
Abstract
This study tested the hypothesis that l-arginine (Arg) may stimulate cell proliferation and prevent lipopolysaccharide (LPS)-induced death of intestinal cells. Intestinal porcine epithelial cells (IPEC-1) were cultured for 4 days in Arg-free Dulbecco’s modified Eagle’s-F12 Ham medium (DMEM-F12) containing 10, 100 or 350 μM Arg and 0 or 20 ng/ml LPS. Cell numbers, protein concentrations, protein synthesis and degradation, as well as mammalian target of rapamycin (mTOR) and Toll-like receptor 4 (TLR4) signaling pathways were determined. Without LPS, IPEC-1 cells exhibited time- and Arg-dependent growth curves. LPS treatment increased cell death and reduced protein concentrations in IPEC-1 cells. Addition of 100 and 350 μM Arg to culture medium dose-dependently attenuated LPS-induced cell death and reduction of protein concentrations, in comparison with the basal medium containing 10 μM Arg. Furthermore, supplementation of 100 and 350 μM Arg increased protein synthesis and reduced protein degradation in both control and LPS-treated IPEC-1 cells. Consistent with the data on cell growth and protein turnover, addition of 100 or 350 μM Arg to culture medium increased relative protein levels for phosphorylated mTOR and phosphorylated ribosomal protein S6 kinase-1, while reducing the relative levels of TLR4 and phosphorylated levels of nuclear factor-κB in LPS-treated IPEC-1 cells. These results demonstrate a protective effect of Arg against LPS-induced enterocyte damage through mechanisms involving mTOR and TLR4 signaling pathways, as well as intracellular protein turnover.
Similar content being viewed by others


Introduction
l-Arginine (Arg) plays an important role in intestinal physiology (Rhoads and Wu 2009). Arg has been studied as a component of an oral rehydration solution to enhance intestinal absorption and villous recovery after injury, and has been shown to be effective in a number of gut injury models (MacKay and Miller 2003; Sukhotnik et al. 2005; Wang et al. 2009). There is evidence showing the promise of Arg as a prophylaxis against necrotizing enterocolitis in premature infants (Amin et al. 2002). Additionally, dietary supplementation of Arg exerts beneficial effects in alleviating gut mucosal injury of LPS-challenged pigs and that the protective effects are associated with decreased expression of intestinal pro-inflammatory cytokines through activating PPARγ expression (Liu et al. 2008a, b).
Arg exerts its protective action on cells through both nitric oxide (NO)-dependent and NO-independent effects (Gualano et al. 2008; Montanez et al. 2008; Suenaga et al. 2008). Arg is the physiological nitrogenous substrate for NO production (Anggard 1994; Wu et al. 2009) and dietary l-Arg supplementation may contribute to the repair of damaged cells and tissues through NO- and polyamine-mediated mechanisms (Tan et al. 2009a; Wu and Meininger 2000). Polyamines (putrescine, spermine and spermidine) are required for growth, differentiation and function of cells, including intestinal mucosal cells (Flynn et al. 2009; Mitchell et al. 1994). Notably, recent studies have demonstrated that Arg stimulates intestinal cell migration and ex vivo intestinal protein synthesis, while enhancing mammalian target of rapamycin (mTOR) activity (Corl et al. 2008; Rhoads et al. 2004) and gaseous signaling (Li et al. 2009). Currently, precise mechanisms responsible for cytoprotective effects of Arg are largely unknown.
We hypothesized that activation of the mTOR pathway and inhibition of oxidative stress may play an important role in protecting intestinal cell death. The present study was designed to test this hypothesis using a model of LPS-induced death of intestinal porcine epithelial cells-1 (IPEC-1; Haynes et al. 2009).
Materials and methods
Reagents
Dulbecco’s modified Eagle’s F12 Ham medium (DMEM-F12), fetal bovine serum (FBS), and antibiotics were procured from Invitrogen (Grand Island, NY, USA). Epidermal growth factor was obtained from BD Biosciences (Bedford, MA, USA), and plastic culture plates were manufactured by Corning Inc. (Corning, NY, USA). Unless indicated, all other chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Cell culture
IPEC-1 cells were grown in serial passage in uncoated plastic culture flasks (100 mm2) in DMEM-F12 containing 17.5 mM D-glucose, 2 mM Gln, 0.7 mM arginine, 15 mM HEPES (pH 7.4), 5% FBS, insulin (5 μg/ml), transferrin (5 μg/ml), selenium (5 ng/ml), epidermal growth factor (5 μg/L), penicillin (50 μg/ml), streptomycin (4 μg/ml), and 0.25 μg/ml amphotericin B (fungizone®). Medium was changed every 2 days. At confluence, cells were trypsinized and seeded in 24-well cell culture plates with approximately 2 × 104 cells per well and maintained at 37°C in a 5% CO2 incubator. After an overnight incubation, the cells were starved for 6 h in Arg-free DMEM. The 5% FBS in the Arg-free DMEM provided 10 μM Arg. The cells were cultured in medium containing 10, 100, 350, or 500 μM Arg. The medium was changed every 2 days. The optimum time and Arg dose for cell growth were determined on the basis of cell numbers after 0, 2, 4 and 6 days incubation.
Determination of cell growth and protein concentrations
To determine effects of Arg on LPS-induced IPEC-1 cell death, cells were seeded in 24-well cell culture plates with approximately 2 × 104 cells per well for cell counting and 50 × 104 cells were seeded in 10-cm dishes for determination of protein concentrations. After an overnight incubation, the cells were starved 6 h in 0.5 ml of Arg-free DMEM. The cells were then incubated in 0.5 ml of medium containing 10, 100 or 350 μM Arg and 0 or 20 ng/ml LPS. The media were changed every 2 days. After a 4-day period of incubation, cells in 24-well cell culture plates were gently washed with calcium-containing Dulbecco’s phosphate buffered saline and viable cells were counted using a counting chamber (Dekaney et al. 2008). Cells were collected, nap-frozen in liquid nitrogen, and stored at −80°C for protein assays and Western blot analysis.
Determination of protein synthesis and degradation
IPEC-1 cells (10 × 104) were seeded in 6-well cell culture plates with 2 ml of DMEM-F12 medium containing 17.5 mM D-glucose, 2 mM Gln, 0.7 mM arginine, 15 mM HEPES (pH 7.4), 5% FBS, insulin (5 μg/ml), transferrin (5 μg/ml), selenium (5 ng/ml), epidermal growth factor (5 μg/L), penicillin (50 μg/ml), streptomycin (4 μg/ml), and 0.25 μg/ml amphotericin B (fungizone®). After overnight culture, the cells were starved 6 h in 2 ml Arg-free DMEM and then used for studies of intracellular protein turnover (protein synthesis and degradation). There were eight replicate sets of wells per Arg dose.
For determining protein synthesis, after a 4-day period of culture in Arg-free DMEM containing 10, 100 or 350 μM Arg and 0 or 20 ng/ml LPS, the medium was removed and the cells were washed once with 2 ml of Arg-free medium. Then, cells were cultured for 3 h in 2 ml DMEM containing 1 mM l-phenylalanine plus 0.8 μCi L-[ring-2,4-3H]phenylalanine (American Radiolabeled Chemicals), 10, 100, or 350 μM Arg, and 0 or 20 ng/ml LPS. At the end of a 3-h culture period, the medium was collected and cells were rapidly washed three times with 2 ml ice-cold PBS. The cell pellet was mixed with 2 ml of 2% trichloroacetic acid (TCA) (Wu and Thompson 1990). Cells in each well were scraped and then the whole solution was collected into a 15-ml tube and centrifuged at 3,000g for 5 min. The supernatant fluid was discarded. The cell pellet was washed three times with 5 ml of 2% TCA and dried in air at 37°C. After the pellet was dissolved in 0.5 ml of 1 M NaOH, 0.4 ml of the solution was transferred to a 20-ml scintillation vial containing 15 ml of Hionic Fluor Scintillation cocktail (PerkinElmer, MA, USA). The 3H radioactivity was determined by a liquid scintillation counter after overnight standing at room temperature. Specific activity of 3H-phenylalanine in medium was used to calculate rate of protein synthesis in cells.
For determining protein degradation, IPEC-1 cells were cultured for 3 days in 2 ml of Arg-free DMEM containing 10, 100 or 350 μM Arg and 0 or 20 ng/ml LPS. Beginning on day 4, cells were cultured for 24 h in 2 ml of Arg-free DMEM containing 0.1 mM l-phenylalanine plus l-[3H]phenylalanine (0.8 μCi/well) and 10, 100 or 350 μM Arg plus 0 or 20 ng/ml LPS. After the 24-h culture to label cellular proteins, cells were washed three times with 2 ml Arg-free medium containing 1 mM l-phenylalanine to deplete intracellular free [3H]phenylalanine (Wu and Thompson 1990). The cells were then cultured for 3 h in 2 ml DMEM containing 1 mM l-phenylalanine and 10, 100, or 350 μM Arg plus 0 or 20 ng/ml LPS. At the end of a 3-h culture period, the medium was collected, the cells were rapidly washed three times with 2 ml ice-cold PBS, and 2 ml of 2% TCA was added to each well. The whole TCA extract was collected into a 15-ml tube and centrifuged at 3,000g for 5 min. The supernatant fluid was removed and the pellet was washed three times with 5 ml of 2% TCA and dried in air at 37°C. After the pellet was dissolved in 0.5 ml of 1 M NaOH, 0.4 ml of the solution was transferred to a 20-ml scintillation vial containing 15 ml Hionic Fluor Scintillation cocktail for 3H measurement.
For determining [3H]phenylalanine released from prelabeled proteins into culture medium, the collected medium was centrifuged at 3,000g for 2 min to remove any dead cells. An aliquot (1 ml) of the supernatant fluid was transferred to a 15-ml tube containing 2 ml of 2% TCA. After the tubes were centrifuged at 3,000g for 5 min, all the supernatant fluid was transferred to a 20-ml Scintillation vial containing 15 ml Hionic Fluor Scintillation cocktail for 3H measurement. The percentage of protein-bound [3H]phenylalanine released into culture medium (namely [3H]phenylalanine in medium/[3H]phenylalanine in cell proteins × 100) was calculated to indicate protein degradation in IPEC-1 cells.
Western blot analysis
Frozen cell samples were homogenized in seven volumes of buffer [20 mM HEPES, pH 7.4, 2 mM EGTA, 50 mM NaF, 100 mM KCl, 0.2 mM EDTA, 50 mM β-glycerophosphate, 1 mM dithiothreitol (DTT), 0.1 mM phenylmethylsulfonyl fluoride (PMSF), 1 mM benzamidine and 0.5 mM sodium vanadate] with a Polytron homogenizer and centrifuged at 10,000g for 10 min at 4°C, as described by Li et al. (2008). Protein concentrations of cell homogenates were measured using the BCA method and bovine serum albumin as standard. All samples were adjusted to an equal concentration. The supernatant fluid (containing cell proteins) were then diluted with 2 × sodiumdodecyl sulfate (SDS) sample buffer (0.63 ml of 0.5 M Tris–HCl pH 6.8, 0.42 ml 75% glycerol, 0.125 g SDS, 0.25 ml β-mercaptoethanol, 0.2 ml 0.05% solution of bromphenol blue and 1 ml water to a final volume of 2.5 ml) and heated in boiling water for 5 min. After the solution was cooled on ice, it was used for Western blot analysis. Aliquots of samples were loaded onto SDS-polyacrylamide gels. After separation on 4–12% gels, proteins were transferred to a nitrocellulose membrane (Bio-Rad, Hercules, CA, USA) under 12 V overnight, using the Bio-Rad Transblot apparatus (Hercules, CA, USA). Membranes were blocked in 5% fat-free dry milk in Tris–Tween buffered saline (TTBS; 20 mM Tris/150 mM NaCl, pH 7.5, and 0.1% Tween-20) for 3 h and then were incubated with the following primary antibodies overnight at 4°C with gentle rocking: mTOR (cell signaling, 1:1,000), phosphorylated mTOR (Ser2448) (cell signaling, 1:1,000), 4EBP1 (cell signaling, 1:1,000), phosphorylated 4EBP1 (Ser65) (cell signaling, 1:1,000), S6K1 (cell signaling, 1:1,000), phosphorylated S6K1 (Thr389) (cell signaling, 1:1,000), TLR4 (Abcam, 1:1,000), NFκB (cell signaling, 1:1,000), phosphorylated NFκB (Ser536) (cell signaling, 1:1,000) or β-actin (cell signaling, 1:1,000). After washing three times with TTBS, the membranes were incubated at room temperature for 3 h with secondary antibodies at 1:50,000 (horseradish peroxidase-conjugated goat anti-rabbit IgG, cell signaling or peroxidase-labeled rabbit anti-goat IgG, Kirkegaard & Perry Lab). Finally, the membranes were washed with TTBS, followed by development using Supersignal West Dura Extended Duration Substrate according to the manufacturer’s instructions (Pierce, Rockford, IL, USA). The signals were detected on Fujifilm LAS-3000 (Tokyo, Japan). All protein measurements were normalized to β-actin and all data were expressed as the relative values to those of LPS-treated IPEC-1 cells cultured with 10 μM Arg.
Statistical analysis
Results are expressed as mean ± SEM. The statistical analysis was performed by one-way or two-way ANOVA using SPSS 13.0 (SPSS Inc., Chicago, IL, USA). P values ≤0.05 were taken to indicate significance.
Results
Cell growth
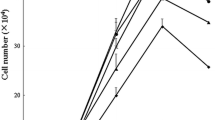
The growth curves of IPEC-1 cells not treated with LPS are illustrated in Fig. 1. On days 0 and 2, the number of IPEC-1 cells did not differ among the 10, 100, 350 and 500 μM Arg groups. On day 4, cell numbers were increased (P < 0.001) with increasing Arg concentrations from 10 to 500 μM. The numbers of IPEC-1 cells in 100, 350 and 500 μM Arg groups were 457, 678 and 818% higher (P < 0.001) than those in the 10 μM Arg group, respectively. On day 6, the number of IPEC-1 cells in 100, 350 and 500 μM Arg groups were also higher (P < 0.001) than those in the 10 μM Arg group, but there was no difference between the 350 and 500 μM Arg groups. The numbers of IPEC-1 cells in 100, 350 and 500 μM Arg groups, but not in the 10 μM Arg group, increased (P < 0.001) on days 4 and 6, compared with those on days 0 and 2.

The number of IPEC-1 cells cultured in Arg-free DMEM containing 10, 100, 350 or 500 μM arginine on day 0, 2, 4 and 6. Data are expressed as means ± SEM, n = 8
Effects of Arg on cell numbers and protein concentrations of normal and LPS-treated IPEC-1 cells
IPEC-1 cells cultured with 20 ng/ml LPS exhibited lower (P < 0.05) survival rate (Fig. 2A) and lower (P < 0.05) protein concentrations (Fig. 2B), compared with cells cultured without LPS, regardless of Arg concentrations in culture medium. The numbers of LPS-treated IPEC-1 cells (Fig. 2A) in 10, 100 and 350 μM Arg groups are 83.3, 21.8 and 14.2% lower (P < 0.05), respectively, than those of IPEC-1 cells not treated with LPS. The IPEC-1 cell protein concentrations (Fig. 2B) in 10, 100 and 350 μM Arg groups are 25.5, 11.8 and 14.4% lower (P < 0.05), respectively, than those of IPEC-1 cells not treated with LPS. Clearly, addition of 100 and 350 μM Arg to culture medium reduced (P < 0.05) LPS-induced cell death and increased (P < 0.05) protein concentrations in cells, compared with the 10 μM Arg group (Fig. 2B).

The cell numbers (A) and protein concentrations (B) of normal and LPS (20 ng/ml)-treated IPEC-1 cells. Cells were cultured for 96 h in Arg-free DMEM containing 10, 100 or 350 μM arginine and 0 or 20 ng/ml LPS. Data are expressed as means ± SEM, n = 8. a–e Means sharing different letters differ (P < 0.05)
Effects of Arg on protein synthesis and degradation in normal- and LPS-treated IPEC-1 cells
Addition of 100 and 350 μM Arg to culture medium increased (P < 0.001) protein synthesis (Fig. 3A) and reduced (P < 0.001) protein degradation (Fig. 3B) in both normal- and LPS-treated IPEC-1 cells. For IPEC-1 cells not treated with LPS, protein synthesis was 96.1 and 97.3% higher (P < 0.001) and protein degradation was 45.1 and 54.7% lower (P < 0.001) in 100 and 350 μM Arg treatments than in the 10 μM Arg group. Supplementing 100 and 350 μM Arg to the culture medium attenuated (P < 0.001) the adverse effects of LPS on inhibition of protein synthesis and activation of protein degradation.

Protein synthesis (A nmol Phe/mg protein/3 h) and protein degradation (B; %) in IPEC-1 cells treated with or without 20 ng/ml LPS. Cells were cultured for 96 h in Arg-free DMEM containing 10, 100, or 350 μM arginine and 0 or 20 ng/ml LPS. Data are expressed as means ± SEM, n = 8. a–d Means sharing different letters differ (P < 0.001)
Effects of Arg on mTOR and TLR4 expression in LPS-treated IPEC-1 cells
Relative protein levels for total mTOR, phosphorylated mTOR, 4EBP1, phosphorylated 4EBP1, S6K1 and phosphorylated S6K1 in IPEC-1 cells are illustrated in Fig. 4. Compared with the 10 μM Arg group, addition of 100 and 350 μM Arg to culture medium increased (P < 0.01) protein levels for phosphorylated mTOR by 75 and 202%, respectively, in LPS-treated IPEC-1 cells. Likewise, addition of 100 and 350 μM Arg to culture medium increased (P < 0.01) protein levels for phosphorylated S6K1 by 89 and 200.6% in LPS-treated cells, respectively. Arginine (100 and 350 μM) had no effect on protein levels for total mTOR, 4EBP1 and S6K1 in LPS-treated IPEC-1 cells but increased the levels of phosphorylated mTOR (P < 0.01) and phosphorylated 4EBP1 (P < 0.05) in these cells.

Relative protein levels for total mTOR (A), phosphorylated mTOR (B), 4EBP1 (C), phosphorylated 4EBP1 (D), S6K1 (E) and phosphorylated S6K1 (F) in IPEC-1 cells. Cells were cultured for 96 h in Arg-free DMEM containing 10, 100 or 350 μM arginine and 20 ng/ml LPS. Data are expressed as means ± SEM, n = 4. a, b Means sharing different letters differ (P < 0.01). Levels of phosphorylated 4EBP1 were higher (P < 0.05) in cells treated with 100 and 350 μM arginine, compared with the control (10 μM arginine)
Arginine at 100 and 350 μM reduced (P < 0.01) total TLR4 levels in LPS-treated IPEC-1 cells, compared with the control (10 μM Arg) (Fig. 5). The Arg treatment (100 and 350 μM) did not affect total NFκB levels but markedly reduced (P < 0.01) the levels of phosphorylated NFκB in LPS-treated cells (Fig. 5). Representative western blots for all the measured proteins are illustrated in Fig. 6.

Relative protein levels for TLR4 (A), NFκB (B) and phosphorylated NFκB (C) in IPEC-1 cells. Cells were cultured for 96 h in Arg-free DMEM containing 10, 100 or 350 μM arginine and 20 ng/ml LPS. Data are expressed as means ± SEM, n = 4. a, b Means sharing different letters differ (P < 0.01)

Representative western blots of mTOR (A), phosphorylated mTOR (Ser2448) (B), 4EBP1 (C), phosphorylated 4EBP1 (Ser65) (D), S6K1 (E), phosphorylated S6K1 (Thr389) (F), TLR4 (G), NFκB (H), phosphorylated NFκB (Ser536) (I), and β-actin (J) in IPEC-1 cells. Cells were cultured for 96 h in Arg-free DMEM containing 20 ng/ml LPS and 10 (column I), 100 (column II) or 350 (column III) μM arginine
Discussion
LPS, a major component of the outer membrane of Gram negative bacteria, has been used to mimic the immunological stress in pigs characterized by symptoms of acute bacterial infection including anorexia, hypersomnia and fever (Liu et al. 2008a). In the current study, we observed the protective effects of Arg on intestinal cells using a cell damage model induced by LPS and found that addition of Arg to culture medium stimulated protein synthesis but reduced protein degradation in LPS-treated IPEC cells (Fig. 3). To our knowledge, this is the first report of the regulation of intracellular protein turnover by Arg in animal cells.
Cell growth depends on the balance between protein synthesis and degradation (Wu 2009). Our results indicate that the mechanisms responsible for the stimulatory effect of Arg on protein accretion in IPEC-1 cells involve activation of cell signaling pathways that regulate the machinery of intracellular protein turnover. Similarly, Arg increases net protein synthesis in skeletal muscle of young pigs (Kim and Wu 2009; Yao et al. 2008), finishing pigs (Tan et al. 2009b), and adult rats (Jobgen et al. 2009a). This effect of Arg can result from the activation of mTOR signaling (increased phosphorylation of mTOR, S6K1 and 4EBP1) (Fig. 4), a master regulator of both protein synthesis and autophage-mediated proteolysis (Phang et al. 2008; Suryawan et al. 2009).
Two key downstream targets of mTOR are p70s6k and 4EBP1 (Liao et al. 2008). Growing evidence shows that Arg can activate mTOR and other kinase-mediated signaling pathways in intestinal epithelial cells (Ban et al. 2004; Rhoads et al. 2007). For example, Arg enhanced cell migration and activated p70(s6k) in porcine enterocytes (Rhoads et al. 2007). Additionally, in severe piglet rotavirus enteritis, in vitro jejunal protein synthetic rate increased twofold, concomitant with a fourfold increase in jejunal p70(s6k) phosphorylation and elevation of total p70(s6k) protein levels (Rhoads et al. 2007). Consistent with these reports, our results indicate that Arg supplementation to culture medium can activate the mTOR signaling pathway partly through S6K1 and 4EBP1 phosphorylation in LPS-treated IPEC cells.
There is evidence suggesting that mTOR has a role of “speed breaker” in suppressing NFκB activation (Mendes Sdos et al. 2009). IκB kinase β (IKKβ), a key activator of the proinflammatory NFκB signaling pathway (Lee et al. 2007), phosphorylates and destabilizes Tuberous Sclerosis Complex 1 (TSC1), thus activating the mTOR complex 1 (mTORC1) in response to inflammatory cytokines (Dan et al. 2008). On the contrary, the mTOR inhibitor, rapamycin, promotes the LPS-induced transcriptional activity of NFκB (Mendes Sdos et al. 2009). LPS binds its specific receptor TLR4 on the plasma membrane, which triggers the MyD88-dependent and independent pathways, resulting in activation of NFκB and apoptosis (Austenaa et al. 2008; Gribar et al. 2008; Lu et al. 2008). Many stimuli activate NFκB, trigger the translocation of NFκB from the cytosol to the nucleus where NFκB binds its consensus sequence on the promoter–enhancer region of different genes and regulates the transcription of specific genes (Steinle et al. 1999).
LPS-induced NFκB nuclear translocation interferes with the DNA binding activity of NFκB and attenuates cell damage (Zhang et al. 2006). Interestingly, Arg supplementation to culture medium-reduced TLR4 expression and phosphorylated NFκB levels in LPS-treated IPEC-1 cells (Fig. 6). These results indicate that Arg possesses a cytoprotective effect on LPS-induced IPEC-1 cells by inhibiting NFκB activation. Likewise, dietary supplementation with arginine attenuates oxidative stress in liver (Jobgen et al. 2009a), skeletal muscle (Ma et al. 2009) and adipose tissue (Jobgen et al. 2009b). These findings help elucidate the mechanisms responsible for the beneficial effect of Arg in treating inflammatory bowel disease and intestinal dysfunction, which are significant problems in preterm infants (Amin et al. 2002) and low-birth-weight neonates (Wang et al. 2008).
In summary, addition of 100 or 350 μM Arg (physiological concentrations in plasma) to culture medium confers a cytoprotective effect on LPS-treated enterocytes. This action of Arg involves increased protein synthesis and reduced protein degradation, as well as mTOR and TLR4 signaling pathways. Results from these in vitro studies have important implications for the use of Arg in the clinical management of endotoxin-infected infants and the rearing of neonatal pigs.
Abbreviations
- 4EBP1:
-
4E-binding protein-1
- Arg:
-
Arginine
- LPS:
-
Lipopolysaccharide
- BSA:
-
Bovine serum albumin
- DMEM-F12:
-
Dulbecco’s modified Eagle’s F12 Ham medium
- FBS:
-
Fetal bovine serum
- IPEC-1:
-
Intestinal porcine epithelial cells-1
- mTOR:
-
Mammalian target of rapamycin
- NFκB:
-
Nuclear factor-κB
- S6K1:
-
Ribosomal protein S6 kinase-1
- TLR4:
-
Toll-like receptor 4
References
Amin HJ, Zamora SA, McMillan DD et al (2002) Arginine supplementation prevents necrotizing enterocolitis in the premature infant. J Pediatr 140:425–431
Anggard E (1994) Nitric oxide: mediator, murderer, and medicine. Lancet 14:1199–1206
Austenaa LM, Carisen H, Hollung K et al (2008) Retinoic acid dampens LPS-induced NF-kB activity. J Nutr Biochem. doi:10.1016/j.jnutbio.2008.07.002
Ban H, Shigemitsu K, Yamatsuji T et al (2004) Arginine and leucine regulate p70S6 kinase and 4E-BP1 in intestinal epithelial cells. Int J Mol Med 13:537–543
Corl BA, Odle J, Niu X et al (2008) Arginine activates intestinal p70(S6 k) and protein synthesis in piglet rotavirus enteritis. J Nutr 138:24–29
Dan HC, Cooper MJ, Cogswell PC et al (2008) Akt-dependent regulation of NF-{kappa}B is controlled by mTOR and Raptor in association with IKK. Genes Dev 22:1490–1500
Dekaney CM, Wu G, Yin YL et al (2008) Regulation of ornithine aminotransferase gene expression and activity by all-trans retinoic acid in Caco-2 intestinal epithelial cells. J Nutr Biochem 19:674–681
Flynn NE, Bird JG, Guthrie AS (2009) Glucocorticoid regulation of amino acid and polyamine metabolism in the small intestine. Amino Acids 37:123–129
Gribar SC, Richardson WM, Sodhi CP et al (2008) No longer an innocent bystander: epithelial Toll-like receptor signaling in the development of mucosal inflammation. Mol Med 14:645–659
Gualano B, Novaes RB, Artioli GG et al (2008) Effects of creatine supplementation on glucose tolerance and insulin sensitivity in sedentary healthy males undergoing aerobic training. Amino Acids 34:245–250
Haynes TE, Li P, Li XL et al (2009) L-Glutamine or L-alanyl-L-glutamine prevents oxidant- or endotoxin-induced death of neonatal enterocytes. Amino Acids 37:131–142
Jobgen WJ, Meininger CJ, Jobgen SC et al (2009a) Dietary L-arginine supplementation reduces white-fat gain and enhances skeletal muscle and brown fat masses in diet-induced obese rats. J Nutr 139:230–237
Jobgen W, Fu WJ, Gao H et al (2009b) High fat feeding and dietary L-arginine supplementation differentially regulate gene expression in rat white adipose tissue. Amino Acids 37:187–198
Kim SW, Wu G (2009) Regulatory role for amino acids in mammary gland growth and milk synthesis. Amino Acids 37:89–95
Lee DF, Kuo HP, Chen CT et al (2007) IKK beta suppression of TSC1 links inflammation and tumor angiogenesis via the mTOR pathway. Cell 130:440–455
Li P, Kim SW, Li XL et al (2008) Dietary supplementation with cholesterol and docosahexaenoic acid increases the activity of the arginine-nitric oxide pathway in tissues of young pigs. Nitric Oxide 19:259–265
Li XL, Bazer FW, Gao H et al (2009) Amino acids and gaseous signaling. Amino Acids 37:65–78
Liao XH, Majithia A, Huang XL et al (2008) Growth control via TOR kinase signaling, an intracellular sensor of amino acids and energy availability, with crosstalk potential to proline metabolism. Amino Acids 35:761–770
Liu YL, Huang JJ, Hou YQ et al (2008a) Dietary arginine supplementation alleviates intestinal mucosal disruption induced by Escherichia coli lipopolysaccharide in weaned pigs. Br J Nutr 100:552–560
Liu YL, Lu J, Shi JX (2008b) Increased expression of the peroxisome proliferator-activated receptor-γ in the immune system of weaned pigs after Escherichia coli lipopolysaccharide injection. Vet Immunol Immunopathol 124:82–92
Lu YC, Yeh WC, Ohashi PS (2008) LPS/TLR4 signaling transduction pathway. Cytokine 42:145–151
Ma XY, Lin YC, Jiang ZY et al (2009) Dietary arginine supplementation enhances antioxidative capacity and improves meat quality of finishing pigs. Amino Acids. doi:10.1007/s00726-008-0213-8
MacKay D, Miller AL (2003) Nutritional support for wound healing. Altern Med Rev 8:359–377
Mendes Sdos S, Candi A, Vansteenbrugge M et al (2009) Microarray analyses of the effects of NF-κB or PI3 K pathway inhibitors on the LPS-induced gene expression profile in RAW264.7 cells Synergistic effects of rapamycin on LPS-induced MMP9-overexpression. Cell Signal 21:1109–1122
Mitchell JB, Wink DA, DeGraff W et al (1994) Hypoxic mammalian cell radiosensitization by nitric oxide. Cancer Res 54:3365–3368
Montanez R, Rodriguez-Caso C, Sanchez-Jimenez F et al (2008) In silico analysis of arginine catabolism as a source of nitric oxide or polyamines in endothelial cells. Amino Acids 34:223–229
Phang JM, Donald SP, Pandhare J et al (2008) The metabolism of proline, a stress substrate, modulates carcinogenic pathways. Amino Acids 35:681–690
Rhoads JM, Wu G (2009) Glutamine, arginine, and leucine signaling in the intestine. Amino Acids 37:111–122
Rhoads JM, Gookin J, Chen W et al (2004) Arginine stimulates intestinal cell migration through a focal adhesion kinase dependent mechanism. Gut 53:514–522
Rhoads JM, Cori BA, Harrell R et al (2007) Intestinal ribosomal p70S6K signaling is increased in piglet rotavirus enteritis. Am J Physiol Gastrointest Liver Physiol 292:G913–G922
Steinle AU, Weidenbach H, Wagner M et al (1999) NF-kappaB/Rel activation in cerulein pancreatitis. Gastroenterology 116:420–430
Suenaga R, Tomonaga S, Yamane H, Kurauchi I et al (2008) Intracerebroventricular injection of L-arginine induces sedative and hypnotic effects under an acute stress in neonatal chicks. Amino Acids 35:139–146
Sukhotnik I, Helou H, Mogilner J et al (2005) Oral arginine improves intestinal recovery following ischemia-reperfusion injury in rat. Pediatr Surg Int 21:191–196
Suryawan A, O’Connor PMJ, Bush JA et al (2009) Differential regulation of protein synthesis by amino acids and insulin in peripheral and visceral tissues of neonatal pigs. Amino Acids 37:97–104
Tan BE, Li XG, Kong XF et al (2009a) Dietary L-arginine supplementation enhances the immune status in early-weaned piglets. Amino Acids 37:323–331
Tan BE, Yin YL, Liu ZQ et al (2009b) Dietary L-arginine supplementation increases muscle gain and reduces body fat mass in growing-finishing pigs. Amino Acids 37:169–175
Wang JJ, Chen LX, Li DF et al (2008) Intrauterine growth restriction affects the proteomes of the small intestine, liver and skeletal muscle in newborn pigs. J Nutr 138:60–66
Wang WW, Qiao SY, Li DF (2009) Amino acids and gut function. Amino Acids 37:105–110
Wu G (2009) Amino acids: metabolism, functions, and nutrition. Amino Acids 37:1–17
Wu G, Meininger CJ (2000) Arginine nutrition and cardiovascular function. J Nutr 130:2626–2629
Wu G, Thompson JR (1990) The effect of glutamine on protein turnover in chick skeletal muscle in vitro. Biochem J 265:593–598
Wu G, Bazer FW, Davis TA et al (2009) Arginine metabolism and nutrition in growth, health and disease. Amino Acids 37:153–168
Yao K, Deng D, Liu ZQ et al (2008) Dietary arginine supplementation increases intracellular mTOR signaling activity in skeletal muscle of neonatal pigs. J Nutr 138:867–872
Zhang H, Li YY, Wu XZ (2006) Effect of Tetrandrine on LPS-induced NF-κB activation in isolated pancreatic acinar cells of rat. World J Gastroenterol 12(26):4232–4236
Acknowledgments
This work was supported by the National Basic Research Program of China (No. 2004CB117502), National Natural Science Foundation of China (No. 30371038 and 30528006), the Outstanding Overseas Chinese Scholars Fund of The Chinese Academy of Sciences (No. 2005-1-4), Texas AgriLife Research (No. H-8200), and National Research Initiative Competitive Grant (No. 2008-35206-18762) from the USDA Cooperative State Research, Education, and Extension Service Animal Growth and Nutrient Utilization Program.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Tan, B., Yin, Y., Kong, X. et al. l-Arginine stimulates proliferation and prevents endotoxin-induced death of intestinal cells. Amino Acids 38, 1227–1235 (2010). https://doi.org/10.1007/s00726-009-0334-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00726-009-0334-8