
Abstract
Spinocerebellar ataxia type 31 (SCA31) is a recently defined subtype of autosomal dominant cerebellar ataxia (ADCA) characterized by adult-onset, pure cerebellar ataxia. The C/T substitution in the 5′-untranslated region of the puratrophin-1 gene (PLEKHG4) or a disease-specific haplotype within the 900-kb SCA31 critical region just upstream of PLEKHG4 has been used for the diagnosis of SCA31. Very recently, a disease-specific insertion containing penta-nucleotide (TGGAA)n repeats has been found in this critical region in SCA31 patients. SCA31 was highly prevalent in Nagano, Japan, where SCA31 accounts for approximately 42% of ADCA families. We screened the insertion in 94 SCA31 patients from 71 families in Nagano. All patients had a 2.6- to 3.7-kb insertion. The size of the insertion was inversely correlated with the age at onset but not associated with the progression rate after onset. (TAGAA)n repeats at the 5′-end of the insertion were variable in number, ranging from 0 (without TAGAA sequence) to 4. The number of (TAGAA)n repeats was inversely correlated to the total size of the insertion. The number of (TAGAA)n repeats was comparatively uniform within patients from the three endemic foci in Nagano. Only one patient, heterozygous for the C/T substitution in PLEKHG4, had the insertions in both alleles; they were approximately 3.0 and 4.3 kb in size. Sequencing and Southern hybridization using biotin-labeled (TGGAA)5 probe strongly indicated that the 3.0-kb insertion, but not the 4.3-kb insertion, contained (TGGAA)n stretch. We also found that 3 of 405 control individuals (0.7%) had the insertions from 1.0 to 3.5 kb in length. They were negative for the C/T substitution in PLEKHG4, and neither of the insertions contained (TGGAA)n stretch at their 5′-end by sequencing. The insertions in normal controls were clearly detected by Southern hybridization using (TAAAA)5 probe, while they were not labeled with (TGGAA)5 or (TAGAA)5 probe. These data indicate that control alleles very rarely have a nonpathogenic large insertion in the SCA31 critical region and that not only the presence of the insertion but also its size is not sufficient evidence for a disease-causing allele. We approve of the view that (TGGAA)n repeats in the insertion are indeed related to the pathogenesis of SCA31, but it remains undetermined whether a large insertion lacking (TGGAA)n is nonpathogenic.
Similar content being viewed by others


Introduction
Spinocerebellar ataxia type 31 (SCA31), formerly known as 16q22.1-linked autosomal dominant cerebellar ataxia (16q-ADCA), is a recently established subtype of ADCA characterized by adult-onset, pure cerebellar ataxia [1–6]. SCA31 accounts for 8–17% of ADCA families and is the third most predominant ADCA subtype after SCA6 and MJD/SCA3 in Japan [7–11]. A single nucleotide substitution (–16C>T) in the 5′-untranslated region (UTR) of the gene encoding puratrophin-1 (PLEKHG4) has been shown to be a disease-specific marker for 16q-ADCA [4, 5]. However, this specific substitution has been found exclusively in the Japanese population [12]; thus, it is still unclear whether SCA31 exists in countries other than Japan.
Two patients with SCA31 have been found not to carry this substitution in PLEKHG4 [5, 13], indicating that it is not a disease-causing mutation for SCA31. Thereafter, Amino et al. [5] narrowed the SCA31 critical region to 900 kb between rs11640843 (SNP04) and PLEKHG4 by fine single nucleotide polymorphism (SNP) typing. Sato et al. [6] identified an inserted sequence in this region, which was confirmed in all SCA31 patients, without exception. The insertion consists of complex penta-nucleotide repeats containing (TGGAA)n, and the size of the insertion is variable, ranging from 2.5 to 3.8 kb in length, among patients [6].
We have shown that SCA31 is the most predominant subtype of ADCA in Nagano, which is located in the central, mountainous district of the main island of Japan [13–15], see Fig. 2]. To date, we have analyzed 168 ADCA families from Nagano and diagnosed 71 families (42%) with SCA31. Thus, the frequency of SCA31 in Nagano is much higher than in other areas of Japan [7–11]. We have found that SCA31 families are highly prevalent in particular areas of Nagano, named as Kiso, Ina, and Saku [13–15], see Fig. 2]. The ratio of SCA31 families to total ADCA families in these areas was 14/16 (88%, Kiso area), 12/18 (67%, Ina area), and 17/25 (68%, Saku area).
Here, we have screened an insertion mutation for SCA31 in 94 patients from 71 families in Nagano.
Materials and methods
Subjects and clinical evaluation
We recruited 94 patients from 71 families with SCA31, and these families most probably originated from Nagano. The diagnosis of SCA31 was based on the presence of the C/T substitution in PLEKHG4 (92 patients from 70 families). Two patients (from two families) without the C/T substitution in PLEKHG4 were also included in this study because they had a disease-specific haplotype in the 900-kb SCA31 critical region [13]. Forty-four of 71 families (62%) originated from the three endemic foci described above. Detailed medical interviews and routine neurological examinations were performed by expert neurologists. Age at onset was determined on the basis of the information provided by the patients or their close relatives. Scales for the assessment and rating of ataxia (SARA) were used for the assessment of cerebellar ataxia. To minimize the interrater variability, SARA was performed by either of two expert neurologists (KY and YS) for 57 patients (67 times).
This research protocol was approved independently by the Ethical Committee of Shinshu University School of Medicine and by the Committee for Ethical Issues at Yokohama City University Graduate School of Medicine.
Molecular analysis
The insertion sequence was amplified by PCR according to the methods described by Sato et al. [6]. PCR products were purified using a PCR purification kit (QIAGEN), digested with HaeIII, and then separated on a 0.8% agarose gel (25 V, 15 h). The size of the HaeIII fragment containing the insertion was calculated with a DNA size marker simultaneously electrophoresed as a reference. HaeIII-undigested PCR products were separated in a 0.8% low-melting agarose gel, and fragments of approximately 3.0 kb were cut out and extracted using a QIA quick® Gel Extraction Kit (QIAGEN). They were then directly sequenced by a standard protocol using BigDye terminator (Applied Biosystems, Foster City, CA) on an ABI PRISM 3100 Genetic analyzer or an ABI PRISM 3500xL Genetic analyzer (Applied Biosystems). HaeIII-undigested PCR products were also separated in a 1% agarose gel, blotted to a nylon membrane (HybondTM- N+, Amersham International plc, Buckinghamshire, UK) using 10× SSC, and subjected to Southern hybridization using biotin-labeled (TGGAA)5, (TAGAA)5, or (TAAAA)5 as a probe. Detection was done with BrightStarTM BioDetectTM Nonisotopic Detection Kit (Ambion Inc., Austin, TX) according to the manufacturer’s instructions.
Statistics
The relationship between the size of the insert and the age at onset was analyzed using Spearman’s correlation coefficient by rank test. Regression analysis for SARA data was also performed. Analysis of the differences in the insert size among the groups was carried out using analysis of variance and the post hoc test of Scheffé. The level of significance was set at p < 0.01.
Results
All of the patients recruited in this study had an insertion ranging from 2.6 to 3.7 kb in length. Direct sequencing confirmed that the insertion contained (TGGAA)n stretch in all of the patients. The average length of the insertions was approximately 3,130 bp (SD, 199 bp) (n = 94). The correlation between the size of the insertion and the age at onset is shown in Fig. 1. The length of the insertion was inversely correlated with the age at onset (n = 89). We observed six intergenerational transmission of a disease-causing allele in five families in our cohort. There was no conspicuous expansion of the insertion size.

Correlation between the insertion size and age at onset (n = 89). Representative PCR screening for the SCA31 insertion (a). Agarose gel electrophoresis of PCR products before and after HaeIII digestion is shown. M GeneRulerTM 1-kb Ladder (Fermentas Life Sciences, Burlington, Canada). The size of the insertion is inversely correlated with the age at onset (b). For 5 of 94 patients, age at onset could not be clearly defined by medical interview
The average size of the insertion was obviously different between patients from the three endemic foci (Fig. 2a, b). The insertion of patients from Kiso area was significantly shorter in length (2,866 ± 132 bp, n = 18) than those from the other two areas (3,263 ± 101 bp, n = 27, Ina area; 3,111 ± 109 bp, n = 18, Saku area).

Distribution of the insertion size in endemic foci in Nagano. The location of the three endemic foci (Kiso, Ina, and Saku) in Nagano prefecture is indicated (a). The distribution of the size of insertions in the three endemic foci is shown (b). The distribution (vertical bar), the average size (horizontal bar), and the standard deviation of the size of insertion (shaded square) in all the patients (n = 94) are shown in left. ## p < 0.01
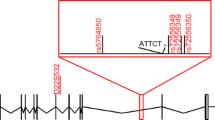
We found the number of penta-nucleotide (TAGAA)n repeats at the 5′-end of the insertion was variable, as well as the subsequent (TGGAA)n repeats. The number of (TAGAA)n repeats ranged from 0 to 4 (Fig. 3); the most predominant number was 1 (50/94, 53%), followed by 2 (20/94, 21%), 3 (14/94, 15%), and 4 (8/94, 9%). There were two patients without (TAGAA) sequence just upstream of (TGGAA)n repeats. The number of (TAGAA)n repeats, if present, was inversely correlated with the size of the insertion (Fig. 4). The repeat size of (TAGAA)n was comparatively uniform within the endemic foci. Seventeen of 18 patients (94%) in Kiso area had three or four (TAGAA)n repeats. Twenty-five of 27 (93%) in Ina area had one repeat, and all 18 patients (100%) in Saku area had one or two repeats. Two patients without (TAGAA) sequence originated from areas outside the three endemic foci. We had 12 families, in which more than two affected members were recruited in this cohort. Intrafamilial variation in (TAGAA)n repeat number was observed only in one family (family ID 166); the sister had four (TAGAA)n repeats and her younger brother had three, but the number of (TAGAA)n repeats was consistent among the family members in the other 11 families.

Sequence of the 5′-end of the insertion. The number of (TAGAA)n repeats (underlined in red) is variable, ranging from 0 to 4

Correlation between the sizes of (TAGAA)n repeats preceding (TGGAA)n repeats and the insertion size. ##p < 0.01; #p < 0.05. n.s. not significant
In our cohort, 93 patients were heterozygous for the insertion, but only one patient aged 87 (patient ID 254) carried the insertions in both alleles (Fig. 5). This patient originated from one of three endemic foci, Kiso area, and developed cerebellar ataxia at age 76. He was still able to walk with a cane, and his SARA score was 15.5 at age 87. The size of the insertions was calculated as approximately 3,040 and 4,280 bp. Direct sequencing showed that only the 3.0-kb insertion, but not the 4.3-kb insertion, contained (TGGAA)n stretch. By Southern hybridization, the 3.0-kb insertion, but not the 4.3-kb insertion, was detected by (TGGAA)5 probe (Fig. 6c). The 4.3-kb insertion was faintly labeled with (TAGAA)5 probe, and the signal intensity was much weaker than the 3.0-kb insertion (Fig. 6d). On the other hand, the 4.3-kb insertion was more intensively labeled with (TAAAA)5 probe than the 3.0-kb insertion (Fig. 6b).

PCR amplification for the insertion. The patient (ID 256, lane 5) had insertions on both alleles, instead of lacking a wild-type 1.5-kb band without the insertion. The 4.3-kb band is indicated by the arrow

Southern hybridization for the insertion. HaeIII-undigested PCR products were separated in a 1% agarose gel, stained with ethidium bromide (a), and then blotted to a nylon membrane. The membrane was hybridized with biotin-labeled (TAAAA)5 probe (b), (TGGAA)5 probe (c), or (TAGAA)5 probe (d). The insertions in SCA31 patients (lanes 1–3) were clearly detected by (TGGAA)5 or (TAGAA)5 probe (c and d) . In patient ID 254 (lane 3), the 3.0-kb insertion (arrow), but not the 4.3-kb insertion (arrowhead), was clearly labeled with (TGGAA)5 probe (c). In contrast, the 4.3-kb insertion (arrowhead), as well as the insertions in normal controls (lanes 4–6), was more intensively labeled with (TAAAA)5 probe than the insertions in SCA31 patients (b). The 1.5-kb fragments derived from a normal allele were visualized by (TAAAA)5 probe (b) because (TAAAA)n repeats are included in the original genomic sequence. Lanes 1–3 ,SCA31 patients (lane 3, patient ID 254); lanes 4–6, control individuals with the insertion (lane 4: control 1; lane 5: control 2; lane 6: control 3 in Table 1); lanes 7 and 8, control individuals without the insertion
Furthermore, we found that 3 of 405 healthy control individuals (0.7%) had the insertions (Fig. 6a, Table 1). Neither of the insertions contained (TGGAA)n stretch at their 5′-end by sequencing. By Southern hybridization, the insertions in control individuals were not detected by (TGGAA)5, or (TAGAA)5 probe (Fig. 6c, d), but were more clearly labeled with (TAAAA)5 probe than the insertions in SCA31 patients (Fig. 6b).
To see the effect of the insertion size on disease progression, we tentatively divided the patients into three groups based on the size of the insertion; groups I (insertion size > 3,300 bp, n = 11), II (3,000–3,300 bp, n = 27), and III (<3,000 bp, n = 17). The correlation between SARA scores and age at examination or duration of illness is shown in Fig. 7. There was no significant difference in the disease progression rate after onset between the groups.

Correlation between SARA and age at examination (a) or duration of disease (b) in SCA31 patients. The patients were divided into three groups based on the size of insertion; groups I (insertion size >3,300 bp, closed square), II (3,000–3,300 bp, open circle), and III (<3,000 bp, open triangle). SARA was performed 14 times in 11 patients from group I, 34 times in 29 patients from group II, and 19 times in 17 patients from group III. Broken, solid, and dotted lines indicate linear regression lines for groups I, II, and III, respectively
Discussion
In the present study, we confirmed that all of the SCA31 patients in our cohort had the insertions of the penta-nucleotide repeats found by Sato et al. [6]. The insertions ranged from 2.6 to 3.7 kb in length, and contained (TGGAA)n stretch at their 5′-end, without exception. We also verified that the size of the insertion was inversely correlated to the age at disease onset in our large cohort. However, the size of the insertion seemed not to be associated with the disease progression rate after onset.
We found that the penta-nucleotide (TAGAA)n repeats at the 5′-end of the insertion were variable in number, as were the subsequent (TGGAA)n repeats. Interestingly, the number of (TAGAA)n repeats, if present, was inversely correlated with the total size of the insertion. Furthermore, the repeat size of (TAGAA)n is comparatively uniform within the endemic foci. From the geographical viewpoint, we previously supposed that there were two major foci in the southwest (Ina–Kiso) and east (Saku) areas in Nagano [15], but patients in Ina and Kiso areas are likely to be different from the viewpoint of population genetics because the number of (TAGAA)n repeats and the size of the insertion were obviously different between the two groups.
In our cohort, only one patient (patient ID 254) was homozygous for the insertion in the SCA31 critical region as determined by PCR analysis (Fig. 5, Table 1). Contrary to our expectation, sequencing showed that only the smaller 3.0-kb insertion had (TGGAA)n stretch. This was also confirmed by Southern hybridization using biotin-labeled (TGGAA)5 probe. The patient developed gait ataxia at approximately age 76 and showed pure cerebellar ataxia by neurological examination at age 87. His clinical features were typical for SCA31 [7–11, 13, 15]. As his parents died a long time ago, we could not obtain reliable information on his parents or their genomic DNA. It is confusing that he was heterozygous for the C/T substitution in PLEKHG4 but was homozygous for a disease-specific G/A substitution at AB473220 (Table 1).
Moreover, three control individuals had the insertions ranging from 1.0 to 3.5 kb in length. Sequencing and Southern hybridization indicated that these insertions did not contain (TGGAA)n or (TAGAA)n repeats. Information on the family history of cerebellar ataxia was not available for these control individuals because they voluntarily participated in this study as anonymous healthy controls. Neither of them carried the C/T substitution in PLEKHG4, but one control individual (control 1 in Table 1) had G/A substitution at AB473220 and a large insertion (3,520 bp) indistinguishable from SCA31 patients. Sato et al. [6] have shown that the insertion in the SCA31 critical region is rarely observed in control individuals but that the insertion in control individuals is shorter in length than SCA31 patients and lacks (TGGAA)n stretch. At present, however, we do not completely exclude the possibility that the 4.3-kb insertion in patient ID 254 and the 3.5-kb insertion in control 1 have some pathogenic effects, although these insertions are likely to lack (TGGAA)n stretch.
In summary, our data clearly indicate that not only the presence of the insertion but also the size of the insertion in the SCA31 critical region is insufficient evidence for the disease-causing allele. They may support the hypothesis by Sato et al. that the presence of (TGGAA)n repeats is important for the pathogenesis of SCA31. However, it is possible that a large insertion without (TGGAA)n repeats may have a pathological significance, requiring further investigation. The control individual with such insertion in this study may potentially develop cerebellar ataxia in the future, considering that SCA31 is a late-onset disease. The insertion sequences in SCA31 patients consist of (TAGAA), (TGGAA), and (TAAAA) penta-nucleotide repeats of variable numbers [6], but the precise pathogenesis by the penta-nucleotide repeat insertion remains unclear in SCA31. Further detailed characterization of the inserted sequence and data on genotype–phenotype correlation will be needed.
References
Takashima M, Ishikawa K, Nagaoka U, Shoji S, Mizusawa H (2001) A linkage disequilibrium at the candidate gene locus for 16q-linked autosomal dominant cerebellar ataxia type III in Japan. J Hum Genet 46:167–171. doi:10.1007/s100380170083
Li M, Ishikawa K, Toru S, Tomimitsu H, Takashima M, Goto J et al (2003) Physical map and haplotype analysis of 16q-linked autosomal dominant cerebellar ataxia (ADCA) type III in Japan. J Hum Genet 48:111–118. doi:10.1007/s100380300017
Hirano R, Takashima H, Okubo R, Tajima K, Okamoto Y, Ishida S et al (2004) Fine mapping of 16q-linked autosomal dominant cerebellar ataxia type III in Japanese families. Neurogenetics 5:215–221. doi:10.1007/s10048-004-0194-z
Ishikawa K, Toru S, Tsunemi T, Li M, Kobayashi K, Yokota T et al (2005) An autosomal dominant cerebellar ataxia linked to chromosome 16q22.1 is associated with a single-nucleotide substitution in the 5′ untranslated region of the gene encoding a protein with spectrin repeat and rho guanine-nucleotide exchange-factor domains. Am J Hum Genet 77:280–296. doi:10.1086/432518
Amino T, Ishikawa K, Toru S, Ishiguro T, Sato N, Tsunemi T et al (2007) Redefining the disease locus of 16q22.1-linked autosomal dominant cerebellar ataxia. J Hum Genet 52:643–649. doi:10.1007/s10038-007-0154-1
Sato N, Amino T, Kobayashi K, Asakawa S, Ishiguro T, Tsunemi T et al (2009) Spinocerebellar ataxia type 31 is associated with “inserted” penta-nucleotide repeats containing (TGGAA)n. Am J Hum Genet 85:1–14. doi:10.1016/j.ajhg.2009.09.019
Ouyang Y, Sakoe K, Shimazaki H, Namekawa M, Ogawa T, Ando Y et al (2006) 16q-linked autosomal dominant cerebellar ataxia: a clinical and genetic study. J Neurol Sci 247:180–186. doi:10.1016/j/jns.2006.04.009
Onodera Y, Aoki M, Mizuno H, Warita H, Shiga Y, Itoyama Y (2006) Clinical features of chromosome 16q22.1 linked autosomal dominant cerebellar ataxia in Japanese. Neurology 67:1300–1302
Nozaki H, Ikeuchi T, Kawakami A, Kimura A, Koide R, Tsuchiya M et al (2007) Clinical and genetic characterizations of 16q-linked autosomal dominant spinocerebellar ataxia (AD-SCA) and frequency analysis of AD-SCA in the Japanese population. Mov Disord 22:857–862. doi:10.1002/mds.21443
Basri R, Yabe I, Soma H, Sasaki H (2007) Spectrum and prevalence of autosomal dominant spinocerebellar ataxia in Hokkaido, the northern island of Japan: a study of 113 Japanese families. J Hum Genet 52:848–855. doi:10.1007/s10038-007-0182-x
Hayashi M, Adachi Y, Mori M, Nakano T, Nakashima K (2007) Clinical and genetic epidemiological study of 16q22.1-linked autosomal dominant cerebellar ataxia in western Japan. Acta Neurol Scand 116:123–127. doi:10.1111/j.1600-0404.2007.00815.x
Wieczorek S, Arning L, Alheite I, Epplen JT (2006) Mutations of the puratrophin-1 (PLEKHG4) gene on chromosome 16q22.1 are not a common genetic cause of cerebellar ataxia in a European population. J Hum Genet 51:363–367. doi:10.1007/s10038-006-0372-y
Ohata T, Yoshida K, Sakai H, Hamanoue H, Mizuguchi T, Shimizu Y et al (2006) A –16C>T substitution in the 5′ UTR of the puratrophin-1 gene is prevalent in autosomal dominant cerebellar ataxia in Nagano. J Hum Genet 51:461–466. doi:10.1007/s10038-006-0385-6
Shimizu Y, Yoshida K, Okano T, Ohara S, Hashimoto T, Fukushima Y et al (2004) Regional features of autosomal-dominant cerebellar ataxia in Nagano: clinical and molecular genetic analysis of 86 families. J Hum Genet 49:610–616. doi:10.1007/s10038-004-0196-6
Yoshida K, Shimizu Y, Morita H, Okano T, Sakai H, Ohata T et al (2009) Severity and progression rate of cerebellar ataxia in 16q-linked autosomal dominant cerebellar ataxia (16q-ADCA) in the endemic Nagano area of Japan. Cerebellum 8:46–51. doi:10.1007/s12311-008-0062-8
Acknowledgements
We are grateful to the clinicians who provided us with information on their patients, and the patients and their families who participated in this study. This work was supported in part by a Grant-in-Aid for Science Research from the Ministry of Education, Science and Culture, Japan (KY and NM); a grant from the Research Committee for Ataxic Diseases, the Ministry of Health, Labour, and Welfare, Japan (KY); and a grant from the Ministry of Health, Labour, and Welfare, Japan (NM).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Haruya Sakai and Kunihiro Yoshida equally contributed to this work.
Disclosure
The authors report no conflicts of interest.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Sakai, H., Yoshida, K., Shimizu, Y. et al. Analysis of an insertion mutation in a cohort of 94 patients with spinocerebellar ataxia type 31 from Nagano, Japan. Neurogenetics 11, 409–415 (2010). https://doi.org/10.1007/s10048-010-0245-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10048-010-0245-6