
Abstract
Virus recognition and induction of interferon (IFN) are critical components of the innate immune system. The Toll-like receptor (TLR) and RIG-I-like receptor families have been characterized as key players in RNA virus detection. Signaling cascades initiated by these receptors are crucial for establishment of an IFN signaling mediated antiviral state in infected and neighboring cells and containment of virus replication as well as initiation of the adaptive immune response. In this review, we focus on the diverse and overlapping functions of these receptors, their physiological importance, and respective viral inducers. We highlight the roles of TRL3, TLR7/8, retinoic acid inducible gene I, melanoma differentiation-associated gene 5, and the RNA molecules responsible for activating these viral sensors.
Similar content being viewed by others

Interferon
The phenomenon of host directed viral interference has been observed for many years, with some descriptions dating back to observations made by Jenner in 1804 in reference to herpes virus infections interfering with vaccinia virus lesion developments. Supporting these initial reports, more controlled studies with numerous bacteria, plant, and animal viruses followed in the 1930s and 1940s and further confirmed the viral interference phenomena. A thorough and detailed review by Henle describes many pioneering studies and provides numerous examples of initial reports of viral interference (Henle 1950). These early studies varied widely in their approach with some using live or inactivated virus as an interfering agent and challenging with either the same or different species of virus. Through hindsight, it becomes clear that many of these initial experiments were not relevant to the action of interferon but could be attributed to other phenomena where the end result was inhibition of viral infection. Nevertheless, numerous important and relevant observations were made in those early years of research and placing these initial findings in the context of current molecular biology knowledge provides a deeper understanding of the field and also highlights areas in need of elucidation.
Experiments done with inactivated influenza virus in embryonated chicken eggs have provided some of the clearest early data relating to the interference phenomenon. From results generated by multiple groups, it became clear that interference can be caused by either inactivated virus particles or live virus grown under specific conditions, such as repeated passage with large inocula or repeated freeze-thawing. It also became apparent that the method of inactivation was highly important for the degree of interference, with UV inactivation being far superior to heat or formalin treatment. The length of UV treatment corresponded to an increase in interfering ability until a peak was reached and decreased with further exposure (Ziegler et al. 1944). Furthermore, the replication ability of a virus was much more sensitive to UV treatment than the interfering ability. Based on these early observations, many proposals as to the mechanism of interference were made, including the conclusion that this phenomenon was caused by a cellular product resulting from primary viral infection (Henle 1950). Further support for this hypothesis came from the now famous work of Isaacs and Lindenmann who coined the term ‘interferon’ and described it as a ‘non-hemagglutinating macromolecular particle which has many different properties from those of heated influenza virus’ (Isaacs and Lindenmann 1957). Over the next 30 years, type I interferon (IFN) was characterized in detail and identified as a family of cytokines encoded by the IFN-β gene and multiple IFN-α genes. After an arduous struggle, human IFN was purified to homogeneity and characterized for its biochemical properties by three individual groups, which identified its acid-stability and amino acid composition (Rubinstein et al. 1978, 1979; Tan et al. 1979; Zoon et al. 1979). Consequently, the availability of purified IFN and subsequent cloning and expression of the IFN-β gene product from E. coli allowed much more detailed analysis of its antiviral action (Nagata et al. 1980). Today, IFN is known as a key component of the innate immune system responsible not only for broad cellular antimicrobial activity in response to primary infection, but also for its role in linking innate and adaptive immune responses (Biron 2001).
Viral inducers
Shortly following the discovery of IFN, viral RNA was proposed to be the inducer of this antiviral response (Isaacs et al. 1963). Many early studies focused on possible nucleic acid inducers and numerous synthetic and biological RNAs were tested for their ability to induce interferon. Common to most of these studies, dsRNA was found to be a potent trigger of the interferon response unlike ssRNA, DNA or RNA:DNA hybrids (Colby and Morgan 1971). Specifically, dsRNA from bacteria, reovirus, vaccinia virus and synthetic polyinosinic:polycytidylic acid poly(I:C) were shown to be potent activators of the antiviral response (Colby and Duesberg 1969; Field et al. 1967, 1968; Lampson et al. 1967; Tytell et al. 1967). Since then many groups have confirmed the strong IFN inducing ability of poly(I:C). The biochemical basis for its high level of activation remains unclear to this day, as it does not appear that stability of the RNA complex directly correlates with its IFN inducing capacity (Colby and Morgan 1971). Based on dsRNA’s induction capacity, the concept of dsRNA as a physiological viral trigger for IFN induction quickly became accepted in the field despite prevailing evidence that the majority of RNA viruses employ mechanisms that protect their RNA from exposure. This conundrum was largely dismissed with the simple explanation that viruses are bound to make mistakes during replication and are therefore likely to expose at least some dsRNA molecules to the cell. However, studies employing dsRNA-specific antibodies have shown that negative-strand RNA viruses do not appear to produce detectible amounts of dsRNA (Weber et al. 2006). Although, it is possible that the threshold amount of dsRNA required to trigger an IFN response is below the antibody detection limit or that the length of dsRNA molecules is not sufficient for antibody recognition. But it is equally plausible that a different molecule serves as the primary recognition motif for RNA viruses.
An important addition to the field was the discovery that a 5′ triphosphate (5′ppp) group on an RNA molecule also served as a potent activator of the interferon response and could provide an alternative/additional trigger to dsRNA (Hornung et al. 2006; Pichlmair et al. 2006). RNA synthesis by RNA polymerases initiates with a triphosphate containing nucleotide and therefore all RNA molecules initially contain a triphosphate moiety on their 5′ end. However, since cells generally process the synthesized RNA by either capping mRNA, removing 5′ppp during RNA processing, folding RNA into complex secondary structures, or packaging it into RNP complexes; exposed 5′ppp are likely uncommon in the cytoplasm and therefore provide an appealing viral recognition motif. The genomes of RNA viruses are known to contain uncapped, 5′ppp-containing RNA, although the question of whether this RNA is ever exposed to the cell during the viral lifecycle remains to be answered. Additionally, it makes sense that if viruses have evolved multiple mechanisms to hide their dsRNA, they are equally likely to protect their 5′ppp from antiviral sensors.
Because early interference experiments relied on inactivated virus as the inducer of interferon response, attempts were made to connect the induction by isolated RNA to that of inactivated virus. An initial report showed that RNA is produced from UV treated Newcastle Disease Virus (NDV) virions even though there is a complete loss of infectivity as measured by plaque assay (Huppert et al. 1969). Thus, it appeared that UV treated virions attempt to replicate and produce at least partially synthesized RNA even in the absence of producing functional viral progeny. This incomplete newly synthesized RNA is thought to base pair with the template resulting in the formation of a dsRNA molecule.
The requirement for viral replication has been supported by numerous studies, even though it does appear that under certain conditions completely inactive virus is capable of inducing IFN (Hidmark et al. 2005). It remains to be determined whether this induction by inactivated virus is due to incomplete inactivation, exposure of viral RNA resulting from physical damage to the virions, or if other viral components are capable of being recognized by the cell. In addition to viral RNA, viral proteins and ribonucleoprotein (RNP) complexes have been implicated in IFN induction. When introduced into cells, purified RNP complexes do induce an IFN response (tenOever et al. 2002, 2004). However, since it is extremely difficult to demonstrate that intact RNPs are introduced into cells, differentiation between RNP recognition and naked RNA recognition remains elusive. A few examples of viral protein recognition do exist. Hepatitis C Virus (HCV) NS5A protein has been shown to activate nuclear factor-κB (NF-κB) when expressed in cells (Waris et al. 2002). In addition, the F protein of respiratory syncytial virus (RSV) is well characterized to be capable of inducing proinflammatory cytokines through TLR4 (Kurt-Jones et al. 2000). However, it is unlikely that viral proteins alone are sufficient for induction of an antiviral response based on their wide diversity and biochemical similarity to cellular proteins. A more plausible scenario is that during a viral infection, multiple signals are recognized by different sensors, which in synergy, alert the cell to the presence of a viral pathogen.
Cellular receptors
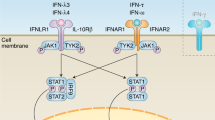
To date, two distinct systems for RNA virus detection and interferon induction have been characterized. One is composed of toll-like receptors (TLRs) and the other is the RIG-I like receptor (RLR) family. Of the 13 mammalian TLR members identified to date, endosomally located TLR3, TLR7 and TLR8 have been characterized as principal sensors of RNA viruses, while other TLRs are responsible for detecting bacteria, fungi, and DNA viruses (Alexopoulou et al. 2001; Diebold et al. 2004). Extracellularly located TLR4 has also been implicated in RNA virus detection through recognition of the F protein of respiratory syncytial virus (Kurt-Jones et al. 2000). RIG-I and MDA5, of the RLR family, are cytoplasmic sensors expressed in majority of cell types and detect intracellular RNA viruses. Viral RNA is thought to function as the pathogen-associated molecular pattern (PAMP) for all intracellular RNA virus pattern-recognition receptors (PRRs), although the exact biochemical nature of inducing molecules remains unclear. Current understanding indicates that TLR3 recognizes any dsRNA in endocytic compartments while MDA5 recognizes long dsRNA in the cytoplasm, TLR7 and 8 are activated by ssRNA rich in G/U residues in endosomes of dendritic cells and RIG-I senses phosphate containing dsRNA in the cytoplasm of majority of cells (Table 1). Upon detection of their corresponding PAMPs, both TLRs and RLRs initiate signaling cascades which converge on activation, and subsequent nuclear localization of three families of transcription factors: NF-κB, interferon regulatory factors (IRFs), and ATF-2/cJun. As can be seen in Fig. 1, the signaling pathways for TLR3 and RLRs utilize adaptors TRIF and MAVS, respectively, and then converge with activation of the canonical (IKKα, β, and γ) and noncanonical (TBK1, IKKε) IKK kinases. Activation of TBK1/IKKε leads to phosphorylation and nuclear translocation of IRF3. Whereas IKKα, β, and γ activate and allow nuclear translocation of NF-κB. TLR7 and 8 in dendritic cells utilize a common TLR adaptor MyD88 to activate a complex of IRAK4/IRAK1/TRAF6, which in turn lead to phosphorylation and nuclear translocation of IRF7. These signaling cascades results in transcription of IFN-β or IFNα genes and production of the first wave of type I interferon (Thompson and Locarnini 2007). Following synthesis, IFN is secreted from the infected cell and initiates an autocrine and paracrine-signaling cascade through Type I IFN receptor (IFNAR) which results in upregulation of more than 100 different genes and creation of an antiviral state in both infected and neighboring uninfected cells. Although the functions of the majority of IFN stimulated genes are not known, some are well characterized and are involved in inhibition of the viral lifecycle by shutting down general cellular processes (Samuel 2001). In addition to its antiviral function, IFN has also been shown to play an important role in modulation of the adaptive immune response through stimulation of MHC class I presentation, activation of natural killer (NK) cells and cytotoxic T cells, and maturation of dendritic cells (DCs) (Biron 2001; Le Bon and Tough 2002; Stetson and Medzhitov 2006).

Endosomal and cytoplasmic pathways for virus recognition and IFN production. In DCs, TLR7 and TLR8 located in endosomal compartments recognize viral ssRNA through either direct infection, autophagocytic uptake of viral material from cytoplasm or phagocytic uptake of other infected cells or vial particles. Both TLR 7 and 8 signal through adaptor MyD88, which through interaction with IRAK4/IRAK1/TRAF6 complex leads to phosphorylation and activation of IRF7 and subsequent IFN transcription. TLR3 located in endosomes of cDCs, macrophages, epithelial cells, and fibroblasts is activated by encountering dsRNA. Following its activation, TLR3 signals through its adaptor, TRIF which leads to activation of noncanonical IKK kinases (TBK1/IKKε) and subsequent phosphorylation and nuclear translocation of IRF3. NF-κB is also activated by TRIF mediated signaling through canonical IKK kinases (IKKα, β, and γ). Cytoplasmically located RIG-I and MDA5 are expressed in most cells and recognize 5′ppp containing dsRNA or long dsRNA, respectively. Both of these cytoplasmic sensors upon activation interact and signal through the mitochondrially located adaptor MAVS. This signaling pathway, analogous to that of TLR3, leads to activation of the canonical and noncanonical IKK kinases and the following nuclear transclocation of NF-κB and IRF3. Concurrent activation of IRF3 and NF-κB in turn allows for transcription of IFN genes and its synthesis and export
TLRs
Toll-like receptors were initially identified through their homology to Drosophila Toll protein, a critical component of Drosophila’s innate immune system (Rock et al. 1998). To date 13 mammalian members of this family have been recognized as receptors involved in recognition of conserved microbial PAMPS (Akira and Hemmi 2003; Janeway 1989). These receptors are composed of an extracellular leucine-rich repeat domain which participates in pathogen recognition, and a conserved cytoplasmic domain with homology to IL-1R which is involved in downstream signaling through MyD88 or TRIF adaptor proteins (Akira and Takeda 2004). The extracellular domains of TLRs exhibit high structural diversity and have been shown to recognize a wide variety of pathogens including bacteria, fungi, protozoa, and viruses. Whereas TLRs responsible for bacterial and fungal recognition are located on the cell surface, those responsible for sensing viral infections are commonly located intracellularly in endosomal compartments (Diebold 2008). Upon activation, TLR3 and TLR7/8 initiate a signaling cascade though adaptors TRIF and MyD88, respectively, leading to expression of IFN and proinflamatory cytokines (Uematsu and Akira 2007).
TLR3
TLR3 was the first characterized receptor to induce IFN production in response to dsRNA. Mice lacking TLR3 were shown to be more resistant to poly(I:C) induced shock and TLR3 deficient macrophages exhibited reduced IFN production in response to poly(I:C) (Alexopoulou et al. 2001). It was therefore proposed that this receptor was primarily responsible for detection of dsRNA generated during viral infections and induction of IFN. However, the physiological importance of TLR3 in IFN induction is questionable in light of a number of studies showing that loss of this receptor does not result in increased viral susceptibility or reduced IFN production in the infected animals. TLR3 knockout mice were shown to recover normally from multiple RNA viruses, including lymphocytic choriomeningitis virus (LCMV), vesicular stomatitis virus (VSV), and reovirus (ReV) (Edelmann et al. 2004; Johansson et al. 2007; Kato et al. 2005). Infections of TLR3 knockout mice with mouse cytomegalovirus (MCMV), a dsDNA virus, has also led to contradictory outcomes with one group reporting normal recovery and IFN production and another reporting increased susceptibility and abrogated IFN levels (Edelmann et al. 2004; Tabeta et al. 2004). TLR3’s role in cytokine production can be observed in TLR3 deficient human lung epithelial cells infected with influenza virus. In response to infection, these cells exhibit a reduced induction of NF-kB dependent genes but not of IRF3 dependent genes, including IFNβ (Le Goffic et al. 2007). In addition to cytokine production, TLR3 signaling has also been implicated in cross-priming of CD8 T cells by DC mediated phagocytosis of infected cells (Schulz et al. 2005), and in activation of NK cells in response to MCMV infection (Tabeta et al. 2004). Consistent with its role in inducing an inflammatory response, one study showed that mice lacking in TLR3 were more resistant to West Nile virus (WNV) associated encephalitis, presumably because of a break down in blood–brain barrier caused by TLR3 mediated inflammation (Wang et al. 2004). However, another more recent study found the opposite effect, with TLR3 knockout mice being more susceptible to WNV infection and having higher viral load in the brain, although IFN levels in these mice were not diminished (Daffis et al. 2008). Influenza A infections in TLR3 deficient mice also resulted in a less pathogenic phenotype despite a higher virus load in the lungs of the animals (Le Goffic et al. 2006). Therefore, while it appears that IFN production in animals does not require TLR3 signaling, most likely because of a functional RLR system, it is clear that this receptor does play a role in the innate and adaptive immune responses. As such, people lacking in TLR3 have been shown to be more susceptible to HSV-1 associated encephalitis (Zhang et al. 2007).
TLR 7 and 8
Unlike TLR3, which is expressed in numerous cell types, TLR7 and 8 are mainly found in endosomal compartments of plasmacytoid dendritic cells (pDCs) and myeloid dendritic cells (mDCs), respectively (Diebold 2008). The two receptors are very closely related and are thought to differ primarily in cell-type specificity and cytokine expression profiles (Gorden et al. 2005). Both TLR 7 and 8 have been determined to be activated by ssRNA rich in guanosine or uridine, and ssRNA from viruses, such as human immunodeficiency virus (HIV), VSV, and influenza A virus (Diebold et al. 2004; Heil et al. 2004; Lund et al. 2004). Interestingly, TLR7 was also shown to be activated by cellular mRNA but not by rRNA or tRNA, highlighting the possible importance of cellular RNA modifications in preventing stimulation of antiviral RNA receptors (Kariko et al. 2005). Based on their endoplasmic location and inaccessibility to cytoplasmically replicating viruses, TLRs can only be activated by viruses through endocytosis, or by phagocytic/autophagocytic uptake of viral RNA from the cytoplasm of infected cells. Therefore, viruses which enter cells through the endosomes, such as influenza virus, might be more easily detected by TLRs than viruses that do not use the endosome for entry (Diebold et al. 2004). On the other hand, autophagy has been shown to be necessary in pDC virus detection of VSV and Sendai virus, which enter cells through direct fusion with the plasma membrane. Accordingly, mice deficient in autophagy related gene 5 (Atg5) or pDCs treated with autophagy inhibitors produced much lower amount of IFN than wild type mice and only responded to replication competent viruses (Lee et al. 2007). An important role for TLR7 signaling came from studies that showed that both TLR7 (and its adaptor MyD88) are essential for IFN production by pDCs following influenza A virus and VSV infections (Kato et al. 2005). The unique dependence of pDCs on TLR7 signaling is intriguing in light of these cells’ characterized ability to produce copious amounts of type I IFN in vitro and their role in IFN production in vivo (Asselin-Paturel et al. 2001). Contrary to pDCs, other cell types have been shown to primarily rely on RLR sensors for RNA virus detection (Kato et al. 2005). A study by Kumagai et al. provides a nice illustration of a possible physiological reason for two parallel IFN inducing systems. In this study, mice were intranasally infected with either wild type or C protein deficient Sendai virus (SeV). The authors went onto show that pDCs, in a MyD88-dependent manner, were the primary IFN producing cells in response to wild type SeV. However, when SeV lacking in C protein were used, the primary IFN producers were alveolar macrophages. These cells did not rely on MyD88-directed signaling for IFN production, but instead depended on MAVS (an adaptor for RIG-I/MDA5 pathway) (Kumagai et al. 2007). Since SeV C protein is known to inhibit RIG-I mediated IFN induction it appears that TLR-dependent pDCs are employed as primary IFN producing cells in the event that MAVS signaling is abrogated by the virus (Strahle et al. 2007). Thus, TLR-mediated recognition of viruses might be important for those pathogens, which have evolved mechanisms that subvert cytoplasmic viral sensors.
TLR7 signaling has also been implicated in having a role in mediating the antibody response to RNA viruses, as MyD88 knockout mice are deficient in B cell IgG class-switching following influenza infection and are deficient in CD4 T cell and B cell response following influenza vaccination (Heer et al. 2007; Koyama et al. 2007). Thus, it is likely that in addition to their role in innate immunity, TLRs serve an important function in triggering signaling pathways leading to establishment of humoral immunity.
RLRs
Discovered in 2004, the RLR family of cytoplasmic viral sensors has become a major focus of research in antiviral innate immunity. The family is composed of three members, RIG-I, MDA5, and Laboratory of Genetic and Physiology 2 (LGP2). Both RIG-I and MDA5 belong to the family of DExD/H RNA helicases and contain a typical ATP-dependent helicase domain. The N-terminus of these proteins is unique in that it encodes two caspase recruitment domains (CARDs), normally associated with cell death and inflammatory signaling pathways. Through numerous studies RIG-I and MDA5 have been found to play a key role in IFN induction following RNA virus infection. Through knockout analysis RIG-I has been shown to be the primary recognition receptor for majority of RNA viruses, while MDA5 is the major receptor for recognition of picornaviruses. Despite their specificities for various viral families, the two sensors often have overlapping roles and individually contribute to IFN production in response to infection. Both sensors appear to be activated by binding to dsRNA, with MDA5 being specific for long dsRNA molecules and RIG-I preferring dsRNA with an exposed 5′ppp group. Ubiquitous expression of RIG-I and MDA5 indicates that these sensors play a role in antiviral innate immunity in majority of tissues and cell types.
The third member of the RLR family, LGP2, also contains a DExD/H helicase domain but is completely lacking in CARDs and is not able to initiate antiviral signaling. Instead, this protein is thought to act as a regulator of RLR signaling; a negative one for RIG-I induced signaling and a positive one for MDA5 induced signaling.
RLR substrates
In line with RIG-I’s prominent role in innate antiviral immunity, the quest for its physiological substrates has been a major focus of research in recent years. Based on the fact that RIG-I is an RNA helicase and was shown early onto directly interact with poly(I:C), the substrate responsible for its induction was initially assumed to be viral dsRNA. However, following up on earlier observations that siRNAs produced by in vitro transcription were capable of inducing an antiviral response, two independent groups showed that in vitro transcribed (IVT) RNA molecules, of at least 19nt, bearing a 5′ppp end can efficiently induce RIG-I (Hornung et al. 2006; Pichlmair et al. 2006). The importance of the 5′ppp was demonstrated by the loss of IFN induction following Calf Intestinal Alkaline Phosphotase (CIP) treatment of the RNA and by the fact that a synthetic ssRNA (with a 5′ OH group) of same sequence was not capable of inducing IFN. Furthermore, RNA isolated from influenza A or rabies viruses also lost IFN stimulation activity following CIP treatment and was unable to induce an IFN response in RIG-I −/− MEFs, thereby confirming that the presence of phosphates on an RNA molecule is a physiologically relevant, RIG-I specific PAMP (Kato et al. 2006). Small RNA generated by measles virus polymerase in vitro (likely containing short leader RNA) also induced IFN-β when transfected into cells (Plumet et al. 2007). Thus, it appeared that the presence of a 5′ppp on an RNA molecule, either single- or double-stranded was sufficient for activation of RIG-I. The concept of a 5′ppp PAMP is appealing in that most RNA viruses contain 5′ppp in their genomes and antigenomes, making this motif an inherent part of a viral lifecycle and unlikely to be mutated under immune system pressure. On the other hand, cellular RNAs lack exposed 5′ppp as a result of mRNA capping or processing of 5′ppp into monophosphates (Nallagatla et al. 2008). In addition to removal of phosphate groups, cellular RNAs are also extensively modified as a result of incorporation of modified nucleotides or methylation. These modifications likely play a role in prevention of an antiviral response to cellular RNA. In fact, incorporation of pseudourudine, 2-thiouridine, or 2′O-methylated uridine into T7 transcripts strongly inhibited IFN production by those RNA molecules (Hornung et al. 2006). As a way to prevent exposure of the 5′ppp to the cell, viruses are thought to hide these molecules in tightly packed nucleocapsids or replicate in cellular compartments physically removed from the sensors (Nallagatla et al. 2008). Members of the bunyaviridae family were shown to remove the triphosphate group from their genomes, thereby creating genomes that no longer interact with RIG-I in vitro (Habjan et al. 2008). Since the discovery of the 5′ppp as a PAMP in 2006, it has become clearly established as a RIG-I specific recognition motif. However, challenging the earlier notion that the 5′ppp moiety was sufficient for RIG-I induction are two recent reports that illustrate a requirement of a double-stranded component in addition to the triphosphate. Both studies made use of previously unavailable, synthetic 5′ppp ssRNA and observed that this molecule was not capable of inducing IFN when introduced into cells. However, the same RNA molecule when generated by T7 in vitro transcription served as a competent activator of the IFN response. The discrepancy appears to result from aberrant transcription events generated by the T7 polymerase. When T7 products were analyzed by gel electrophoresis and sequencing, it was observed that the RNA mixture contained a significant number of RNA molecules of double-stranded nature. After polyacrylamide gel separation, the products corresponding to the true ssRNA size were no longer capable of inducing an IFN response upon transfection into cells. Further characterization of RIG-I activation requirements showed that dsRNA complementarity of at least 10–18 nt was required at the 5′ppp containing end in order to induce RIG-I activity. The nature of these types of RNA molecules fits nicely with the structure of RNA virus panhandles and the ends of copy back defective interfering (DIs) genomes from negative strand RNA viruses, which are characterized as very potent IFN inducers (Strahle et al. 2006). However, since very short synthetic (19–24 nt) RNA molecules were analyzed in these studies, it remains to be seen whether dsRNA structures complementary to the 5′ppp end of an RNA molecule will be required for RIG-I activation with its natural substrates. Since longer products of T7 transcription also induce IFN, it should also be determined whether these RNAs are produced with similar 5′ dsRNA characteristics. Based on numerous studies, it is very likely that all dsRNA molecules regardless of length, sequence, phosphates, or overhangs are capable of binding RIG-I. The question of why some of those molecules induce RIG-I mediated IFN induction while others do not has been recently addressed. In a study by Takahasi et al. (2008), RIG-I was found to be able to bind any dsRNA molecule regardless of presence of 3′ or 5′ overhangs, contrary to some earlier findings by Marques et al. (2006), and ssRNA molecules containing a 5′-ppp, but not ssRNA containing a 5′-OH group or a 5′-monophosphate. This group also found that dsRNA molecules which possessed even a single monophosphate on one RNA strand were still able to activate RIG-I and induce IFN. Taking into account reports by Schlee et al. and Schmidt et al. on the nature of T7 transcribed RNA products it becomes more challenging to interpret this data since it is unclear whether true ssRNA species were analyzed. In addition, the induction of IFN by short dsRNA with a monophosphate is in disagreement with reports of Schlee et al. and Schmidt et al. which showed that a 5′ monophosphate group on a dsRNA molecule was not sufficient for IFN induction. Therefore, it appears that slight differences between the RNA molecules or cells used in the three studies might account for the discrepancy of whether a 5′ppp is required in the context of a short dsRNA, or if a single phosphate is sufficient for RIG-I activation. Nevertheless, it is clear that at least one phosphate group is required for induction of RIG-I signaling, when short dsRNA is used as substrate. The study by Takahasi et al. also provides some insightful information on the discrepancy between poly(I:C) binding and signaling. Poly(I:C) has been shown to bind RIG-I with very high affinity but has been characterized by many groups to signal through MDA5. To address this apparent discrepancy, partial protease digestion of RIG-I/poly(I:C) complex was performed and revealed that this interaction is different from that of RIG-I with 5′ppp-RNA, as different cleavage products were generated (Takahasi et al. 2008). It has not been established whether any viral RNAs can recapitulate the poly(I:C) phenotype of being able to bind RIG-I but not activate its signaling.
Another study which provided some clarity concerning poly(I:C) and dsRNA showed that it was possible to convert poly(I:C) from an MDA5 substrate into a RIG-I substrate by subjecting it to RNAse III digestion, thereby producing shorter poly(I:C) molecules. The length of the resultant poly(I:C) molecules directly correlated with their dependence on either RIG-I or MDA5, with shorter fragments becoming more dependent on RIG-I. The poly(I:C) cleavage products contain 5′ monophosphate ends, supporting the possibility that in the context of some dsRNA molecules a single phosphate might be sufficient for activation of RIG-I. Generation of capped dsRNA products of increasing lengths confirmed the relationship between length dependent activation of RIG-I and MDA5. Whereas dsRNA of 1 kb was entirely dependent on RIG-I for IFN induction, increasing the length to 4 kb progressively led to dual MDA5 and RIG-I dependence. The authors also examined the specificity of RIG-I and MDA5 for the different genomic segments of ReV, a dsRNA virus, previously characterized to be sensed by both sensors. They found that the smallest segment was primarily recognized by RIG-I and the larger ones relied more extensively on both RIG-I and MDA5 (Kato et al. 2008). In addition, the authors demonstrated that RNA isolated from VSV infected cells did not completely lose its ability to induce IFN following CIP treatment, unlike RNA from influenza A infected cells. However, combined digestion of this RNA with CIP and dsRNA-specific RNAse III, led to complete loss of IFN induction. By utilization of a dsRNA specific antibody the authors were able to determine that the size of this molecule in VSV infected cells corresponded to approximately 2.2 kb, whereas dsRNA from the EMCV infected cells, a virus dependent on MDA5 for recognition, was much longer. The effect of poly(I:C) length on RIG-I or MDA5 specificity was confirmed by another group which showed that increasing the length of poly(I:C) correlated with MDA5 specific detection (Ranjith-Kumar et al. 2009). Supporting the claim that RIG-I recognizes shorter dsRNA is yet another study which found that RIG-I was responsible for detection of dsRNA produced by coinfection of cells with Sendai viruses expressing GFP and antisense GFP (Hausmann et al. 2008). It is important to keep in mind that in the above studies short dsRNA refers to RNA species of a few kilobases and the size at which this RNA is no longer capable of being a RIG-I substrate has not been determined.
In addition to 5′ppp and dsRNA, a possible novel PAMP was proposed by Saito et al. in a report demonstrating that RNAs with a high U/A composition induced IFN more efficiently than those without (Saito et al. 2008). In this work, the genomic and replicative intermediate RNAs of HCV were analyzed for their relative IFN inducing ability. The authors found that the 3′NTR region of the HCV genome was a much more potent inducer of IFN than other regions of the genome. This region of HCV genome is particularly rich in polyuradine tracks and upon further examination this polyU sequence composition in conjunction with a 5′ppp proved to lead to increased RIG-I activation. Addition of a triphosphate to a different region of the genome did not improve induction, showing that sequence components other than the triphosphate group determine the extent of RIG-I activation (Saito et al. 2008). The conclusions of this work are, however, confounded by another recent study in which the authors demonstrated that the uridine-rich 3′UTR of fulminant HCV from strain JFH-1 was a relatively weak inducer of IFN, contrary to the HCV strain used in the previous paper (Uzri and Gehrke 2009). Although it appears that stretches of U or A residues do stimulate the activity of RIG-I, additional, yet unknown sequence characteristics determine the RNA’s immunostimulatory potential. The authors did show that the poly-U region could be separated from the 5′ppp by as much as 300 nt and still retain signaling activity, thus possibly explaining how a U-rich 3′ RNA sequence can contribute to RIG-I activation and again implicating the helicase domain in PAMP recognition.
An interesting possibility for RIG-I activation could involve production of cellular RNAs capable of acting as substrates following the initial detection of viral infection. This type of mechanism would act to stimulate IFN production under conditions where viral substrates were limiting, as would presumably be the case early in infection. Indeed, one example of such a mechanism appears to be the production of stimulatory RNAs by RNAse L digestion of cellular mRNA. The resultant small RNAs, with possible double-stranded composition and 3′ monophosphates, induced an IFN-β reporter in a RIG-I and MDA5 dependent manner (Malathi et al. 2007). Another example of cell-mediated synthesis of a RIG-I substrates comes from two recent studies examining the previously reported (Cheng et al. 2007; Rasmussen et al. 2007, 2009; Samanta et al. 2006) involvement of the RIG-I pathway in response to DNA viruses and intracellular bacteria (Ablasser et al. 2009; Chiu et al. 2009). Both reports demonstrate that poly(dA:dT) when introduced into cells served as a template for DNA Polymerase III synthesis of 5′ppp containing dsRNA molecules which in turn activated RIG-I signaling. Inhibition of Pol III activity in infected cells led to loss of IFN induction following infection with DNA viruses, such as Epstein–Barr virus, herpes simplex virus 1 and adenovirus, and intracellular bacterium Legionella pneumophila. Thus, in addition to being the primary receptor for RNA viruses, RIG-I might potentially play an important role in recognition of some DNA viruses and intracellular bacteria.
Identification of MDA5 specific substrates and its mode of RNA recognition have proven very challenging and its specificity for long dsRNA is not understood. Currently, it appears that while RIG-I can recognize a wide size range of dsRNA molecules, MDA5 is not able to be activated by RNA shorter than approximately 2 kb (Kato et al. 2008). The distinction by RIG-I and MDA5 of such large RNA molecules is difficult to explain since the proteins are likely to interact with only a few dozen bases at a time. It is possible that time spent in translocation mode and the concurrent ATP hydrolysis could be critical for MDA5 specific signaling.
RIG-I
Initially identified by Yoneyama et al. (2004) through a cDNA library screen for its ability to induce an IRF reporter upon poly(I:C) treatment, RIG-I has proven to be a key sensor of RNA virus infections and activator of the signaling cascade leading to production of type I IFN. Through a number of studies, RIG-I has been demonstrated to be the main recognition receptor for multiple RNA viruses including Newcastle disease virus (NDV), vesicular stomatitis virus (VSV), Sendai Virus, HCV, Japanese encephalitis virus (JEV), influenza A virus, rabies virus, measles virus, and respiratory syncytial virus (RSV) (Foy et al. 2005; Hornung et al. 2006; Kato et al. 2005, 2006; Liu et al. 2007; Melchjorsen et al. 2005; Pichlmair et al. 2006; Rothenfusser et al. 2005). The physiological importance of RIG-I is highlighted by ex vivo studies from RIG-I knockout MEFs which show drastically reduced interferon levels in response to NDV, VSV, SeV, and influenza ∆NS1 virus infections (Kato et al. 2005, 2006). Infection of RIG-I −/− mice confirm these findings, as levels of IFN and survival of the mice are greatly reduced upon infection with JEV and VSV (Kato et al. 2006).
The signaling cascade of RIG-I continues to be resolved and has proven to be distinct than that of the TLR system. Similar to TLR induction, RIG-I mediated signaling cascade leads to activation of IRF3 and NF-kB (Yoneyama et al. 2004). However, it was shown early on that knockout MEFs of key TLR adaptors MyD88 and TRIF, do not have a defect in RIG-I mediated induction of IFN (Kato et al. 2005; Yoneyama et al. 2004). The critical adaptor for RIG-I signaling was simultaneously identified by four groups as a mitochondrially located, CARD containing protein MAVS (also known as IPS-1, VISA, or Cardif). This adaptor is activated via CARD-CARD association with RIG-I and initiates a signaling cascade leading to activation of IFN-β transcription factors and subsequent production of IFN (Kawai et al. 2005; Meylan et al. 2005; Seth et al. 2005; Xu et al. 2005).
RIG-I can be divided into three basic domains, the N-terminal CARD, central helicase domain, and C-terminal regulatory domain (Fig. 2). The function of these individual domains has been carefully dissected by biochemical and structural studies. The N-terminal tandem CARD domains are required for interaction with the MAVS CARD domain and downstream signaling. Even though only the terminal CARD forms the physical interaction with MAVS, both CARDs are required for signaling and constructs lacking either domain are dominant negative (Saito et al. 2007). When expressed alone, the RIG-I CARD domain induces IFN production in a constitutive, substrate-independent manner. Interestingly, this phenomenon is only observed in the presence of wt RIG-I. When RIGI −/− cells are transfected with the CARD construct no signaling is initiated (Saito et al. 2007). Lack of IFN induction upon overexpression of the full length RIG-I indicates that RIG-I’s native conformation is in an inactive state and requires appropriate viral stimulus to undergo a conformational change required for signaling initiation (Yoneyama et al. 2004, 2005).

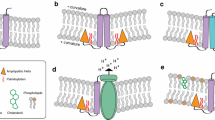
RLR domains and their function. The RLR proteins can be divided into three basic domains. (1) The N-terminal CARD domain, composed of two tandem CARDs. (2) The central helicase domain, belonging to the DExD/H family of RNA helicases. (3) The unique C-terminal domain containing multiple regulatory functions (RD). The CARD domain, present in RIG-I and MDA5 but absent in LGP2, is required for interaction with MAVS and downstream signaling. CARD1 (C1) is involved in physical interaction with the CARD domain of MAVS, whereas CARD2 (C2) of RIG-I undergoes ubiquitination required for RIG-I activation. The helicase domain contains six conserved DExD/H helicase motifs and is involved in translocation/unwinding of RNA and ATP hydrolysis required for RLR function. The helicase domain is also implicated in RNA binding for all three RLR members. The RD is required for recognition and binding of RNA substrates. This domain provides specificity for either 5′ppp containing RNA (RIG-I) or dsRNA (MDA5, LGP2). RD is also required for homo- (RIG-I, MDA5) and hetero- (LGP2) dimer formation, necessary for signaling by these receptors. The RD of RIG-I additionally provides a unique function of autorepression, and RIG-I constructs lacking the RD domain constitutively induce IFN in the absence of RNA stimuli. *Activity has not been shown directly and is assumed based on sequence similarity to the helicase domain of RIG-I
The carboxy-terminal regulatory domain (RD), also referred to as the repressor domain or carboxy-terminal domain (CTD) of RIG-I has proven to contain multiple diverse functions critical to RIG-I activity. Through mutational analysis, this domain was identified to posses the repressor activity responsible for self-inhibition, and constructs lacking the RD are constitutively active. The repression of signaling likely occurs through intramolecular association between the RD and both the CARD and helicase domains (Saito et al. 2007b; Takahasi et al. 2008). A conformational change induced by RNA binding leads to the unfolding of the molecule and exposure of the CARD allowing for downstream signaling. Surprisingly, the RD domain and not the helicase domain was also identified as the primary RNA recognition domain of RIG-I (Cui et al. 2008; Takahasi et al. 2008). Structural studies of the RD have revealed a basic groove located on one side of this domain and an acidic surface on the opposite side. The basic groove is believed to serve as a site of 5′ppp-RNA recognition since mutation of key residues (K858, K888, and H830) within this region led to a loss of RNA binding in vitro and inability of the mutant RIG-I to rescue the phenotype of RIG-I −/− MEFs (Takahasi et al. 2008). The acidic surface on the opposite side of the RD presents a suitable area for interaction with the CARD domain of RIG-I. In addition to signaling repression and RNA recognition, the RD domain has also been characterized as being required for RIG-I dimerization. Like full length RIG-I, the RD alone forms dimers in vitro in the presence of 5′ppp-RNA; unlike RIG-I-∆RD which is unable to dimerize in the presence of 5′ppp-RNA or synthetic dsRNA. Complex formation is also observed between wild-type RIG-I and the RD alone or in conjunction with the helicase domain, providing a mechanism for the dominant negative phenotype of those mutants (Cui et al. 2008; Saito et al. 2007b; Yoneyama et al. 2004).
The exact role of the helicase domain in RIG-I activity has been the most challenging to elucidate. The biochemical roles of this domain can be separated into two related but separate enzymatic functions, ATPase activity and helicase/translocase activity. As with other helicases of the DExD/H family, ATP hydrolysis is required for the helicase function of RIG-I. In support of the ATPase requirement are mutational studies illustrating that walker-type ATP binding site mutants possess a dominant-negative phenotype (Bamming and Horvath 2009; Yoneyama et al. 2004). Additionally, a direct relationship between ATPase activity and immunostimulatory potential of RNA molecules is illustrated by in vitro biochemical analysis with purified RIG-I protein. The same biochemical studies, however, also highlight the fact that while ATP hydrolysis is required for RIG-I signaling it is not sufficient as a large number of RNA molecules are capable of inducing ATPase activity in vitro while failing to induce IFN production upon transfection into cells (Schlee et al. 2009; Schmidt et al. 2009; Takahasi et al. 2008). A critical role of the RD domain in ATP hydrolysis has also been demonstrated, as the helicase domain alone possesses much lower ATPase activity in vitro in the presence of in vitro transcribed (IVT) RNA or synthetic dsRNA than RD with helicase domain (Cui et al. 2008). The role of the helicase/translocase function of RIG-I remains poorly understood. It is not clear whether RIG-I unwinds dsRNA duplexes in vivo or simply translocates on the RNA molecule, leaving it intact. Like all characterized helicases, RIG-I is capable of unwinding dsRNA in vitro (Takahasi et al. 2008). However, the rate of helicase activity of RIG-I in vitro inversely correlated with the immunostimulatory potential of the RNA substrate. As RNA molecules which induced highest helicase activity possessed a 3′ overhang or were complexed with DNA in a heteroduplex, it is difficult to ascertain the relationship of these types of molecules to the viral lifecycle. On the other hand, the lack of unwinding by IFN inducing dsRNA could indicate that RIG-I does not unwind dsRNA in vivo but simply moves along it. Translocation activity of RIG is reported in a study by Myong et al. which illustrated that RIG-I movement on dsRNA does not involve unwinding of the RNA duplex (Myong et al. 2009). The same study also found that the rate of translocation and ATPase activity by full length RIG-I is much higher on 5′ppp containing RNA than on dsRNA with a 5′OH group. The rate of ATP hydrolysis and translocation was similar between full length RIG-I on 5′ppp containing RNA with RIG-I-∆CARD on synthetic dsRNA, implying that the displacement of CARD by 5′ppp binding of the RD allows for more rapid RIG-I movement and associated ATPase activity. Examination of whether change in ssRNA length or dsRNA length had an effect on translocation rate indicated that RIG-I translocates on the dsRNA portion of the molecule. It is important to keep in mind that under infection conditions the RNA molecules recognized by RIG-I are likely to be complexed with nucleoprotein in RNP structures. The demonstrated ability of many helicases to displace protein from RNP complexes during movement (Jankowsky and Fairman 2007) could provide an interesting mechanism for RIG-I substrate recognition where upon binding to any exposed dsRNA the helicase could proceed to move along the dsRNA and displace nucleoprotein until a 5′ppp group was found at which time the CARDs would be displaced and signaling could initiate.
Apart from translocation and ATP hydrolysis, the helicase domain also appears to have an important role in RNA binding. In vitro RNA binding assays show that the RNA affinity of purified RD alone is not as strong as that of the full-length protein. Interestingly, analysis of helicase deletion mutants demonstrated impaired binding affinity to dsRNA but had little effect on in vitro transcribed RNA binding, suggesting that the RD and helicase domains likely recognize different PAMPs within the same RNA molecule (Bamming and Horvath 2009). The contribution of the helicase domain to RNA recognition and binding is further clarified by competition experiments which show that while the RD domain alone has a preference for any 5′ppp molecule (either double or single stranded) the full length protein prefers to bind dsRNA with 5′OH groups than 5′ppp ssRNA (Schmidt et al. 2009). However, in vitro binding analysis of the purified helicase domain, failed to show dsRNA interaction. Therefore, it appears that the RD and the helicase domain within the full length protein exhibit cooperative binding properties, as neither domain alone possesses the complete RNA affinity of the full length molecule (Takahasi et al. 2008). Together these findings support the notion that the helicase domain may only recognize dsRNA molecules, while the RD domain has specificity to both dsRNA and the 5′ppp.
A large number of proteins have been characterized as regulators of RIG-I activity in recent years. One of the best characterized to date is an E3 ubiquitin and ISG15 ligase tripartite motif protein 25 (TRIM25). TRIM25 acts as a positive regulator of RIG-I by adding a critical K172 K63-linked ubiquitin group to the RIG-I CARD domain. The loss of lysine 172 (and subsequent lack of ubiquitination) is correlated with loss of RIG-I/MAVS interaction and lack of IFN production. Supporting an important role of TRIM25 as a positive regulator of IFN response is the reduced ability of TRIM25 −/− MEFs to produce IFN following Sendai virus infection (Gack et al. 2007). The unique roles of RIG-I tandem CARD domains was partially deciphered when it was shown that CARD1 is required for TRIM25 binding while CARD2 serves as a target for TRIM25 mediated ubiquitination. Therefore, the presence of both CARDs is necessary for interaction with MAVS. Interestingly, a RIG-I splice variant lacking residues 36–80 in its first CARD domain was identified as being produced following viral infection. As this splice variant is unable to undergo TRIM25-mediated ubiquitination, it possesses dominant negative activity and is proposed to play a role in negative feedback regulation of RIG-I signaling (Gack et al. 2008). Supporting an important role of TRIM25 in activation of RIG-I is a recent study describing inhibition of TRIM25 activity by influenza A NS1 protein, a well-characterized viral antagonist of the innate immune system (Egorov et al. 1998; Garcia-Sastre et al. 1998). In this study, NS1 was shown to prevent oligomerization of TRIM25 by direct interaction and therefore prevent the ability of TRIM25 to ubiquitinate and activate RIG-I. Chimeric viruses with NS1 mutations that lack the ability to bind TRIM25 resulted in an attenuated viral phenotype (Gack et al. 2009). In addition to TRIM25, a number of other ubiquitinases and deubiquitinases have been proposed to regulate RIG-I activity. Riplet/RNF135/REUL, an E3 ligase was identified by two independent groups and shown to play a positive role in RIG-I signaling. However, the two reports diverged on whether the C- or N-terminal region of RIG-I was being ubiquitinated (Gao et al. 2009; Oshiumi et al. 2009). Another E3 ligase, RNF125 has been proposed to negatively regulate RIG-I and target it for proteosomal degradation (Arimoto et al. 2007). CYLD, a deubiquitinase, has been proposed to remove polyubiquitin chains from RIG-I and have a negative effect on RIG-I signaling (Friedman et al. 2008). RIG-I signaling has also been shown to be negatively regulated by gC1qR, a multifunctional ubiquitously expressed protein. Following viral infection gC1qR was shown to translocate to the mitochondria and through its interaction with MAVS inhibits RIG-I/MAVS association (Xu et al. 2009). ER localized, stimulator of IFN genes (STING) is the first ER resident protein shown to interact with RIG-I and be required for its full activity; possibly implicating ER associated functions, such as translation or stress response in RIG-I signaling (Ishikawa and Barber 2008).
MDA5
MDA5 was identified as a DExD/H helicase family member during a screening of genes, which were upregulated by IFN treatment and at the same time involved in growth suppression of melanoma cells. Similar to RIG-I, MDA5 contains two N-terminal CARD domains, a dsRNA-dependent ATPase motif within a central helicase domain, and a regulatory C terminal domain (Kang et al. 2002). The initial report which implicated MDA5 as an antiviral protein showed that the interferon antagonist V proteins of Simian Virus 5 (SV5) and of other paramyxoviruses interact with MDA5 (Andrejeva et al. 2004). Later, it was demonstrated that V proteins of all paramyxoviruses directly bind to MDA5 and prevent its dsRNA binding and self-association, thereby inhibiting downstream signaling (Childs et al. 2009). Similarly to RIG-I in the presence of RNA activators, overexpression of MDA5 in the presence of poly(I:C) induces activation of an IFN-β reporter construct and knockdown of endogenous MDA5 by siRNA inhibits IFN induction following poly(I:C) transfection (Andrejeva et al. 2004). Like RIG-I, the truncated CARD domain of MDA5 is capable of inducing an antiviral response independently of stimuli, and the helicase domain when expressed alone possesses a dominant negative phenotype (Andrejeva et al. 2004). However, unlike RIG-I, the RD of MDA5 does not appear to contain autoinhibitory activity, since expression of full length MDA5 and MDA5-∆RD induce the same amount of IFN-reporter activation (Saito et al. 2007b). The negative regulation of MDA5 in uninfected cells is proposed to be maintained by dihydroxyacetone kinase (DAK), a protein which specifically inhibits MDA5 but not RIG-I mediated signaling, and most likely by other yet undiscovered regulators (Diao et al. 2007). The signaling cascade of MDA5 appears to be identical to that of RIG-I, leading to the conclusion that the two sensors act in parallel after being triggered by their respective viral PAMPS (Yoneyama et al. 2005). Studies in MDA5 −/− mice show this receptor to be specific for in vivo recognition of poly(I:C) and picornaviruses, including the encephalomyocarditis virus (EMCV) and mengovirus (Gitlin et al. 2006; Kato et al. 2006). These knockout mice respond normally to JEV and VSV infections, supporting the importance of a RIG-I dependent recognition of those viruses. The inability of picornaviruses to be recognized by RIG-I has been attributed to the lack of 5′ppp in the genome of these viruses. However, a recent study showing that picornavirus proteinase 3C(pro) specifically degrades RIG-I, suggests that this sensor may also play a role in picornavirus infections (Barral et al. 2009). In addition to recognition of picornaviruses, MDA5 has been shown to play a major role in recognition of a coronavirus Mouse Hepatitis Virus (MHV) in brain macrophages (Roth-Cross et al. 2008) and a murine norovirus in DCs (McCartney et al. 2008). Viruses such as Dengue virus type 2 (DEN2), type 3 Dearing (T3D) reovirus, and type 1 Lang (T1L) reovirus were shown to be recognized by both RIG-I and MDA5, illustrating the sometimes overlapping functions of these two receptors (Loo et al. 2007). The relative contribution of RIG-I and MDA5 to recognition of any particular virus appears to be highly cell-type specific. For example, Sendai virus is clearly shown to rely on RIG-I sensing in MEFs (Kato et al. 2006) but is recognized primarily by MDA5 in DCs (Yount et al. 2008). Since Sendai virus is known to express both MDA5 and RIG-I specific inhibitors, the ability of this virus to be recognized by both sensors is not surprising (Strahle et al. 2007). It is unclear whether this cell-type specific recognition of viruses is a result of differential expression of the sensors or whether cell-type specific differences in viral replication lead to different modes of recognition. Together, the above data lead to a model where MDA5 and RIG-I may possess both overlapping and distinct roles in RNA virus detection. For the vast majority of viruses and cell types, deletion of one of the sensors does not completely abrogate IFN induction (Diao et al. 2007; Kato et al. 2006), as opposed to deletion of the common adaptor MAVS, which leads to a much more severe phenotype (Kawai et al. 2005; Meylan et al. 2005; Seth et al. 2005; Xu et al. 2005). It is plausible that many viruses produce RNA molecules detected by both sensors and the relative abundance of these molecules dictates which receptor will play a predominant role in IFN production. An interesting question is whether most viral infections result in production of multiple distinct PAMPs (i.e. misformed RNPs and double stranded replicative intermediates) or whether the same basic PAMP is being recognized by both RIG-I and MDA5 depending on the abundance of these receptors and the substrates.
Structural studies of RD domains of RIG-I, MDA5, and LGP2 have revealed very similar overall architecture (Cui et al. 2008; Li et al. 2009a; Murali et al. 2008; Pippig et al. 2009; Takahasi et al. 2008, 2009). All RDs contain four conserved cysteine residues which participate in zinc binding and appear necessary for RD function. A basic surface on one side of the RD is proposed to play a role in RNA biding for all three receptors, with minor structural differences accounting for the difference in specificities between the molecules (Cui et al. 2008; Pippig et al. 2009; Takahasi et al. 2008). Binding studies show that unlike RDs of RIG-I and LGP2, the RD of MDA5 associates very weakly or not at all with dsRNA or IVT RNA (Cui et al. 2008; Li et al. 2009a; Takahasi et al. 2008, 2009). These differences in binding are in agreement with values obtained for full-length proteins, with LGP2 having highest affinity for dsRNA and MDA5 the weakest (Takahasi et al. 2009; Yoneyama et al. 2005). The weak affinity of MDA5 to dsRNA and poly(I:C) is puzzling in light of its role in dsRNA detection, and the precise mechanism of MDA5 activation by dsRNA remains to be understood.
LGP2
The third member of the RLR family, LGP2, has been implicated as a negative regulator of RIG-I and a positive regulator of MDA; however, the exact role of this molecule in viral infection remains controversial. Like RIG-I and MDA5, LGP2 contains a DExH/D box helicase domain and a carboxy-terminal RD. However, unlike those receptors, it lacks the CARD domain, and therefore, it is unable to signal through MAVS. Initial studies of LGP2 function demonstrated that upon overexpression this molecule had a negative effect on IFN production following Sendai virus or NDV infection, or poly(I:C) transfection. LGP2 activity was confirmed to be specific to RLR signaling as its expression had no effect on TLR3 mediated IFN induction. (Rothenfusser et al. 2005; Yoneyama et al. 2005). The role of LGP2 as a negative regulator of RIG-I signaling was confirmed in Lgp2 −/− mice infected with VSV (Venkataraman et al. 2007). The exact mechanism by which LGP2 interferes with RIG-I signaling has not been resolved. One possibility is that LGP2 simply sequesters dsRNA from RIG-I. This potential mechanism is supported by LGP2’s high binding affinity for poly(I:C) and synthetic dsRNA in vitro, and the high expression level of this protein following virus infection (Yoneyama et al. 2005). Inhibition of RIG-I dimer formation has also been proposed as the possible mode of LGP2 inhibitory activity. This mechanism is supported by observed complex formation between LGP2 and RIG-I in infected cells and the structural similarity of LGP2 to RIG-I ∆CARD (Rothenfusser et al. 2005; Saito et al. 2007a). Finally, observed association of LGP2 with MAVS in virus infected cells leads to a possible third mechanism for its activity. Komuro et al. demonstrated that LGP2 was able to compete with IKKε for MAVS binding, therefore inhibiting a downstream step in RIG-I mediated activation pathway (Komuro and Horvath 2006). The RD of LGP2 when expressed alone is sufficient to inhibit RIG-I mediated signaling, but it is not as efficient as full length LGP2 (Murali et al. 2008). Similarly to RIG-I RD, LGP2 RD is able to bind RNA and form dimers in vitro. The structure of LGP2 RD is very similar to that of the RIG-I RD (Pippig et al. 2009). Surprisingly, binding assays with purified protein have revealed that LGP2 possesses no specificity to 5′ppp-RNA, and instead has very high affinity for any dsRNA, with the presence of phosphate groups appearing irrelevant. (Murali et al. 2008; Pippig et al. 2009). The RD of LGP2 has been shown to bind to the blunt-end of dsRNA and not to the phosphate backbone of the molecule (Li et al. 2009b). Since RIG-I interacts with both 5′ppp-RNA and dsRNA, this finding still allows LGP2 to inhibit RIG-I signaling by dsRNA sequesteration or inhibition of RIG-I duplex formation. A recent study showed that LGP2 defective in RNA binding inhibited RIG-I to the same degree as wild-type LGP2; supporting the hypothesis that dsRNA sequesteration is not the primary mode of LGP2 mediated inhibition (Li et al. 2009b).
Unlike its negative regulation of RIG-I induced signaling, LGP2 appears to have an enhancing effect on MDA5 specific IFN induction. Similarly to its complex formation with RIG-I, LGP2 has been found to interact with MDA5 in infected cells (Saito et al. 2007a). The initial observation that LGP2 might act as a positive regulator of MDA5 came from an observation that LGP2 knockout mice infected with EMCV exhibited reduced levels of IFN in sera and increased mortality. In agreement with the in vivo results, LGP2 knockout MEFs were also severely limited in IFN production after poly(I:C) transfection (Venkataraman et al. 2007). Since the same animals were more resistant to infections with RIG-I specific viruses, these results suggested a differential role of LGP2 in regulation of RIG-I and MDA5 signaling. Supporting this hypothesis is a recent study showing that poly(I:C) activation of MDA5 peaked in the presence of an equal ratio of LGP2, and that LGP2 constructs deficient in RNA binding or individual domains alone were not capable of augmenting MDA5 directed signaling (Pippig et al. 2009). Also supporting the possible unique link between MDA5 and LGP2 is a study showing that paramyxovirus V proteins interact with both MDA5 and LGP2, but not RIG-I. This helicase domain mediated interaction leads to reduction of ATPase activity of both receptors (Parisien et al. 2009). The possible role of LGP2 as a negative regulator of RIG-I and a positive regulator of MDA5 is very puzzling since the two receptors are thought to act in parallel to induce IFN following virus infection. Possible abundance and expression kinetic differences of these three proteins in various cell types may account for this intriguing observation.
Conclusions
Proteins involved in the recognition of viral infection have only recently been identified and the initial characterization of their roles in this intricate system is just now starting to be understood. Identification of the TLR and RLR family of sensors within the last several years has provided critically important and necessary information concerning cellular recognition of RNA viruses. To date, these two receptor families are the only known systems sufficient for IFN induction in response to RNA viruses; although evidence from knockout studies point to the existence of other undiscovered receptors. Other previously described viral sensors, such as Protein Kinase R (PKR) and 2′-5′oligoadenylate synthetase (2-5 OAS) (together with RNAse L) are not sufficient for IFN production but likely play a role in mediating the IFN response. Indeed, PKR is a known dsRNA-dependent inducer of NFκB, and RNAse L has recently been shown to be responsible for generation of endogenous RNA molecules that may be stimulatory to IFN production (Kumar et al. 1994; Malathi et al. 2007). In addition, it has been shown that cells lacking in PKR produce lower amounts on IFN-α after infection with EMCV, supporting its modulating role in IFN production (Der and Lau 1995). PKR’s ability to be activated by 5′ppp RNA presents another interesting addition to PAMP/PRR interaction story (Nallagatla et al. 2007).
The search for viral recognition motifs has been a focus of virology research for the past 50 years. With the recent identification of proteins involved in viral recognition, the characteristics of viral motifs that stimulate an antiviral response are now rapidly being discovered. In addition to dsRNA, which has become a paradigm molecule for viral detection, the recent discovery of 5′ppp RNA as a possible viral PAMP adds an important piece to this puzzle. Yet, it remains to be shown whether these molecules are indeed physiological triggers of the innate immune system, and if so whether their presence is sufficient for initiation of the antiviral response. It is also important to understand if these RNA molecules are products of viral errors in replication or are an inherent part of the viral life cycle. By convention, virologists have assumed that generation of viral PAMPs is a byproduct of errors in viral replication, and although this type of PAMP generation likely contributes to viral recognition, it is also possible that a component of the virus structure or lifecycle becomes recognized by the cell very early on in viral infection. Indeed, phosphorylation of Ik-B can be observed as early as five minutes after infection of glial cells with measles virus indicating activation of upstream components almost immediately after virus entry (Dhib-Jalbut et al. 1999). It is always possible that differences in virus preparation, cells, and infection conditions could account for observations which would not be relevant to naturally occurring viruses and infections but it is also likely that yet undiscovered sensors or viral PAMPs ensure that infection is recognized immediately in the invaded cell leading to a more rapid innate immune response.
Modulation of the innate immune response offers promising and novel approaches for the treatment of infectious agents, cancer, allergies, and autoimmune disorders. Incorporation of TLR and RLR agonists as adjuvants in vaccines is likely to offer significant increase in vaccine potency and efficiency. Currently the only approved TLR agonist is imiquimod (a TLR7 agonist from 3 M Pharmaceuticals) which has been used for treatment of various skin disorders for over 10 years. However, many other TLR agonists are being actively investigated in animal studies and clinical trials (Panter et al. 2009). Liposome-nucleic acid complexes which specifically target TLR3, TLR7/8, and TLR9 have also shown to be very effective in treatment of certain cancers and acute viral and bacterial infections (Dow 2008). Since TLR and RLR signaling often leads to activation of many different cytokines, it is possible that the use of an array of multiple agonists and antagonists could potentially be used to finely regulate the immune response. Moreover, comprehensive screening and evaluation of molecules which act as antagonists of TLRs and RLRs could lead to new insights for the development of potent therapeutics for treatment of autoimmune disorders.
Despite the wealth of new and exciting information concerning TLRs and RLR pathways, many questions remain to be answered. Specifically, what is the physiological significance of each of these systems and which viral triggers are responsible for their activation. The problem of how a cell manages to differentiate between self and non-self remains one of the fundamental questions in virology and immunology.
References
Ablasser A, Bauernfeind F, Hartmann G, Latz E, Fitzgerald KA, Hornung V (2009) RIG-I-dependent sensing of poly(dA:dT) through the induction of an RNA polymerase III-transcribed RNA intermediate. Nat Immunol 10:1065–1072
Akira S, Hemmi H (2003) Recognition of pathogen-associated molecular patterns by TLR family. Immunol Lett 85:85–95
Akira S, Takeda K (2004) Functions of toll-like receptors: lessons from KO mice. C R Biol 327:581–589
Alexopoulou L, Holt AC, Medzhitov R, Flavell RA (2001) Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature 413:732–738
Andrejeva J, Childs KS, Young DF, Carlos TS, Stock N, Goodbourn S, Randall RE (2004) The V proteins of paramyxoviruses bind the IFN-inducible RNA helicase, mda-5, and inhibit its activation of the IFN-beta promoter. Proc Natl Acad Sci USA 101:17264–17269
Arimoto K, Takahashi H, Hishiki T, Konishi H, Fujita T, Shimotohno K (2007) Negative regulation of the RIG-I signaling by the ubiquitin ligase RNF125. Proc Natl Acad Sci USA 104:7500–7505
Asselin-Paturel C, Boonstra A, Dalod M, Durand I, Yessaad N, Dezutter-Dambuyant C, Vicari A, O’Garra A, Biron C, Briere F et al (2001) Mouse type I IFN-producing cells are immature APCs with plasmacytoid morphology. Nat Immunol 2:1144–1150
Bamming D, Horvath CM (2009) Regulation of signal transduction by enzymatically inactive antiviral RNA helicase proteins MDA5, RIG-I, and LGP2. J Biol Chem 284:9700–9712
Barral PM, Sarkar D, Fisher PB, Racaniello VR (2009) RIG-I is cleaved during picornavirus infection. Virology 391:171–176
Biron CA (2001) Interferons alpha and beta as immune regulators—a new look. Immunity 14:661–664
Cheng G, Zhong J, Chung J, Chisari FV (2007) Double-stranded DNA and double-stranded RNA induce a common antiviral signaling pathway in human cells. Proc Natl Acad Sci USA 104:9035–9040
Childs KS, Andrejeva J, Randall RE, Goodbourn S (2009) Mechanism of mda-5 Inhibition by paramyxovirus V proteins. J Virol 83:1465–1473
Chiu YH, Macmillan JB, Chen ZJ (2009) RNA Polymerase III Detects Cytosolic DNA and Induces Type I Interferons through the RIG-I Pathway. Cell 138:576–591
Colby C, Duesberg PH (1969) Double-stranded RNA in vaccinia virus infected cells. Nature 222:940–944
Colby C, Morgan MJ (1971) Interferon induction and action. Annu Rev Microbiol 25:333–360
Cui S, Eisenacher K, Kirchhofer A, Brzozka K, Lammens A, Lammens K, Fujita T, Conzelmann KK, Krug A, Hopfner KP (2008) The C-terminal regulatory domain is the RNA 5′-triphosphate sensor of RIG-I. Mol Cell 29:169–179
Daffis S, Samuel MA, Suthar MS, Gale M Jr, Diamond MS (2008) Toll-like receptor 3 has a protective role against West Nile virus infection. J Virol 82:10349–10358
Der SD, Lau AS (1995) Involvement of the double-stranded-RNA-dependent kinase PKR in interferon expression and interferon-mediated antiviral activity. Proc Natl Acad Sci USA 92:8841–8845
Dhib-Jalbut S, Xia J, Rangaviggula H, Fang YY, Lee T (1999) Failure of measles virus to activate nuclear factor-kappa B in neuronal cells: implications on the immune response to viral infections in the central nervous system. J Immunol 162:4024–4029
Diao F, Li S, Tian Y, Zhang M, Xu LG, Zhang Y, Wang RP, Chen D, Zhai Z, Zhong B et al (2007) Negative regulation of MDA5—but not RIG-I-mediated innate antiviral signaling by the dihydroxyacetone kinase. Proc Natl Acad Sci USA 104:11706–11711
Diebold SS (2008) Recognition of viral single-stranded RNA by Toll-like receptors. Adv Drug Deliv Rev 60:813–823
Diebold SS, Kaisho T, Hemmi H, Akira S, Reis e Sousa C (2004) Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science 303:1529–1531
Dow S (2008) Liposome-nucleic acid immunotherapeutics. Expert Opin Drug Deliv 5:11–24
Edelmann KH, Richardson-Burns S, Alexopoulou L, Tyler KL, Flavell RA, Oldstone MB (2004) Does Toll-like receptor 3 play a biological role in virus infections? Virology 322:231–238
Egorov A, Brandt S, Sereinig S, Romanova J, Ferko B, Katinger D, Grassauer A, Alexandrova G, Katinger H, Muster T (1998) Transfectant influenza A viruses with long deletions in the NS1 protein grow efficiently in Vero cells. J Virol 72:6437–6441
Field AK, Tytell AA, Lampson GP, Hilleman MR (1967) Inducers of interferon and host resistance. II. Multistranded synthetic polynucleotide complexes. Proc Natl Acad Sci USA 58:1004–1010
Field AK, Tytell AA, Lampson GP, Hilleman MR (1968) Inducers of interferon and host resistance, V. In vitro studies. Proc Natl Acad Sci USA 61:340–346
Foy E, Li K, Sumpter R Jr, Loo YM, Johnson CL, Wang C, Fish PM, Yoneyama M, Fujita T, Lemon SM et al (2005) Control of antiviral defenses through hepatitis C virus disruption of retinoic acid-inducible gene-I signaling. Proc Natl Acad Sci USA 102:2986–2991
Friedman CS, O’Donnell MA, Legarda-Addison D, Ng A, Cardenas WB, Yount JS, Moran TM, Basler CF, Komuro A, Horvath CM et al (2008) The tumour suppressor CYLD is a negative regulator of RIG-I-mediated antiviral response. EMBO Rep 9:930–936
Gack MU, Shin YC, Joo CH, Urano T, Liang C, Sun L, Takeuchi O, Akira S, Chen Z, Inoue S et al (2007) TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 446:916–920
Gack MU, Kirchhofer A, Shin YC, Inn KS, Liang C, Cui S, Myong S, Ha T, Hopfner KP, Jung JU (2008) Roles of RIG-I N-terminal tandem CARD and splice variant in TRIM25-mediated antiviral signal transduction. Proc Natl Acad Sci USA 105:16743–16748
Gack MU, Albrecht RA, Urano T, Inn KS, Huang IC, Carnero E, Farzan M, Inoue S, Jung JU, Garcia-Sastre A (2009) Influenza A virus NS1 targets the ubiquitin ligase TRIM25 to evade recognition by the host viral RNA sensor RIG-I. Cell Host Microbe 5:439–449
Gao D, Yang YK, Wang RP, Zhou X, Diao FC, Li MD, Zhai ZH, Jiang ZF, Chen DY (2009) REUL is a novel E3 ubiquitin ligase and stimulator of retinoic-acid-inducible gene-I. PLoS ONE 4:e5760
Garcia-Sastre A, Egorov A, Matassov D, Brandt S, Levy DE, Durbin JE, Palese P, Muster T (1998) Influenza A virus lacking the NS1 gene replicates in interferon-deficient systems. Virology 252:324–330
Gitlin L, Barchet W, Gilfillan S, Cella M, Beutler B, Flavell RA, Diamond MS, Colonna M (2006) Essential role of mda-5 in type I IFN responses to polyriboinosinic:polyribocytidylic acid and encephalomyocarditis picornavirus. Proc Natl Acad Sci USA 103:8459–8464
Gorden KB, Gorski KS, Gibson SJ, Kedl RM, Kieper WC, Qiu X, Tomai MA, Alkan SS, Vasilakos JP (2005) Synthetic TLR agonists reveal functional differences between human TLR7 and TLR8. J Immunol 174:1259–1268
Habjan M, Andersson I, Klingstrom J, Schumann M, Martin A, Zimmermann P, Wagner V, Pichlmair A, Schneider U, Muhlberger E et al (2008) Processing of genome 5’ termini as a strategy of negative-strand RNA viruses to avoid RIG-I-dependent interferon induction. PLoS ONE 3:e2032
Hausmann S, Marq JB, Tapparel C, Kolakofsky D, Garcin D (2008) RIG-I and dsRNA-induced IFNbeta activation. PLoS ONE 3:e3965
Heer AK, Shamshiev A, Donda A, Uematsu S, Akira S, Kopf M, Marsland BJ (2007) TLR signaling fine-tunes anti-influenza B cell responses without regulating effector T cell responses. J Immunol 178:2182–2191
Heil F, Hemmi H, Hochrein H, Ampenberger F, Kirschning C, Akira S, Lipford G, Wagner H, Bauer S (2004) Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science 303:1526–1529
Henle W (1950) Interference phenomena between animal viruses: a review. J Immunol 64:203–236
Hidmark AS, McInerney GM, Nordstrom EK, Douagi I, Werner KM, Liljestrom P, Karlsson Hedestam GB (2005) Early alpha/beta interferon production by myeloid dendritic cells in response to UV-inactivated virus requires viral entry and interferon regulatory factor 3 but not MyD88. J Virol 79:10376–10385
Hornung V, Ellegast J, Kim S, Brzozka K, Jung A, Kato H, Poeck H, Akira S, Conzelmann KK, Schlee M et al (2006) 5′-Triphosphate RNA is the ligand for RIG-I. Science 314:994–997
Huppert J, Hillova J, Gresland L (1969) Viral RNA synthesis in chicken cells infected with ultraviolet irradiated Newcastle Disease Virus. Nature 223:1015–1017
Isaacs A, Lindenmann J (1957) Virus interference. I. The interferon. Proc R Soc Lond B Biol Sci 147:258–267
Isaacs A, Cox RA, Rotem Z (1963) Foreign nucleic acids as the stimulus to make interferon. Lancet 2:113–116
Ishikawa H, Barber GN (2008) STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 455:674–678
Janeway CA Jr (1989) Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb Symp Quant Biol 54(Pt 1):1–13
Jankowsky E, Fairman ME (2007) RNA helicases—one fold for many functions. Curr Opin Struct Biol 17:316–324
Johansson C, Wetzel JD, He J, Mikacenic C, Dermody TS, Kelsall BL (2007) Type I interferons produced by hematopoietic cells protect mice against lethal infection by mammalian reovirus. J Exp Med 204:1349–1358
Kang DC, Gopalkrishnan RV, Wu Q, Jankowsky E, Pyle AM, Fisher PB (2002) mda-5: An interferon-inducible putative RNA helicase with double-stranded RNA-dependent ATPase activity and melanoma growth-suppressive properties. Proc Natl Acad Sci USA 99:637–642
Kariko K, Buckstein M, Ni H, Weissman D (2005) Suppression of RNA recognition by Toll-like receptors: the impact of nucleoside modification and the evolutionary origin of RNA. Immunity 23:165–175
Kato H, Sato S, Yoneyama M, Yamamoto M, Uematsu S, Matsui K, Tsujimura T, Takeda K, Fujita T, Takeuchi O et al (2005) Cell type-specific involvement of RIG-I in antiviral response. Immunity 23:19–28
Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ et al (2006) Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 441:101–105
Kato H, Takeuchi O, Mikamo-Satoh E, Hirai R, Kawai T, Matsushita K, Hiiragi A, Dermody TS, Fujita T, Akira S (2008) Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J Exp Med 205:1601–1610
Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H, Ishii KJ, Takeuchi O, Akira S (2005) IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat Immunol 6:981–988
Komuro A, Horvath CM (2006) RNA- and virus-independent inhibition of antiviral signaling by RNA helicase LGP2. J Virol 80:12332–12342
Koyama S, Ishii KJ, Kumar H, Tanimoto T, Coban C, Uematsu S, Kawai T, Akira S (2007) Differential role of TLR- and RLR-signaling in the immune responses to influenza A virus infection and vaccination. J Immunol 179:4711–4720
Kumagai Y, Takeuchi O, Kato H, Kumar H, Matsui K, Morii E, Aozasa K, Kawai T, Akira S (2007) Alveolar macrophages are the primary interferon-alpha producer in pulmonary infection with RNA viruses. Immunity 27:240–252
Kumar A, Haque J, Lacoste J, Hiscott J, Williams BR (1994) Double-stranded RNA-dependent protein kinase activates transcription factor NF-kappa B by phosphorylating I kappa B. Proc Natl Acad Sci USA 91:6288–6292
Kurt-Jones EA, Popova L, Kwinn L, Haynes LM, Jones LP, Tripp RA, Walsh EE, Freeman MW, Golenbock DT, Anderson LJ et al (2000) Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nat Immunol 1:398–401
Lampson GP, Tytell AA, Field AK, Nemes MM, Hilleman MR (1967) Inducers of interferon and host resistance. I. Double-stranded RNA from extracts of Penicillium funiculosum. Proc Natl Acad Sci USA 58:782–789
Le Bon A, Tough DF (2002) Links between innate and adaptive immunity via type I interferon. Curr Opin Immunol 14:432–436
Le Goffic R, Balloy V, Lagranderie M, Alexopoulou L, Escriou N, Flavell R, Chignard M, Si-Tahar M (2006) Detrimental contribution of the Toll-like receptor (TLR)3 to influenza A virus-induced acute pneumonia. PLoS Pathog 2:e53
Le Goffic R, Pothlichet J, Vitour D, Fujita T, Meurs E, Chignard M, Si-Tahar M (2007) Cutting Edge: Influenza A virus activates TLR3-dependent inflammatory and RIG-I-dependent antiviral responses in human lung epithelial cells. J Immunol 178:3368–3372
Lee HK, Lund JM, Ramanathan B, Mizushima N, Iwasaki A (2007) Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science 315:1398–1401
Li X, Lu C, Stewart M, Xu H, Strong RK, Igumenova T, Li P (2009a) Structural basis of double-stranded RNA recognition by the RIG-I like receptor MDA5. Arch Biochem Biophys 488:23–33
Li X, Ranjith-Kumar CT, Brooks MT, Dharmaiah S, Herr AB, Kao C, Li P (2009b) The RIG-I like receptor LGP2 recognizes the termini of double-stranded RNA. J Biol Chem 284:13881–13891
Liu P, Jamaluddin M, Li K, Garofalo RP, Casola A, Brasier AR (2007) Retinoic acid-inducible gene I mediates early antiviral response and Toll-like receptor 3 expression in respiratory syncytial virus-infected airway epithelial cells. J Virol 81:1401–1411
Loo YM, Fornek J, Crochet N, Bajwa G, Perwitasari O, Martinez-Sobrido L, Akira S, Gill MA, Garcia-Sastre A, Katze MG et al (2007) Distinct RIG-I and MDA5 Signaling by RNA Viruses in Innate Immunity. J Virol 82:335–345
Lund JM, Alexopoulou L, Sato A, Karow M, Adams NC, Gale NW, Iwasaki A, Flavell RA (2004) Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proc Natl Acad Sci USA 101:5598–5603
Malathi K, Dong B, Gale M Jr, Silverman RH (2007) Small self-RNA generated by RNase L amplifies antiviral innate immunity. Nature 448:816–819
Marques JT, Devosse T, Wang D, Zamanian-Daryoush M, Serbinowski P, Hartmann R, Fujita T, Behlke MA, Williams BR (2006) A structural basis for discriminating between self and nonself double-stranded RNAs in mammalian cells. Nat Biotechnol 24:559–565
McCartney SA, Thackray LB, Gitlin L, Gilfillan S, Virgin HW, Colonna M (2008) MDA-5 recognition of a murine norovirus. PLoS Pathog 4:e1000108
Melchjorsen J, Jensen SB, Malmgaard L, Rasmussen SB, Weber F, Bowie AG, Matikainen S, Paludan SR (2005) Activation of innate defense against a paramyxovirus is mediated by RIG-I and TLR7 and TLR8 in a cell-type-specific manner. J Virol 79:12944–12951
Meylan E, Curran J, Hofmann K, Moradpour D, Binder M, Bartenschlager R, Tschopp J (2005) Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 437:1167–1172
Murali A, Li X, Ranjith-Kumar CT, Bhardwaj K, Holzenburg A, Li P, Kao CC (2008) Structure and function of LGP2, a DEX(D/H) helicase that regulates the innate immunity response. J Biol Chem 283:15825–15833
Myong S, Cui S, Cornish PV, Kirchhofer A, Gack MU, Jung JU, Hopfner KP, Ha T (2009) Cytosolic viral sensor RIG-I is a 5’-triphosphate-dependent translocase on double-stranded RNA. Science 323:1070–1074
Nagata S, Taira H, Hall A, Johnsrud L, Streuli M, Ecsodi J, Boll W, Cantell K, Weissmann C (1980) Synthesis in E. coli of a polypeptide with human leukocyte interferon activity. Nature 284:316–320
Nallagatla SR, Hwang J, Toroney R, Zheng X, Cameron CE, Bevilacqua PC (2007) 5’-triphosphate-dependent activation of PKR by RNAs with short stem-loops. Science 318:1455–1458
Nallagatla SR, Toroney R, Bevilacqua PC (2008) A brilliant disguise for self RNA: 5’-end and internal modifications of primary transcripts suppress elements of innate immunity. RNA Biol 5:140–144
Oshiumi H, Matsumoto M, Hatakeyama S, Seya T (2009) Riplet/RNF135, a RING finger protein, ubiquitinates RIG-I to promote interferon-beta induction during the early phase of viral infection. J Biol Chem 284:807–817
Panter G, Kuznik A, Jerala R (2009) Therapeutic applications of nucleic acids as ligands for Toll-like receptors. Curr Opin Mol Ther 11:133–145
Parisien JP, Bamming D, Komuro A, Ramachandran A, Rodriguez JJ, Barber G, Wojahn RD, Horvath CM (2009) A Shared Interface Mediates Paramyxovirus Interference with Antiviral RNA Helicases MDA5 and LGP2. J Virol 83:7252–7260
Pichlmair A, Schulz O, Tan CP, Naslund TI, Liljestrom P, Weber F, Reis e Sousa C (2006) RIG-I-mediated antiviral responses to single-stranded RNA bearing 5’-phosphates. Science 314:997–1001
Pippig DA, Hellmuth JC, Cui S, Kirchhofer A, Lammens K, Lammens A, Schmidt A, Rothenfusser S, Hopfner KP (2009) The regulatory domain of the RIG-I family ATPase LGP2 senses double-stranded RNA. Nucleic Acids Res 37:2014–2025
Plumet S, Herschke F, Bourhis JM, Valentin H, Longhi S, Gerlier D (2007) Cytosolic 5′-triphosphate ended viral leader transcript of measles virus as activator of the RIG I-mediated interferon response. PLoS ONE 2:e279
Ranjith-Kumar CT, Murali A, Dong W, Srisathiyanarayanan D, Vaughan R, Ortiz-Alacantara J, Bhardwaj K, Li X, Li P, Kao CC (2009) Agonist and antagonist recognition by RIG-I, a cytoplasmic innate immunity receptor. J Biol Chem 284:1155–1165
Rasmussen SB, Sorensen LN, Malmgaard L, Ank N, Baines JD, Chen ZJ, Paludan SR (2007) Type I interferon production during herpes simplex virus infection is controlled by cell-type-specific viral recognition through Toll-like receptor 9, the mitochondrial antiviral signaling protein pathway, and novel recognition systems. J Virol 81:13315–13324
Rasmussen SB, Jensen SB, Nielsen C, Quartin E, Kato H, Chen ZJ, Silverman RH, Akira S, Paludan SR (2009) Herpes simplex virus infection is sensed by both Toll-like receptors and retinoic acid-inducible gene- like receptors, which synergize to induce type I interferon production. J Gen Virol 90:74–78
Rock FL, Hardiman G, Timans JC, Kastelein RA, Bazan JF (1998) A family of human receptors structurally related to Drosophila Toll. Proc Natl Acad Sci USA 95:588–593
Roth-Cross JK, Bender SJ, Weiss SR (2008) Murine coronavirus mouse hepatitis virus is recognized by MDA5 and induces type I interferon in brain macrophages/microglia. J Virol 82:9829–9838
Rothenfusser S, Goutagny N, DiPerna G, Gong M, Monks BG, Schoenemeyer A, Yamamoto M, Akira S, Fitzgerald KA (2005) The RNA helicase Lgp2 inhibits TLR-independent sensing of viral replication by retinoic acid-inducible gene-I. J Immunol 175:5260–5268
Rubinstein M, Rubinstein S, Familletti PC, Gross MS, Miller RS, Waldman AA, Pestka S (1978) Human leukocyte interferon purified to homogeneity. Science 202:1289–1290
Rubinstein M, Rubinstein S, Familletti PC, Miller RS, Waldman AA, Pestka S (1979) Human leukocyte interferon: production, purification to homogeneity, and initial characterization. Proc Natl Acad Sci USA 76:640–644
Saito T, Hirai R, Loo YM, Owen D, Johnson CL, Sinha SC, Akira S, Fujita T, Gale M Jr (2007) Regulation of innate antiviral defenses through a shared repressor domain in RIG-I and LGP2. Proc Natl Acad Sci USA 104:582–587
Saito T, Owen DM, Jiang F, Marcotrigiano J, Gale M Jr (2008) Innate immunity induced by composition-dependent RIG-I recognition of hepatitis C virus RNA. Nature 454:523–527
Samanta M, Iwakiri D, Kanda T, Imaizumi T, Takada K (2006) EB virus-encoded RNAs are recognized by RIG-I and activate signaling to induce type I IFN. EMBO J 25:4207–4214
Samuel CE (2001) Antiviral actions of interferons. Clin Microbiol Rev 14:778–809 table of contents
Schlee M, Roth A, Hornung V, Hagmann CA, Wimmenauer V, Barchet W, Coch C, Janke M, Mihailovic A, Wardle G et al (2009) Recognition of 5′ triphosphate by RIG-I helicase requires short blunt double-stranded RNA as contained in panhandle of negative-strand virus. Immunity 31:25–34
Schmidt A, Schwerd T, Hamm W, Hellmuth JC, Cui S, Wenzel M, Hoffmann FS, Michallet MC, Besch R, Hopfner KP et al (2009) 5′-triphosphate RNA requires base-paired structures to activate antiviral signaling via RIG-I. Proc Natl Acad Sci USA 106:12067–12072
Schulz O, Diebold SS, Chen M, Naslund TI, Nolte MA, Alexopoulou L, Azuma YT, Flavell RA, Liljestrom P, Reis e Sousa C (2005) Toll-like receptor 3 promotes cross-priming to virus-infected cells. Nature 433:887–892
Seth RB, Sun L, Ea CK, Chen ZJ (2005) Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 122:669–682
Stetson DB, Medzhitov R (2006) Type I interferons in host defense. Immunity 25:373–381
Strahle L, Garcin D, Kolakofsky D (2006) Sendai virus defective-interfering genomes and the activation of interferon-beta. Virology 351:101–111
Strahle L, Marq JB, Brini A, Hausmann S, Kolakofsky D, Garcin D (2007) Activation of the IFN{beta} promoter by unnatural Sendai virus infection requires RIG-I and is inhibited by the viral C proteins. J Virol 81:12227–12237
Tabeta K, Georgel P, Janssen E, Du X, Hoebe K, Crozat K, Mudd S, Shamel L, Sovath S, Goode J et al (2004) Toll-like receptors 9 and 3 as essential components of innate immune defense against mouse cytomegalovirus infection. Proc Natl Acad Sci USA 101:3516–3521
Takahasi K, Yoneyama M, Nishihori T, Hirai R, Kumeta H, Narita R, Gale M Jr, Inagaki F, Fujita T (2008) Nonself RNA-sensing mechanism of RIG-I helicase and activation of antiviral immune responses. Mol Cell 29:428–440
Takahasi K, Kumeta H, Tsuduki N, Narita R, Shigemoto T, Hirai R, Yoneyama M, Horiuchi M, Ogura K, Fujita T, et al. (2009) Solution structures of cytosolic RNA sensors MDA5 and LGP2 C-terminal domains: Identification of the RNA recognition loop in RIG-I like receptors. J Biol Chem
Tan YH, Barakat F, Berthold W, Smith-Johannsen H, Tan C (1979) The isolation and amino acid/sugar composition of human fibroblastoid interferon. J Biol Chem 254:8067–8073
tenOever BR, Servant MJ, Grandvaux N, Lin R, Hiscott J (2002) Recognition of the measles virus nucleocapsid as a mechanism of IRF-3 activation. J Virol 76:3659–3669
tenOever BR, Sharma S, Zou W, Sun Q, Grandvaux N, Julkunen I, Hemmi H, Yamamoto M, Akira S, Yeh WC et al (2004) Activation of TBK1 and IKKvarepsilon kinases by vesicular stomatitis virus infection and the role of viral ribonucleoprotein in the development of interferon antiviral immunity. J Virol 78:10636–10649
Thompson AJ, Locarnini SA (2007) Toll-like receptors, RIG-I-like RNA helicases and the antiviral innate immune response. Immunol Cell Biol 85:435–445
Tytell AA, Lampson GP, Field AK, Hilleman MR (1967) Inducers of interferon and host resistance. 3. Double-stranded RNA from reovirus type 3 virions (reo 3-RNA). Proc Natl Acad Sci USA 58:1719–1722
Uematsu S, Akira S (2007) Toll-like receptors and Type I interferons. J Biol Chem 282:15319–15323
Uzri D, Gehrke L (2009) Nucleotide sequences and modifications that determine RIG-I/RNA binding and signaling activities. J Virol 83:4174–4184
Venkataraman T, Valdes M, Elsby R, Kakuta S, Caceres G, Saijo S, Iwakura Y, Barber GN (2007) Loss of DExD/H box RNA helicase LGP2 manifests disparate antiviral responses. J Immunol 178:6444–6455
Wang T, Town T, Alexopoulou L, Anderson JF, Fikrig E, Flavell RA (2004) Toll-like receptor 3 mediates West Nile virus entry into the brain causing lethal encephalitis. Nat Med 10:1366–1373
Waris G, Tardif KD, Siddiqui A (2002) Endoplasmic reticulum (ER) stress: hepatitis C virus induces an ER-nucleus signal transduction pathway and activates NF-kappaB and STAT-3. Biochem Pharmacol 64:1425–1430
Weber F, Wagner V, Rasmussen SB, Hartmann R, Paludan SR (2006) Double-stranded RNA is produced by positive-strand RNA viruses and DNA viruses but not in detectable amounts by negative-strand RNA viruses. J Virol 80:5059–5064
Xu LG, Wang YY, Han KJ, Li LY, Zhai Z, Shu HB (2005) VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol Cell 19:727–740
Xu L, Xiao N, Liu F, Ren H, Gu J (2009) Inhibition of RIG-I and MDA5-dependent antiviral response by gC1qR at mitochondria. Proc Natl Acad Sci USA 106:1530–1535
Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, Taira K, Akira S, Fujita T (2004) The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol 5:730–737
Yoneyama M, Kikuchi M, Matsumoto K, Imaizumi T, Miyagishi M, Taira K, Foy E, Loo YM, Gale M Jr, Akira S et al (2005) Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J Immunol 175:2851–2858
Yount JS, Gitlin L, Moran TM, Lopez CB (2008) MDA5 participates in the detection of paramyxovirus infection and is essential for the early activation of dendritic cells in response to Sendai Virus defective interfering particles. J Immunol 180:4910–4918
Zhang SY, Jouanguy E, Ugolini S, Smahi A, Elain G, Romero P, Segal D, Sancho-Shimizu V, Lorenzo L, Puel A et al (2007) TLR3 deficiency in patients with herpes simplex encephalitis. Science 317:1522–1527
Ziegler J, Lavin G, Horsfall F (1944) Interference between the influenza Viruses. II. The effect of virus rendered non-infective by ultraviolet radiation upon the multiplication of influenza viruses in the chick embryo. J Exper Med 79:379–400
Zoon KC, Smith ME, Bridgen PJ, zur Nedden D, Anfinsen CB (1979) Purification and partial characterization of human lymphoblast interferon. Proc Natl Acad Sci USA 76:5601–5605
Acknowledgments
Work in the laborotary of AG-S is being supported by NIH grants R01AI46954, P01AI58113, P01AI82325, U01AI70469, U19AI62623, U19AI83025, U54AI57168 and by CRIP (Center for Research in Influenza Pathogenesis), an NIAID funded Center of Excellence for Influenza Research and Surveillance, HHSN266200700010C.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
An erratum to this article can be found at http://dx.doi.org/10.1007/s00726-011-0844-z
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Baum, A., García-Sastre, A. Induction of type I interferon by RNA viruses: cellular receptors and their substrates. Amino Acids 38, 1283–1299 (2010). https://doi.org/10.1007/s00726-009-0374-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00726-009-0374-0