter JS, Goate A, Marder K, Fiske B, Sutherland M, Xiromerisiou G, Myers RH, Clark LN, Stefansson K, Hardy JA, Heutink P, Chen H, Wood NW, Houlden H, Payami H, Brice A, Scott WK, Gasser T, Bertram L, Eriksson N, Foroud T, Singleton AB (2014) Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat Genet 46(9):989–993. doi:10.1038/ng.3043
Stefanis L (2012) Alpha-synuclein in Parkinson’s disease. Cold Spring Harb Perspect Med 2(2):a009399. doi:10.1101/cshperspect.a009399
Pihlstrom L, Toft M (2011) Genetic variability in SNCA and Parkinson’s disease. Neurogenetics 12(4):283–293. doi:10.1007/s10048-011-0292-7
Deng H, Yuan L (2014) Genetic variants and animal models in SNCA and Parkinson disease. Ageing Res Rev 15C:161–176. doi:10.1016/j.arr.2014.04.002
Nuytemans K, Meeus B, Crosiers D, Brouwers N, Goossens D, Engelborghs S, Pals P, Pickut B, Van den Broeck M, Corsmit E, Cras P, De Deyn PP, Del-Favero J, Van Broeckhoven C, Theuns J (2009) Relative contribution of simple mutations vs. copy number variations in five Parkinson disease genes in the Belgian population. Hum Mutat 30(7):1054–1061. doi:10.1002/humu.21007
Beyer K, Domingo-Sabat M, Lao JI, Carrato C, Ferrer I, Ariza A (2008) Identification and characterization of a new alpha-synuclein isoform and its role in Lewy body diseases. Neurogenetics 9(1):15–23. doi:10.1007/s10048-007-0106-0
Beyer K, Domingo-Sabat M, Humbert J, Carrato C, Ferrer I, Ariza A (2008) Differential expression of alpha-synuclein, parkin, and synphilin-1 isoforms in Lewy body disease. Neurogenetics 9(3):163–172. doi:10.1007/s10048-008-0124-6
McLean JR, Hallett PJ, Cooper O, Stanley M, Isacson O (2012) Transcript expression levels of full-length alpha-synuclein and its three alternatively spliced variants in Parkinson’s disease brain regions and in a transgenic mouse model of alpha-synuclein overexpression. Mol Cell Neurosci 49(2):230–239. doi:10.1016/j.mcn.2011.11.006
Cardo LF, Coto E, de Mena L, Ribacoba R, Mata IF, Menendez M, Moris G, Alvarez V (2014) Alpha-synuclein transcript isoforms in three different brain regions from Parkinson’s disease and healthy subjects in relation to the SNCA rs356165/rs11931074 polymorphisms. Neurosci Lett 562:45–49. doi:10.1016/j.neulet.2014.01.009
Beyer K, Lao JI, Carrato C, Mate JL, Lopez D, Ferrer I, Ariza A (2004) Differential expression of alpha-synuclein isoforms in dementia with Lewy bodies. Neuropathol Appl Neurobiol 30(6):601–607. doi:10.1111/j.1365-2990.2004.00572.x
Beyer K, Humbert J, Ferrer A, Lao JI, Carrato C, Lopez D, Ferrer I, Ariza A (2006) Low alpha-synuclein 126 mRNA levels in dementia with Lewy bodies and Alzheimer disease. Neuroreport 17(12):1327–1330. doi:10.1097/01.wnr.0000224773.66904.e7
McCarthy JJ, Linnertz C, Saucier L, Burke JR, Hulette CM, Welsh-Bohmer KA, Chiba-Falek O (2011) The effect of SNCA 3′ region on the levels of SNCA-112 splicing variant. Neurogenetics 12(1):59–64. doi:10.1007/s10048-010-0263-4
Kalivendi SV, Yedlapudi D, Hillard CJ, Kalyanaraman B (2010) Oxidants induce alternative splicing of alpha-synuclein: implications for Parkinson’s disease. Free Radic Biol Med 48(3):377–383. doi:10.1016/j.freeradbiomed.2009.10.045
Rhinn H, Qiang L, Yamashita T, Rhee D, Zolin A, Vanti W, Abeliovich A (2012) Alternative alpha-synuclein transcript usage as a convergent mechanism in Parkinson’s disease pathology. Nat Commun 3:1084. doi:10.1038/ncomms2032
Beyer K (2006) Alpha-synuclein structure, posttranslational modification and alternative splicing as aggregation enhancers. Acta Neuropathol 112(3):237–251. doi:10.1007/s00401-006-0104-6
Bungeroth M, Appenzeller S, Regulin A, Volker W, Lorenzen I, Grotzinger J, Pendziwiat M, Kuhlenbaumer G (2014) Differential aggregation properties of alpha-synuclein isoforms. Neurobiol Aging 35(8):1913–1919. doi:10.1016/j.neurobiolaging.2014.02.009
Burre J, Sharma M, Tsetsenis T, Buchman V, Etherton MR, Sudhof TC (2010) Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 329(5999):1663–1667. doi:10.1126/science.1195227
Rideout HJ, Stefanis L (2014) The neurobiology of LRRK2 and its role in the pathogenesis of Parkinson’s disease. Neurochem Res 39(3):576–592. doi:10.1007/s11064-013-1073-5
Johnson J, Paisan-Ruiz C, Lopez G, Crews C, Britton A, Malkani R, Evans EW, McInerney-Leo A, Jain S, Nussbaum RL, Foote KD, Mandel RJ, Crawley A, Reimsnider S, Fernandez HH, Okun MS, Gwinn-Hardy K, Singleton AB (2007) Comprehensive screening of a North American Parkinson’s disease cohort for LRRK2 mutation. Neurodegener Dis 4(5):386–391. doi:10.1159/000105160
Skipper L, Shen H, Chua E, Bonnard C, Kolatkar P, Tan LC, Jamora RD, Puvan K, Puong KY, Zhao Y, Pavanni R, Wong MC, Yuen Y, Farrer M, Liu JJ, Tan EK (2005) Analysis of LRRK2 functional domains in nondominant Parkinson disease. Neurology 65(8):1319–1321. doi:10.1212/01.wnl.0000180517.70572.37
Zabetian CP, Samii A, Mosley AD, Roberts JW, Leis BC, Yearout D, Raskind WH, Griffith A (2005) A clinic-based study of the LRRK2 gene in Parkinson disease yields new mutations. Neurology 65(5):741–744. doi:10.1212/01.wnl.0000172630.22804.73
Shojaee S, Sina F, Farboodi N, Fazlali Z, Ghazavi F, Ghorashi SA, Parsa K, Sadeghi H, Shahidi GA, Ronaghi M, Elahi E (2009) A clinic-based screening of mutations in exons 31, 34, 35, 41, and 48 of LRRK2 in Iranian Parkinson’s disease patients. Mov Disord 24(7):1023–1027. doi:10.1002/mds.22503
Di Fonzo A, Tassorelli C, De Mari M, Chien HF, Ferreira J, Rohe CF, Riboldazzi G, Antonini A, Albani G, Mauro A, Marconi R, Abbruzzese G, Lopiano L, Fincati E, Guidi M, Marini P, Stocchi F, Onofrj M, Toni V, Tinazzi M, Fabbrini G, Lamberti P, Vanacore N, Meco G, Leitner P, Uitti RJ, Wszolek ZK, Gasser T, Simons EJ, Breedveld GJ, Goldwurm S, Pezzoli G, Sampaio C, Barbosa E, Martignoni E, Oostra BA, Bonifati V, Italian Parkinson’s Genetics N (2006) Comprehensive analysis of the LRRK2 gene in sixty families with Parkinson’s disease. Eur J Hum Genet 14(3):322–331. doi:10.1038/sj.ejhg.5201539
Grimes DA, Racacho L, Han F, Panisset M, Bulman DE (2007) LRRK2 screening in a Canadian Parkinson’s disease cohort. Can J Neurol Sci 34(3):336–338
Paisan-Ruiz C, Nath P, Washecka N, Gibbs JR, Singleton AB (2008) Comprehensive analysis of LRRK2 in publicly available Parkinson’s disease cases and neurologically normal controls. Hum Mutat 29(4):485–490. doi:10.1002/humu.20668
Lesage S, Condroyer C, Lannuzel A, Lohmann E, Troiano A, Tison F, Damier P, Thobois S, Ouvrard-Hernandez AM, Rivaud-Pechoux S, Brefel-Courbon C, Destee A, Tranchant C, Romana M, Leclere L, Durr A, Brice A, French Parkinson’s Disease Genetics Study G (2009) Molecular analyses of the LRRK2 gene in European and North African autosomal dominant Parkinson’s disease. J Med Genet 46(7):458–464. doi:10.1136/jmg.2008.062612
Trabzuni D, Ryten M, Emmett W, Ramasamy A, Lackner KJ, Zeller T, Walker R, Smith C, Lewis PA, Mamais A, de Silva R, Vandrovcova J, International Parkinson Disease Genomics C, Hernandez D, Nalls MA, Sharma M, Garnier S, Lesage S, Simon-Sanchez J, Gasser T, Heutink P, Brice A, Singleton A, Cai H, Schadt E, Wood NW, Bandopadhyay R, Weale ME, Hardy J, Plagnol V (2013) Fine-mapping, gene expression and splicing analysis of the disease associated LRRK2 locus. PLoS ONE 8(8):e70724. doi:10.1371/journal.pone.0070724
Giesert F, Hofmann A, Burger A, Zerle J, Kloos K, Hafen U, Ernst L, Zhang J, Vogt-Weisenhorn DM, Wurst W (2013) Expression analysis of Lrrk1, Lrrk2 and Lrrk2 splice variants in mice. PLoS ONE 8(5):e63778. doi:10.1371/journal.pone.0063778
Sheng D, Qu D, Kwok KH, Ng SS, Lim AY, Aw SS, Lee CW, Sung WK, Tan EK, Lufkin T, Jesuthasan S, Sinnakaruppan M, Liu J (2010) Deletion of the WD40 domain of LRRK2 in zebrafish causes parkinsonism-like loss of neurons and locomotive defect. PLoS Genet 6(4):e1000914. doi:10.1371/journal.pgen.1000914
Vilarino-Guell C, Wider C, Ross OA, Dachsel JC, Kachergus JM, Lincoln SJ, Soto-Ortolaza AI, Cobb SA, Wilhoite GJ, Bacon JA, Behrouz B, Melrose HL, Hentati E, Puschmann A, Evans DM, Conibear E, Wasserman WW, Aasly JO, Burkhard PR, Djaldetti R, Ghika J, Hentati F, Krygowska-Wajs A, Lynch T, Melamed E, Rajput A, Rajput AH, Solida A, Wu RM, Uitti RJ, Wszolek ZK, Vingerhoets F, Farrer MJ (2011) VPS35 mutations in Parkinson disease. Am J Hum Genet 89(1):162–167. doi:10.1016/j.ajhg.2011.06.001
Zimprich A, Benet-Pages A, Struhal W, Graf E, Eck SH, Offman MN, Haubenberger D, Spielberger S, Schulte EC, Lichtner P, Rossle SC, Klopp N, Wolf E, Seppi K, Pirker W, Presslauer S, Mollenhauer B, Katzenschlager R, Foki T, Hotzy C, Reinthaler E, Harutyunyan A, Kralovics R, Peters A, Zimprich F, Brucke T, Poewe W, Auff E, Trenkwalder C, Rost B, Ransmayr G, Winkelmann J, Meitinger T, Strom TM (2011) A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. Am J Hum Genet 89(1):168–175. doi:10.1016/j.ajhg.2011.06.008
Verstraeten A, Wauters E, Crosiers D, Meeus B, Corsmit E, Elinck E, Mattheijssens M, Peeters K, Cras P, Pickut B, Vandenberghe R, Engelborghs S, De Deyn PP, Van Broeckhoven C (1844) Theuns J (2012) Contribution of VPS35 genetic variability to LBD in the Flanders–Belgian population. Neurobiol Aging 33(8):e1811–e1843. doi:10.1016/j.neurobiolaging.2012.01.006
Bonifati V (2012) Autosomal recessive parkinsonism. Parkinsonism Relat Disord 18(1):S4–S6. doi:10.1016/S1353-8020(11)70004-9
Lucking CB, Durr A, Bonifati V, Vaughan J, De Michele G, Gasser T, Harhangi BS, Meco G, Denefle P, Wood NW, Agid Y, Brice A, French Parkinson’s Disease Genetics Study G, European Consortium on Genetic Susceptibility in Parkinson’s D (2000) Association between early-onset Parkinson’s disease and mutations in the parkin gene. N Engl J Med 342(21):1560–1567. doi:10.1056/NEJM200005253422103
Oliveira SA, Scott WK, Martin ER, Nance MA, Watts RL, Hubble JP, Koller WC, Pahwa R, Stern MB, Hiner BC, Ondo WG, Allen FH Jr, Scott BL, Goetz CG, Small GW, Mastaglia F, Stajich JM, Zhang F, Booze MW, Winn MP, Middleton LT, Haines JL, Pericak-Vance MA, Vance JM (2003) Parkin mutations and susceptibility alleles in late-onset Parkinson’s disease. Ann Neurol 53(5):624–629. doi:10.1002/ana.10524
Illarioshkin SN, Periquet M, Rawal N, Lucking CB, Zagorovskaya TB, Slominsky PA, Miloserdova OV, Markova ED, Limborska SA, Ivanova-Smolenskaya IA, Brice A (2003) Mutation analysis of the parkin gene in Russian families with autosomal recessive juvenile parkinsonism. Mov Disord 18(8):914–919. doi:10.1002/mds.10467
Pigullo S, De Luca A, Barone P, Marchese R, Bellone E, Colosimo A, Scaglione C, Martinelli P, Di Maria E, Pizzuti A, Abbruzzese G, Dallapiccola B, Ajmar F, Mandich P (2004) Mutational analysis of parkin gene by denaturing high-performance liquid chromatography (DHPLC) in essential tremor. Parkinsonism Relat Disord 10(6):357–362. doi:10.1016/j.parkreldis.2004.04.012
Scherfler C, Khan NL, Pavese N, Eunson L, Graham E, Lees AJ, Quinn NP, Wood NW, Brooks DJ, Piccini PP (2004) Striatal and cortical pre- and postsynaptic dopaminergic dysfunction in sporadic parkin-linked parkinsonism. Brain 127(Pt 6):1332–1342. doi:10.1093/brain/awh150
Bertoli-Avella AM, Giroud-Benitez JL, Akyol A, Barbosa E, Schaap O, van der Linde HC, Martignoni E, Lopiano L, Lamberti P, Fincati E, Antonini A, Stocchi F, Montagna P, Squitieri F, Marini P, Abbruzzese G, Fabbrini G, Marconi R, Dalla Libera A, Trianni G, Guidi M, De Gaetano A, Boff Maegawa G, De Leo A, Gallai V, de Rosa G, Vanacore N, Meco G, van Duijn CM, Oostra BA, Heutink P, Bonifati V, Italian Parkinson Genetics N (2005) Novel parkin mutations detected in patients with early-onset Parkinson’s disease. Mov Disord 20(4):424–431. doi:10.1002/mds.20343
Bardien S, Keyser R, Yako Y, Lombard D, Carr J (2009) Molecular analysis of the parkin gene in South African patients diagnosed with Parkinson’s disease. Parkinsonism Relat Disord 15(2):116–121. doi:10.1016/j.parkreldis.2008.04.005
Pankratz N, Kissell DK, Pauciulo MW, Halter CA, Rudolph A, Pfeiffer RF, Marder KS, Foroud T, Nichols WC, Parkinson Study Group PI (2009) Parkin dosage mutations have greater pathogenicity in familial PD than simple sequence mutations. Neurology 73(4):279–286. doi:10.1212/WNL.0b013e3181af7a33
La Cognata V, Iemmolo R, D’Agata V, Scuderi S, Drago F, Zappia M, Cavallaro S (2014) Increasing the coding potential of genomes through alternative splicing: the case of PARK2 gene. Curr Genomics 15(3):203–216. doi:10.2174/1389202915666140426003342
Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N (1998) Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392(6676):605–608
Matsumine H, Saito M, Shimoda-Matsubayashi S, Tanaka H, Ishikawa A, Nakagawa-Hattori Y, Yokochi M, Kobayashi T, Igarashi S, Takano H, Sanpei K, Koike R, Mori H, Kondo T, Mizutani Y, Schaffer AA, Yamamura Y, Nakamura S, Kuzuhara S, Tsuji S, Mizuno Y (1997) Localization of a gene for an autosomal recessive form of juvenile parkinsonism to chromosome 6q25.2–27. Am J Hum Genet 60(3):588–596
Xu L, Lin DC, Yin D, Koeffler HP (2014) An emerging role of PARK2 in cancer. J Mol Med (Berl) 92(1):31–42. doi:10.1007/s00109-013-1107-0
Ikeuchi K, Marusawa H, Fujiwara M, Matsumoto Y, Endo Y, Watanabe T, Iwai A, Sakai Y, Takahashi R, Chiba T (2009) Attenuation of proteolysis-mediated cyclin E regulation by alternatively spliced parkin in human colorectal cancers. Int J Cancer 125(9):2029–2035. doi:10.1002/ijc.24565
Kitada T, Asakawa S, Minoshima S, Mizuno Y, Shimizu N (2000) Molecular cloning, gene expression, and identification of a splicing variant of the mouse parkin gene. Mamm Genome 11(6):417–421
Humbert J, Beyer K, Carrato C, Mate JL, Ferrer I, Ariza A (2007) Parkin and synphilin-1 isoform expression changes in Lewy body diseases. Neurobiol Dis 26(3):681–687. doi:10.1016/j.nbd.2007.03.007
Tan EK, Shen H, Tan JM, Lim KL, Fook-Chong S, Hu WP, Paterson MC, Chandran VR, Yew K, Tan C, Yuen Y, Pavanni R, Wong MC, Puvan K, Zhao Y (2005) Differential expression of splice variant and wild-type parkin in sporadic Parkinson’s disease. Neurogenetics 6(4):179–184. doi:10.1007/s10048-005-0001-5
Dagata V, Cavallaro S (2004) Parkin transcript variants in rat and human brain. Neurochem Res 29(9):1715–1724
Sunada Y, Saito F, Matsumura K, Shimizu T (1998) Differential expression of the parkin gene in the human brain and peripheral leukocytes. Neurosci Lett 254(3):180–182
Solano SM, Miller DW, Augood SJ, Young AB, Penney JB Jr (2000) Expression of alpha-synuclein, parkin, and ubiquitin carboxy-terminal hydrolase L1 mRNA in human brain: genes associated with familial Parkinson’s disease. Ann Neurol 47(2):201–210
Kasap M, Akpinar G, Sazci A, Idrisoglu HA, Vahaboglu H (2009) Evidence for the presence of full-length PARK2 mRNA and parkin protein in human blood. Neurosci Lett 460(3):196–200. doi:10.1016/j.neulet.2009.05.079
Pawlyk AC, Giasson BI, Sampathu DM, Perez FA, Lim KL, Dawson VL, Dawson TM, Palmiter RD, Trojanowski JQ, Lee VM (2003) Novel monoclonal antibodies demonstrate biochemical variation of brain parkin with age. J Biol Chem 278(48):48120–48128. doi:10.1074/jbc.M306889200
Horowitz JM, Myers J, Stachowiak MK, Torres G (1999) Identification and distribution of Parkin in rat brain. Neuroreport 10(16):3393–3397
Stichel CC, Augustin M, Kuhn K, Zhu XR, Engels P, Ullmer C, Lubbert H (2000) Parkin expression in the adult mouse brain. Eur J Neurosci 12(12):4181–4194
Gu WJ, Abbas N, Lagunes MZ, Parent A, Pradier L, Bohme GA, Agid Y, Hirsch EC, Raisman-Vozari R, Brice A (2000) Cloning of rat parkin cDNA and distribution of parkin in rat brain. J Neurochem 74(4):1773–1776
Huynh DP, Dy M, Nguyen D, Kiehl TR, Pulst SM (2001) Differential expression and tissue distribution of parkin isoforms during mouse development. Brain Res Dev Brain Res 130(2):173–181
D’Agata V, Grimaldi M, Pascale A, Cavallaro S (2000) Regional and cellular expression of the parkin gene in the rat cerebral cortex. Eur J Neurosci 12(10):3583–3588
Pogson JH, Ivatt RM, Whitworth AJ (2011) Molecular mechanisms of PINK1-related neurodegeneration. Curr Neurol Neurosci Rep 11(3):283–290. doi:10.1007/s11910-011-0187-x
Nuytemans K, Theuns J, Cruts M, Van Broeckhoven C (2010) Genetic etiology of Parkinson disease associated with mutations in the SNCA, PARK2, PINK1, PARK7, and LRRK2 genes: a mutation update. Hum Mutat 31(7):763–780. doi:10.1002/humu.21277
Marongiu R, Brancati F, Antonini A, Ialongo T, Ceccarini C, Scarciolla O, Capalbo A, Benti R, Pezzoli G, Dallapiccola B, Goldwurm S, Valente EM (2007) Whole gene deletion and splicing mutations expand the PINK1 genotypic spectrum. Hum Mutat 28(1):98. doi:10.1002/humu.9472
Samaranch L, Lorenzo-Betancor O, Arbelo JM, Ferrer I, Lorenzo E, Irigoyen J, Pastor MA, Marrero C, Isla C, Herrera-Henriquez J, Pastor P (2010) PINK1-linked parkinsonism is associated with Lewy body pathology. Brain 133(Pt 4):1128–1142. doi:10.1093/brain/awq051
Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Healy DG, Albanese A, Nussbaum R, Gonzalez-Maldonado R, Deller T, Salvi S, Cortelli P, Gilks WP, Latchman DS, Harvey RJ, Dallapiccola B, Auburger G, Wood NW (2004) Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 304(5674):1158–1160. doi:10.1126/science.1096284
Silvestri L, Caputo V, Bellacchio E, Atorino L, Dallapiccola B, Valente EM, Casari G (2005) Mitochondrial import and enzymatic activity of PINK1 mutants associated to recessive parkinsonism. Hum Mol Genet 14(22):3477–3492. doi:10.1093/hmg/ddi377
Beilina A, Van Der Brug M, Ahmad R, Kesavapany S, Miller DW, Petsko GA, Cookson MR (2005) Mutations in PTEN-induced putative kinase 1 associated with recessive parkinsonism have differential effects on protein stability. Proc Natl Acad Sci U S A 102(16):5703–5708. doi:10.1073/pnas.0500617102
d’Amora M, Angelini C, Marcoli M, Cervetto C, Kitada T, Vallarino M (2011) Expression of PINK1 in the brain, eye and ear of mouse during embryonic development. J Chem Neuroanat 41(2):73–85. doi:10.1016/j.jchemneu.2010.11.004
Lin W, Kang UJ (2008) Characterization of PINK1 processing, stability, and subcellular localization. J Neurochem 106(1):464–474. doi:10.1111/j.1471-4159.2008.05398.x
Deas E, Plun-Favreau H, Gandhi S, Desmond H, Kjaer S, Loh SH, Renton AE, Harvey RJ, Whitworth AJ, Martins LM, Abramov AY, Wood NW (2011) PINK1 cleavage at position A103 by the mitochondrial protease PARL. Hum Mol Genet 20(5):867–879. doi:10.1093/hmg/ddq526
Lockhart PJ, Lincoln S, Hulihan M, Kachergus J, Wilkes K, Bisceglio G, Mash DC, Farrer MJ (2004) DJ-1 mutations are a rare cause of recessively inherited early onset parkinsonism mediated by loss of protein function. J Med Genet 41(3):e22
Moore DJ, West AB, Dawson VL, Dawson TM (2005) Molecular pathophysiology of Parkinson’s disease. Annu Rev Neurosci 28:57–87. doi:10.1146/annurev.neuro.28.061604.135718
Bonifati V, Rizzu P, Squitieri F, Krieger E, Vanacore N, van Swieten JC, Brice A, van Duijn CM, Oostra B, Meco G, Heutink P (2003) DJ-1( PARK7), a novel gene for autosomal recessive, early onset parkinsonism. Neurol Sci 24(3):159–160. doi:10.1007/s10072-003-0108-0
van Duijn CM, Dekker MC, Bonifati V, Galjaard RJ, Houwing-Duistermaat JJ, Snijders PJ, Testers L, Breedveld GJ, Horstink M, Sandkuijl LA, van Swieten JC, Oostra BA, Heutink P (2001) Park7, a novel locus for autosomal recessive early-onset parkinsonism, on chromosome 1p36. Am J Hum Genet 69(3):629–634. doi:10.1086/322996
Corti O, Lesage S, Brice A (2011) What genetics tells us about the causes and mechanisms of Parkinson’s disease. Physiol Rev 91(4):1161–1218. doi:10.1152/physrev.00022.2010
Tarantino P, Civitelli D, Annesi F, De Marco EV, Rocca FE, Pugliese P, Nicoletti G, Carrideo S, Provenzano G, Annesi G, Quattrone A (2009) Compound heterozygosity in DJ-1 gene non-coding portion related to parkinsonism. Parkinsonism Relat Disord 15(4):324–326. doi:10.1016/j.parkreldis.2008.07.001
Hedrich K, Djarmati A, Schafer N, Hering R, Wellenbrock C, Weiss PH, Hilker R, Vieregge P, Ozelius LJ, Heutink P, Bonifati V, Schwinger E, Lang AE, Noth J, Bressman SB, Pramstaller PP, Riess O, Klein C (2004) DJ-1 (PARK7) mutations are less frequent than parkin (PARK2) mutations in early-onset Parkinson disease. Neurology 62(3):389–394
Lev N, Roncevic D, Ickowicz D, Melamed E, Offen D (2006) Role of DJ-1 in Parkinson’s disease. J Mol Neurosci 29(3):215–225
Ariga H, Takahashi-Niki K, Kato I, Maita H, Niki T, Iguchi-Ariga SM (2013) Neuroprotective function of DJ-1 in Parkinson’s disease. Oxid Med Cell Longev 2013:683920. doi:10.1155/2013/683920
Xu J, Zhong N, Wang H, Elias JE, Kim CY, Woldman I, Pifl C, Gygi SP, Geula C, Yankner BA (2005) The Parkinson’s disease-associated DJ-1 protein is a transcriptional co-activator that protects against neuronal apoptosis. Hum Mol Genet 14(9):1231–1241. doi:10.1093/hmg/ddi134
Bandopadhyay R, Kingsbury AE, Cookson MR, Reid AR, Evans IM, Hope AD, Pittman AM, Lashley T, Canet-Aviles R, Miller DW, McLendon C, Strand C, Leonard AJ, Abou-Sleiman PM, Healy DG, Ariga H, Wood NW, de Silva R, Revesz T, Hardy JA, Lees AJ (2004) The expression of DJ-1 (PARK7) in normal human CNS and idiopathic Parkinson’s disease. Brain 127(Pt 2):420–430. doi:10.1093/brain/awh054
Besong Agbo D, Klafki H, Poschmann G, Seyfarth K, Genius J, Janssen C, Stuhler K, Wurst W, Meyer HE, Klingenspor M, Wiltfang J (2013) Development of a capillary isoelectric focusing immunoassay to measure DJ-1 isoforms in biological samples. Anal Biochem 443(2):197–204. doi:10.1016/j.ab.2013.09.013
Kumaran R, Kingsbury A, Coulter I, Lashley T, Williams D, de Silva R, Mann D, Revesz T, Lees A, Bandopadhyay R (2007) DJ-1 (PARK7) is associated with 3R and 4R tau neuronal and glial inclusions in neurodegenerative disorders. Neurobiol Dis 28(1):122–132. doi:10.1016/j.nbd.2007.07.012
Kumaran R, Vandrovcova J, Luk C, Sharma S, Renton A, Wood NW, Hardy JA, Lees AJ, Bandopadhyay R (2009) Differential DJ-1 gene expression in Parkinson’s disease. Neurobiol Dis 36(2):393–400. doi:10.1016/j.nbd.2009.08.011
Lin X, Cook TJ, Zabetian CP, Leverenz JB, Peskind ER, Hu SC, Cain KC, Pan C, Edgar JS, Goodlett DR, Racette BA, Checkoway H, Montine TJ, Shi M, Zhang J (2012) DJ-1 isoforms in whole blood as potential biomarkers of Parkinson disease. Sci Rep 2:954. doi:10.1038/srep00954
Zhong N, Kim CY, Rizzu P, Geula C, Porter DR, Pothos EN, Squitieri F, Heutink P, Xu J (2006) DJ-1 transcriptionally up-regulates the human tyrosine hydroxylase by inhibiting the sumoylation of pyrimidine tract-binding protein-associated splicing factor. J Biol Chem 281(30):20940–20948. doi:10.1074/jbc.M601935200
Vilarino-Guell C, Soto AI, Lincoln SJ, Ben Yahmed S, Kefi M, Heckman MG, Hulihan MM, Chai H, Diehl NN, Amouri R, Rajput A, Mash DC, Dickson DW, Middleton LT, Gibson RA, Hentati F, Farrer MJ (2009) ATP13A2 variability in Parkinson disease. Hum Mutat 30(3):406–410. doi:10.1002/humu.20877
Murphy KE, Cottle L, Gysbers AM, Cooper AA, Halliday GM (2013) ATP13A2 (PARK9) protein levels are reduced in brain tissue of cases with Lewy bodies. Acta Neuropathol Commun 1(1):11. doi:10.1186/2051-5960-1-11
Ugolino J, Fang S, Kubisch C, Monteiro MJ (2011) Mutant Atp13a2 proteins involved in parkinsonism are degraded by ER-associated degradation and sensitize cells to ER-stress induced cell death. Hum Mol Genet 20(18):3565–3577. doi:10.1093/hmg/ddr274
Lu CS, Lai SC, Wu RM, Weng YH, Huang CL, Chen RS, Chang HC, Wu-Chou YH, Yeh TH (2012) PLA2G6 mutations in PARK14-linked young-onset parkinsonism and sporadic Parkinson’s disease. Am J Med Genet B Neuropsychiatr Genet 159B(2):183–191. doi:10.1002/ajmg.b.32012
Di Fonzo A, Dekker MC, Montagna P, Baruzzi A, Yonova EH, Correia Guedes L, Szczerbinska A, Zhao T, Dubbel-Hulsman LO, Wouters CH, de Graaff E, Oyen WJ, Simons EJ, Breedveld GJ, Oostra BA, Horstink MW, Bonifati V (2009) FBXO7 mutations cause autosomal recessive, early-onset parkinsonian-pyramidal syndrome. Neurology 72(3):240–245. doi:10.1212/01.wnl.0000338144.10967.2b
Deng H, Liang H, Jankovic J (2013) F-box only protein 7 gene in parkinsonian-pyramidal disease. JAMA Neurol 70(1):20–24. doi:10.1001/jamaneurol.2013.572
Gomez-Garre P, Jesus S, Carrillo F, Caceres-Redondo MT, Huertas-Fernandez I, Bernal-Bernal I, Bonilla-Toribio M, Vargas-Gonzalez L, Carballo M, Mir P (2014) Systematic mutational analysis of FBXO7 in a Parkinson’s disease population from southern Spain. Neurobiol Aging 35(3):727. e5–7. doi:10.1016/j.neurobiolaging.2013.09.011
Ilyin GP, Rialland M, Pigeon C, Guguen-Guillouzo C (2000) cDNA cloning and expression analysis of new members of the mammalian F-box protein family. Genomics 67(1):40–47. doi:10.1006/geno.2000.6211
Zhao T, De Graaff E, Breedveld GJ, Loda A, Severijnen LA, Wouters CH, Verheijen FW, Dekker MC, Montagna P, Willemsen R, Oostra BA, Bonifati V (2011) Loss of nuclear activity of the FBXO7 protein in patients with parkinsonian-pyramidal syndrome (PARK15). PLoS ONE 6(2):e16983. doi:10.1371/journal.pone.0016983
Nolte D, Niemann S, Muller U (2003) Specific sequence changes in multiple transcript system DYT3 are associated with X-linked dystonia parkinsonism. Proc Natl Acad Sci U S A 100(18):10347–10352. doi:10.1073/pnas.1831949100
Herzfeld T, Nolte D, Grznarova M, Hofmann A, Schultze JL, Muller U (2013) X-linked dystonia parkinsonism syndrome (XDP, lubag): disease-specific sequence change DSC3 in TAF1/DYT3 affects genes in vesicular transport and dopamine metabolism. Hum Mol Genet 22(5):941–951. doi:10.1093/hmg/dds499
Korvatska O, Strand NS, Berndt JD, Strovas T, Chen DH, Leverenz JB, Kiianitsa K, Mata IF, Karakoc E, Greenup JL, Bonkowski E, Chuang J, Moon RT, Eichler EE, Nickerson DA, Zabetian CP, Kraemer BC, Bird TD, Raskind WH (2013) Altered splicing of ATP6AP2 causes X-linked parkinsonism with spasticity (XPDS). Hum Mol Genet 22(16):3259–3268. doi:10.1093/hmg/ddt180
Myhre R, Klungland H, Farrer MJ, Aasly JO (2008) Genetic association study of synphilin-1 in idiopathic Parkinson’s disease. BMC Med Genet 9:19. doi:10.1186/1471-2350-9-19
Keyser RJ, Oppon E, Carr JA, Bardien S (2011) Identification of Parkinson’s disease candidate genes using CAESAR and screening of MAPT and SNCAIP in South African Parkinson’s disease patients. J Neural Transm 118(6):889–897. doi:10.1007/s00702-011-0591-z
Marx FP, Holzmann C, Strauss KM, Li L, Eberhardt O, Gerhardt E, Cookson MR, Hernandez D, Farrer MJ, Kachergus J, Engelender S, Ross CA, Berger K, Schols L, Schulz JB, Riess O, Kruger R (2003) Identification and functional characterization of a novel R621C mutation in the synphilin-1 gene in Parkinson’s disease. Hum Mol Genet 12(11):1223–1231
Eyal A, Szargel R, Avraham E, Liani E, Haskin J, Rott R, Engelender S (2006) Synphilin-1A: an aggregation-prone isoform of synphilin-1 that causes neuronal death and is present in aggregates from alpha-synucleinopathy patients. Proc Natl Acad Sci U S A 103(15):5917–5922. doi:10.1073/pnas.0509707103
Eyal A, Engelender S (2006) Synphilin isoforms and the search for a cellular model of Lewy body formation in Parkinson’s disease. Cell Cycle 5(18):2082–2086
Szargel R, Rott R, Engelender S (2008) Synphilin-1 isoforms in Parkinson’s disease: regulation by phosphorylation and ubiquitylation. Cell Mol Life Sci 65(1):80–88. doi:10.1007/s00018-007-7343-0
Lei P, Ayton S, Finkelstein DI, Adlard PA, Masters CL, Bush AI (2010) Tau protein: relevance to Parkinson’s disease. Int J Biochem Cell Biol 42(11):1775–1778. doi:10.1016/j.biocel.2010.07.016
Spatola M, Wider C (2014) Genetics of Parkinson’s disease: the yield. Parkinsonism Relat Disord 20(1):S35–S38. doi:10.1016/S1353-8020(13)70011-7
Svobodova E, Mrazova L, Luksan O, Elstein D, Zimran A, Stolnaya L, Minks J, Eberova J, Dvorakova L, Jirsa M, Hrebicek M (2011) Glucocerebrosidase gene has an alternative upstream promoter, which has features and expression characteristic of housekeeping genes. Blood Cells Mol Dis 46(3):239–245. doi:10.1016/j.bcmd.2010.12.011
Jakubauskiene E, Janaviciute V, Peciuliene I, Soderkvist P, Kanopka A (2012) G/A polymorphism in intronic sequence affects the processing of MAO-B gene in patients with Parkinson disease. FEBS Lett 586(20):3698–3704. doi:10.1016/j.febslet.2012.08.028
Thomas T (2000) Monoamine oxidase-B inhibitors in the treatment of Alzheimer’s disease. Neurobiol Aging 21(2):343–348
Stern G (1998) Neuroprotection by selegiline and other MAO inhibitors. J Neural Transm Suppl 52:99–107
Ho SL, Kapadi AL, Ramsden DB, Williams AC (1995) An allelic association study of monoamine oxidase B in Parkinson’s disease. Ann Neurol 37(3):403–405. doi:10.1002/ana.410370318
Kurth JH, Kurth MC, Poduslo SE, Schwankhaus JD (1993) Association of a monoamine oxidase B allele with Parkinson’s disease. Ann Neurol 33(4):368–372. doi:10.1002/ana.410330406
Sobell JL, Lind TJ, Hebrink DD, Heston LL, Sommer SS (1997) Screening the monoamine oxidase B gene in 100 male patients with schizophrenia: a cluster of polymorphisms in African-Americans but lack of functionally significant sequence changes. Am J Med Genet 74(1):44–49
Shehadeh LA, Yu K, Wang L, Guevara A, Singer C, Vance J, Papapetropoulos S (2010) SRRM2, a potential blood biomarker revealing high alternative splicing in Parkinson’s disease. PLoS ONE 5(2):e9104. doi:10.1371/journal.pone.0009104
Stamper C, Siegel A, Liang WS, Pearson JV, Stephan DA, Shill H, Connor D, Caviness JN, Sabbagh M, Beach TG, Adler CH, Dunckley T (2008) Neuronal gene expression correlates of Parkinson’s disease with dementia. Mov Disord 23(11):1588–1595. doi:10.1002/mds.22184
Soreq L, Bergman H, Israel Z, Soreq H (2012) Exon arrays reveal alternative splicing aberrations in Parkinson’s disease leukocytes. Neurodegener Dis 10(1–4):203–206. doi:10.1159/000332598
Soreq L, Bergman H, Israel Z, Soreq H (2013) Deep brain stimulation modulates nonsense-mediated RNA decay in Parkinson’s patients leukocytes. BMC Genomics 14:478. doi:10.1186/1471-2164-14-478
Potashkin JA, Santiago JA, Ravina BM, Watts A, Leontovich AA (2012) Biosignatures for Parkinson’s disease and atypical parkinsonian disorders patients. PLoS ONE 7(8):e43595. doi:10.1371/journal.pone.0043595
Santiago JA, Scherzer CR, Harvard Biomarker S, Potashkin JA (2013) Specific splice variants are associated with Parkinson’s disease. Mov Disord 28(12):1724–1727. doi:10.1002/mds.25635
Soreq L, Guffanti A, Salomonis N, Simchovitz A, Israel Z, Bergman H, Soreq H (2014) Long non-coding RNA and alternative splicing modulations in Parkinson’s leukocytes identified by RNA sequencing. PLoS Comput Biol 10(3):e1003517. doi:10.1371/journal.pcbi.1003517
Soreq L, Salomonis N, Bronstein M, Greenberg DS, Israel Z, Bergman H, Soreq H (2013) Small RNA sequencing-microarray analyses in Parkinson leukocytes reveal deep brain stimulation-induced splicing changes that classify brain region transcriptomes. Front Mol Neurosci 6:10. doi:10.3389/fnmol.2013.00010
Acknowledgments
This study was supported by the international Ph.D. program in Neuroscience of the University of Catania. We also gratefully acknowledge Cristina Calì, Alfia Corsino, Maria Patrizia D’Angelo, and Francesco Marino for their administrative and technical support.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
La Cognata, V., D’Agata, V., Cavalcanti, F. et al. Splicing: is there an alternative contribution to Parkinson’s disease?. Neurogenetics 16, 245–263 (2015). https://doi.org/10.1007/s10048-015-0449-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10048-015-0449-x