
Abstract
The phosphonate-substituted zirconium oxo clusters Zr6O2(OBu)12(O3PPh)4 and Zr7O2(OiPr)12(O3PCH2CH2CH2Br)6, with octahedrally coordinated Zr atoms, were synthesized by reaction of zirconium alkoxides with phosphonic acid bis(trimethylsilyl) esters. The basic structural motif are Zr3O(µ2-OR)3(OR)3 units which are connected in different ways.
Graphical abstract

Similar content being viewed by others

Introduction
Many phosphonate-substituted zirconium compounds with 1D, layered 2D, and 3D interconnected structures have been reported [1–3], including coordination polymers with bis- or tetra-phosphonate ligands [4–11]. Surprisingly, no zirconium oxo clusters with phosphonate ligands are known, although zirconium oxo clusters with a variety of other bi- or multidentate ligands have been prepared and zirconia nanoparticles are frequently stabilized by phosphonate groups [12, 13]. In this article, we report the preparation and structural characterization of the first phosphonate-substituted zirconium oxo clusters.
Phosphonate-substituted metal compounds are commonly prepared from the corresponding phosphonic acids or their metal salts. We have recently shown that titanium oxo clusters can be easily obtained from the reaction of titanium alkoxides with phosphonic acid bis(trimethylsilyl) esters [14, 15]. The esters have the advantage of being soluble in organic solvents. Their reaction with alcohol added to the reaction mixture liberates phosphonic acid which substitutes part of the OR groups of Ti(OR)4 in a relatively fast reaction. Oxo groups are generated in situ either by water originating from esterification of (coordinated or non-coordinated) phosphonic acid or by non-hydrolytic processes.
Results and discussion
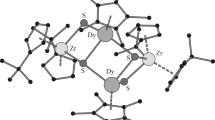
Crystals of Zr6O2(OBu)12(O3PPh)4 (1, Fig. 1) were obtained when Zr(OBu)4 was reacted with bis(trimethylsilyl)phenylphosphonate in a 2:1 ratio.

Molecular structure of Zr6(μ 3-O)2(μ 2-OBu)6(OBu)6(O3PPh)4 (1). Hydrogen atoms are omitted for clarity. Selected bond lengths/pm and angles/°: Zr(1)-O(1) 211.6(5), Zr(1)-O(2) 214.8(5), Zr(1)-O(3) 214.5(5), Zr(1)-O(5) 192.9(5), Zr(1)-O(8) 209.2(5), Zr(1)-O(11) 209.9(5), Zr(2)-O(1) 208.1(5), Zr(2)-O(2) 215.8(5), Zr(2)-O(4) 215.5(8), Zr(2)-O(6) 192.6(5), Zr(2)-O(9) 210.4(5), Zr(2)-O(13) 207.7(5), Zr(3)-O(1) 208.7(5), Zr(3)-O(3) 215.3(5), Zr(3)-O(4) 215.3(9), Zr(3)-O(7) 193.4(5), Zr(3)-O(10) 209.2(5), Zr(3)-O(12) 209.4(5); Zr(1)-O(1)-Zr(2) 106.1(2), Zr(1)-O(1)-Zr(3) 106.0(2), Zr(2)-O(1)-Zr(3) 109.6(2), Zr(1)-O(2)-Zr(2) 102.3(2), Zr(1)-O(3)-Zr(3) 102.7(2), Zr(2)-O(4)-Zr(3) 104.5(4)
The basic structural motif in centrosymmetric 1 are two Zr3O(μ 2-OBu)3(OBu)3 moieties (Zr3O). They are interconnected with each other through four bridging phenylphosphonate ligands which are arranged up, up, down, down. Each phosphonate ligand binds to two zirconium atoms of one Zr3O triangle and one zirconium atom of the other Zr3O triangle and is, therefore, coordinating 3.111 (w.xyz refers to the number of metal atoms to which the phosphonate ligand is coordinated [w], and the number of metal atoms to which each oxygen is coordinated [x, y, z] [16]). The crystallographic symmetry of 1 is retained in solution since the 31P NMR spectrum in CD2Cl2 showed only one signal at 6.57 ppm. The expected number of signals with the expected shifts was observed in the 1H NMR spectrum.
The most surprising feature of 1 is that all zirconium atoms are octahedrally coordinated. This is remarkable since higher coordination numbers (7–9) are mostly found in zirconium oxo clusters. The structure of 1 is different from that of oxo clusters obtained from reactions of Ti(OiPr)4 with bis(trimethylsilyl) phosphonates although Ti is also six-coordinated there. M3O(μ 2-OR)3(OR)3 units are the basic structural motif in both cases. While two Zr3O units are directly connected with each other in 1, the two Ti3O units in Ti7O2(OiPr)12(O3PR)6 (R=CH2CH2CH2Cl or benzyl) are connected through a central Ti atom [14]. In the case of titanium, structures Ti4(µ3-O)(µ2-OiPr)3(OiPr)5(O3PR)3L (L = neutral ligand) and dimers thereof were also obtained, where the Ti3O unit is capped by a Ti(OiPr)2L group.
A zirconium oxo cluster isostructural to Ti7O2(OiPr)12(O3PR)6, viz. Zr7O2(µ 2-OiPr)6(OiPr)6(O3PCH2CH2CH2Br)6 (2, Fig. 2), was, however, obtained in another experiment, i.e., reaction of Zr(OiPr)4 with bis(trimethyl)silyl(3-bromopropyl)phosphonate, methacrylic acid, and water. Since water generation by esterification of phosphonic acid (as in the first experiment) is relatively slow, water was deliberately added. Methacrylic acid was added anticipating an oxo cluster with a mixed ligand sphere as had been the case for analogous reactions with Ti(OR)4 [15, 17]. No mixed ligand cluster was obtained, however, in the reaction of Zr(OiPr)4.

Molecular structure of Zr7O2(µ 2-OiPr)6(OiPr)6(O3PCH2CH2CH2Br)6 (2). Hydrogen atoms are omitted for clarity. Selected bond lengths/pm and angles/°: O(1)-Zr(1) 207.9(4), O(1)-Zr(2) 209.3(4), O(1)-Zr(3) 208.2(4), O(2)-Zr(5) 208.9(4), O(2)-Zr(6) 209.2(4), O(2)-Zr(7) 208.8(4), O(5)-Zr(2) 216.4(4), O(5)-Zr(3) 217.0(4), O(7)-Zr(5) 218.6(4), O(7)-Zr(7) 216.5(5), O(9)-Zr(1) 194.2(5), O(10)-Zr(2) 193.1(5), O(13)-Zr(6) 192.2(5), O(18)-Zr(1) 210.1(4), O(21)-Zr(2) 211.7(4), O(23)-Zr(4) 206.0(4), O(25)-Zr(6) 211.1(4), O(26)-Zr(4) 207.4(4), O(28)-Zr(7) 210.6(5); Zr(1)-O(1)-Zr(2) 108.09(18), Zr(7)-O(7)-Zr(5) 101.9(2)
The symmetry of 2 is retained in solution as only one signal at 30.6 ppm was observed in the 31P NMR spectrum in C6D6. The 1H NMR spectrum shows only two doublets for the isopropoxo CH3 groups as well as two multiplets of the CH groups. Therefore, all terminal as well as all bridging isopropoxo ligands are symmetry related in solution.
Conclusions
The coordination chemistry of titanium and zirconium, including that of metal oxo clusters, is usually quite different even if the same reaction conditions and stoichiometric ratios of the reactants are employed. This is due to the different coordination numbers.
The surprising outcome of the work reported in this article is that oxo clusters were obtained in the reaction of M(OR)4 (M = Ti, Zr) with bis(trimethyl)silylphosphonates where the coordination numbers and geometries of both Ti and Zr were the same. For this reason, the structures of the obtained Zr clusters were the same as those of Ti oxo clusters (for 2) or very closely related (for 1). A possible reason for this feature might be that the M3O(μ 2-OR)3(OR)3 moiety appears to be a very robust building block, as already postulated earlier [14].
Experimental
All operations were carried out in a moisture- and oxygen-free argon atmosphere using Schlenk techniques. 2-Propanol and 1-butanol were dried by distilling twice from sodium metal. The phosphonates were prepared as previously reported [14, 15]. Zirconium isopropoxide and zirconium n-butoxide were obtained from Sigma-Aldrich and used without further purification.
Zr 6 O 2 Cluster Zr 6 (μ 3 -O) 2 (μ 2 -OBu) 6 (OBu) 6 (O 3 PPh) 4 (1, C72H128O26P4Zr6)
Bis(trimethylsilyl) phenylphosphonate (100 mm3, 0.33 mmol) was added to 302 mm3 of Zr(OBu)4 (0.66 mmol) in 2 cm3 of BuOH. After 16 weeks at room temperature part of the solvent was removed from the clear solution. Crystals of 1 were obtained after 5 additional weeks at −20 °C. Yield 50 mg (22 %); 1H NMR (CD2Cl2, 250 MHz): δ = 0.61–1.07 (m, 36H, CH3), 1.08–1.86 (m, 48H, CH2), 3.50–4.38 (m, 24H, CH2O), 7.30–7.53 (m, 12H, CH), 7.68–8.05 (m, 8H, CH) ppm; 13C NMR (CD2Cl2, 62.9 MHz): δ = 13.65, 13.93 (CH3), 18.88, 19.10 (CH2CH3), 34.98, 35.60, 36.08 (CH2CH2O), 69.66, 69.92, 70.12 (CH2O), 127.58, 127.82, 130.87, 131.02 (CH) ppm; 31P NMR (CD2Cl2, 101.2 MHz): δ = 6.57 ppm.
Zr 7 O 2 Cluster Zr 7 O 2 (µ 2 -OiPr) 6 (OiPr) 6 (O 3 PCH 2 CH 2 CH 2 Br) 6 (2, C54H120Br6O32P6Zr7)
Methacrylic acid (33.8 mm3, 0.4 mmol) was added to a solution of 465 mg of Zr(OiPr)4 (1.2 mmol) in 2 cm3 of 2-propanol followed by addition of 120 mm3 of bis(trimethyl)silyl(3-bromopropyl)phosphonate (0.4 mmol). After 5 min of vigorous stirring, 10.8 mm3 of water in 1 cm3 of 2-propanol were added quickly. Crystals of 2 were obtained after 2 weeks. Yield 20 mg (12 %); 1H NMR (C6D6, 250 MHz): δ = 1.37 (d 3 J H,H = 6.10 Hz, 36H, CH3), 1.59 (d, 3 J H,H = 6.24 Hz, 36H, CH3), 1.81–1.95 (m, 12H, CH2P), 2.28–2.41 (m, 12H, CH 2 CH2P), 3.57 (t, 3 J H,H = 6.55 Hz, CH2Br), 4.37 (m, 6H, CH), 4.97 (m, 6H, CH) ppm; 31P NMR (C6D6, 101.2 MHz): δ = 30.58 ppm.
X-Ray structure analyses
All measurements were performed using MoK α radiation (λ = 71.073 pm). Data were collected on a Bruker AXS Smart Apex II four-circle diffractometer with κ-geometry at 100 K with φ and ω-scans and 0.5° frame width (Table 1). The data were corrected for polarization and Lorentz effects, and an empirical absorption correction (SADABS) was applied. The cell dimensions were refined with all unique reflections. Saint Plus software (Bruker Analytical X-ray Instruments, 2007) was used to integrate the frames. Symmetry was checked with the program PLATON.
The structure was solved by the Patterson method (SHELXS97 [18]). Refinement was performed by the full-matrix least-squares method based on F with anisotropic thermal parameters for all non-hydrogen atoms. Hydrogen atoms were inserted in calculated positions and refined riding with the corresponding atom. Four of the six crystallographic independent butoxo ligands in 1 were disordered and refined with about 50 % for each position. The same treatment was done for three of the 3-bromopropyl moieties and two isopropoxo ligands in 2. Furthermore, one 3-bromopropyl moiety was refined using three different positions with 42, 36, and 21 % occupancy.
CCDC-1402779 (for 1) and 1402780 (for 2) contain the supplementary crystallographic data. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
References
Poojary DM, Bhardwaj C, Clearfield A (1995) J Mater Chem 5:171
Vermeulen LA, Fateen RZ, Robinson PD (2002) Inorg Chem 41:2310
Costantino F, Sassi P, Geppi M, Taddei M (2012) Cryst Growth Des 12:5462
Byrd H, Clearfield A, Poojary D, Reis KP, Thompson ME (1996) Chem Mater 8:2239
Alberti G, Vivani R, Murcia Mascarós S (1998) J Mol Struct 470:81
Vivani R, Costantino F, Nocchetti M, Gatta GD (2004) J Solid State Chem 177:4013
Vivani R, Costantino F, Costantino U, Nocchetti M (2006) Inorg Chem 45:2388
Taddei M, Costantino F, Vivani R (2010) Inorg Chem 49:9664
Taddei M, Costantino F, Manuali V, Vivani R (2011) Inorg Chem 50:10835
Donnadio A, Pica M, Taddei M, Vivani R (2012) J Mater Chem 22:5098
Taddei M, Vivani R, Costantino F (2013) Dalton Trans 42:9671
Feichtenschlager B, Pabisch S, Peterlik H, Kickelbick G (2012) Langmuir 28:741
Lomoschitz CJ, Feichtenschlager B, Moszner N, Puchberger M, Müller K, Abele M, Kickelbick G (2011) Langmuir 27:3534
Czakler M, Artner C, Schubert U (2013) Eur J Inorg Chem 5790
Czakler M, Artner C, Schubert U (2014) Eur J Inorg Chem 2038
Chandrasekhar V, Senapati T, Dey A, Hossain S (2011) Dalton Trans 40:5394
Czakler M, Artner C, Schubert U (2014) Monatsh Chem. doi:10.1007/s00706-015-1444-5
Sheldrick GM (1997) Program for Crystal Structure Determination. University of Göttingen, Germany
Acknowledgments
This work was supported by the Fonds zur Förderung der wissenschaftlichen Forschung (FWF), Austria (Project P22915). The X-ray measurements were carried out at the X-ray Center of Vienna University of Technology.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Czakler, M., Schubert, U. Phosphonate-substituted zirconium oxo clusters. Monatsh Chem 146, 1371–1374 (2015). https://doi.org/10.1007/s00706-015-1519-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00706-015-1519-3