
Abstract
Pigeon circovirus (PiCV) is one of four viruses in the family Circoviridae that affect young pigeons around the world. We collected 158 serum or tissue samples from six poultry farms in eastern China to investigate the prevalence and genetic characteristics of PiCV in Chinese pigeons. We tested for PiCV using a PCR assay and found that PiCV was present in 80.7 % (88/109) of diseased pigeons and 63.3 % (31/49) of healthy pigeons; overall, 75.3 % (119/158) of samples were PiCV positive. One PiCV-positive sample from each poultry farm was randomly chosen for amplification of the complete PiCV genome by inverse primer PCR (IP-PCR). The six genomic PiCV strains were designated as AHBZ (KJ704801), HBLF-E2 (KJ704802), JSJN (KJ704803), NJPK-21 (KJ704804), SDDZ (KJ704805) and SHWH-AB4 (KJ704806). We compared these new PiCV genomes to six publicly available PiCV genomes and found that the Rep and Cap genes had sequence identity ranging from 93.8 % to 100 % and 79.1 % to 100 %, respectively. In a phylogenetic analysis, PiCV and eight other members of the genus Circovirus were sister to chicken anemia virus (CAV), the only member of genus Gyrovirus. The results of this study provide evidence that PiCV is present in Chinese pigeons at a high rate and that PiCV is a viral lineage that is distinct from CAV.
Similar content being viewed by others


Introduction
Pigeon circovirus (PiCV), also known as columbid circovirus (CoCV), is a small, circular, non-enveloped, single-stranded DNA virus that belongs to the genus Circovirus of the family Circoviridae [1]. PiCV was first recognized in the USA in 1993 [2], and it was later reported in Australia [3] and several European countries, including Northern Ireland [4], England [5], Germany [6], France [7], Italy [8] and Belgium [9]. PiCV infection has been identified in other parts of the world in recent years [10], but little is known about the pathogenesis of the virus. It was reported recently that PiCV is responsible for a complex and variable immune suppression syndrome that mostly affects young pigeons, called “young pigeon disease syndrome” (YPDS) [11]. PiCV infection is generally characterized by a wide range of clinical symptoms, including lethargy, anorexia, weight loss, respiratory diseases, diarrhoea, and other non-characteristic behaviours [12, 13].
The complete genome of PiCV was first cloned from pigeon tissues in 2000 [14]. The average PiCV genome size is 2.0 kb, with minor length variations among different isolates [15]. Viral genomes are characterized by two major open reading frames (ORF), namely ORF V1, composed of 318 amino acids (nt 41–994), encoding the replication-associated protein (Rep), and ORF C1 in the opposite orientation, composed of 274 amino acids (nt 1165–1988) [16], encoding the only structural protein of the virion—capsid protein (Cap) [17]. An additional ORF, ORF C2, is composed of 250 amino acids (nt 411–791), encoding a hypothetical protein [17, 18]. The functions of the other ORFs are unknown.
The family Circoviridae includes two genera: Gyrovirus and Circovirus [19]. Presently, the chicken anemia virus (CAV) is the only member of the genus Gyrovirus, while the genus Circovirus includes several members, including pigeon circovirus (PiCV), porcine circovirus (PCV), psittacine beak and feather disease virus (BFDV) and canary circovirus (CaCV) [20, 21]. Thus far, there are no available culture methods for PiCV, but a PCR assay based on specific PiCV DNA sequences can be used to detect the presence of the virus [22]. In this study, we detected PiCV in Chinese pigeons by PCR and amplified complete PiCV genomes by inverse primer PCR (IP-PCR). We used bioinformatics software to align and analyse the complete PiCV genome sequences from the samples collected in this study as well as those of nine other members of the family Circoviridae available in the GenBank database. We also estimated a phylogeny of PiCV based on analysis of Rep and Cap genes.
Materials and methods
Sample sources
A total of 109 infected pigeons displaying clinical symptoms (such as respiratory signs, weight loss, and diarrhea) and 49 healthy pigeons were collected from poultry farms in the six provinces (Shandong, Anhui, Zhejiang, Shanghai, Hebei and Jiangsu) with the highest pigeon population densities in eastern China (Table 1). Serum or tissue samples were collected from pigeons and immediately frozen at −80 °C.
Reagents
DNA and gel extraction kits were purchased from Geneaid (USA). DL2000 DNA Marker, Pfu DNA polymerase, Taq DNA polymerase, 10× buffer, dNTP mixture and a pMD18-T vector kit were purchased from TaKaRa (Dalian, China). DH5α competent cells were purchased from Tiangen (Beijing, China). All reagents were high-purity and intended for analytical use.
Primer design and synthesis
Two pairs of primers, one for PiCV detection (PiCV-1/2) and the other for amplification of complete genomes (PiCV-JST1/2), were designed according to the published genome sequence of PiCV available in the GenBank database (Table 2). Primers were synthesized by Invitrogen (Shanghai, China).
PCR detection of PiCV
Genomic DNA was extracted from 200 μl of serum (10 %, w/v) or from approximately 25 mg of tissue samples using a commercial DNA extraction kit according to the manufacturer’s instructions. The extracted DNA was eluted twice with 25 μl of sterile distilled water to give a final extract volume of 50 μl.
The genomic DNA was used as template to perform PCR for the detection of PiCV. PCR reaction mixtures (25 μL) contained 18 μL of ddH2O, 2.5 μL of 10× buffer, 1 μL each of primer pairs PiCV-1 and PiCV-2 (10 μM each), 2 μL of DNA template, and 0.5 μL of Taq DNA polymerase (5 U/μL). PCR was initiated by heating for 5 min at 94 °C, followed by 40 cycles of 30 s at 94 °C, 30 s at 60 °C, 40 s at 72 °C and a final extension for 10 min at 72 °C. The amplification products (2 μL) were analyzed by electrophoresis in a 2 % TAE agarose gel and visualized under ultraviolet light.
Cloning and sequencing of the PiCV genome
Complete PiCV genomes were amplified by IP-PCR using the inverse primers PiCV-JST1 and PiCV-JST2. Templates were randomly chosen PiCV-positive DNA samples from six poultry farms (one from each geographic region). IP-PCR reaction mixtures (50 μL) contained 21 μL of Pfu DNA polymerase, 4 μL of DNA template, 2 μL each of the primers, and 21 μL of ddH2O. The PCR protocol was the same as described above. PCR amplification products with the expected sizes were purified using a commercial gel extraction kit according to the manufacturer’s instructions.
The recovered DNA was cloned into the pMD18-T vector and used to transform DH5α competent cells using the heat shock method. The positive clones were confirmed by PCR and Sanger sequencing.
Sequence alignment and analysis
Sequences were first identified by BLAST search against the NCBI database. DNASTAR 7.1 and ClustalX 1.8 software were used to perform alignments and sequence comparisons [23]. Phylogenies were inferred from distance matrices using the neighbor-joining method with a bootstrap test of 1000 iterations in MEGA5 [24].
Results
PCR detection of PiCV infection
An approximately 338-bp specific fragment was amplified. The specific PCR products were identified as PiCV by sequencing and BLAST analysis. Of the 158 samples, 119 were PiCV positive (positive rate, 75.3 %). Among the samples, 88 samples from the 109 unhealthy pigeons (80.7 %) and 31 samples from the 49 healthy pigeons (63.3 %) were PiCV positive (Table 3). PiCV was detected in samples from both diseased and healthy pigeons.
Amplification of PiCV genomes
An approximately 2040-bp complete genome fragment was obtained from PiCV-positive samples from six poultry farms. BLAST alignment results confirmed that six of the recovered genomes were PiCV, and they were designated as the AHBZ, HBLF-E2, JSJN, NJPK-21, SDDZ and SHWH-AB4 strains. The GenBank IDs of these six PiCV complete genomes were KJ704801, KJ704802, KJ704803, KJ704804, KJ704805, and KJ704806, respectively.
Sequence analysis of PiCV genomes
PiCV is a member of the genus Circovirus and the family Circovirdae, so we included nine publicly available genomes to represent the diversity of the family Circovirdae. The genomes were downloaded from GenBank and included chicken anemia virus (CAV), porcine circovirus (PCV), duck circovirus (DuCV), goose circovirus (GoCV), beak and feather disease virus (BFDV), canary circovirus (CaCV), gull circovirus (GuCV), starling circovirus (StCV), raven circovirus (RaCV), and six PiCV strians (PiCV-zj1, NI7050, Ger1, Bel936, FraA4042, US93A) from China, the USA, and four European countries (Table 4).
Sequence analysis demonstrated that the PiCV genome contained two main ORFs, ORF V1 (ORF 1) and ORF C1 (ORF2). As in other members of the genus Circovirus, these genes encode the viral replication-associated protein (Rep) and the viral capsid protein (Cap), respectively. ORF V1 corresponds to nt 41–994 in the nucleotide sequence and encodes 318 amino acids. ORF C1 corresponds to nt 1165–1988 in the nucleotide sequence and encodes 274 amino acids. Notably, the PiCV coding sequence region of ORF V1 and ORF C1 had a conserved nine-nucleotide sequence (TAGTATTAC; Table 5).
Sequence similarity of PiCV genomes
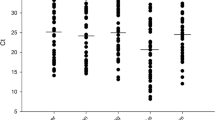
The 12 complete PiCV genome sequences were analysed to assess sequence similarity. The percent sequence similarity among the six complete PiCV genomes in this study ranged from 88.0 % to 100 %, and from 86.9 % to 100 % in a comparison with six other PiCV genomes (Fig. 1), suggesting that PiCV strains maintain an evolutionary signal despite their distribution, and there were no obvious geographical differences in these strains.

Percent identity of 12 PiCV genome sequences based on amino acids. Samples 1–6 are PiCV sequences obtained in this study. Samples 7–12 are PiCV sequences from GenBank
We also assessed the sequence similarity of Rep and Cap genes of the 12 complete PiCV genomes. The sequence similarity of the six Rep and Cap datasets generated in this study ranged from 93.9 % to 100 % and 83.1 % to 98.5 %, respectively. The sequence similarity ranged from 93.8 % to 100 % and 79.1 % to 100 % in a comparison with the six other Rep and Cap sequences available from GenBank (Fig. 2 and Fig. 3). The PiCV Cap sequences were more variable than the relatively conserved Rep sequences.

Percent identity of 12 PiCV strains based on Rep protein amino acid sequences. Samples 1–6 are PiCV sequences obtained in this study. Samples 7–12 are PiCV sequences from GenBank

Percent identity of 12 PiCV strains based on Cap protein amino acid sequences. Samples 1–6 are PiCV sequences obtained in this study. Samples 7–12 are PiCV sequences from GenBank
Sequence analysis of circoviruses
The sequence similarity rate of the 11 circovirus genome sequences from GenBank (Table 4) ranged from 30.2 % to 82.7 %, and the sequence similarity between the 11 circoviruses and the 12 PiCV genomes ranged from 31.4 % to 65.0 % (Fig. 4). It was notable that the sequence similarity between the CAV genome and 22 other circovirus genomes ranged from 30.2 % to 35.7 %, indicating that the only member of the genus Gyrovirus, CAV, is distantly related to the other circoviruses.

Percent identity of 23 circovirus genomes. Samples 1–6 are PiCV strains obtained in this study. Samples 7–12 are PiCV strains and samples 13–23 are circovirus sequences from GenBank
In addition, we constructed a phylogenetic tree based on the 23 complete genomic sequences of the family Circoviridae listed in Table 4 (Fig. 5). In the phylogenetic tree, the 23 circovirus genomes formed two distinct groups. The first branch included the 22 genomes of circoviruses from different hosts, suggesting that they had a close genetic relationship. As the only member of the genus Gyrovirus, one CAV strain was grouped into the second branch alone.

Neighbor-joining tree of 23 circovirus genomes. “●”indicates PiCV genomes obtained in this study, “○” indicates PiCV sequences from GenBank. The 12 PiCV strains are within the shaded box
Discussion
As with other avian circoviruses, PiCV was detected in a variety of tissues and is therefore likely to be present in many secretions [25]. However, there is little information on the prevalence of PiCV infection [26]. We found that PiCV is widely distributed in eastern Chinese pigeons, and there are no obvious differences in the geographical distribution of this virus. The fact that PiCV was detected in both healthy and diseased pigeons implies a potential threat to the pigeon-breeding industry. Histopathological examinations have revealed lymphocyte depletion in the primary and secondary lymphoid tissues in PiCV-infected pigeons, and PiCV infection is thought to cause immune suppression, like other circovirus infections [27]. When the immune system is compromised, secondary infections determine the clinical signs of illness [28, 29], and the initial PiCV infection may be overlooked.
There are currently no serological tests to diagnose PiCV, and the virus has not yet been grown in vitro [30]. Original diagnoses of PiCV infection were dependent on histopathological examination and electron microscopy, but recent diagnostic approaches have focused on detecting the specific PiCV DNA, and a number of PCR methods have been developed and applied [31–33]. The pairwise comparison of 12 complete PiCV genomes as well as Cap and Rep genes showed that regardless of where the strains were collected, the level of nt sequence identity was very similar, and differences in geographic origin play only a minor role in their genetic relationships. It is notable that the sequence similarity of Rep genes (93.8 %–100 %) was much higher than that of Cap genes (79.1 %–100 %) among the PiCV genomes. It is well known that ORF V1 is located on the sense strand, while ORF C1 is located on the complementary strand [34]. The 5′ intergenic region is located between the start sites of the two ORFs and contains a highly conserved nucleotide sequence (TAGTATTAC), the initiation site of rolling-circle replication [35]. Thus, conserved nucleotide sequences and 5′ intergenic regions within the PiCV Rep genes would be suitable for inclusion in diagnostic PCR primers, while variable sequences within the Cap genes should be considered for phylogenetic studies and for selecting virus isolates for vaccine development [36].
Our study confirms that PiCV should be included as a member of the family Circoviridae based on the close relationship of PiCV strains to other circoviruses, the size of the virus, and associated pathologic features [37]. Further research on the family Circoviridae will likely identify more members of this interesting viral lineage. In this study, we included 10 members of the family Circoviridae whose genome sequences were available in GenBank, including nine circoviruses and one gyrovirus [38, 39]. Members of the genus Circovirus formed a group in the phylogenetic tree, and there were no discernible evolutionary patterns related to host differences or geographical distribution (Fig. 5). The two genera of the Circoviridae family, Circovirus and Gyrovirus, are reciprocally monophyletic according to these preliminary data (Fig. 4) and have a different evolution process. The 12 PiCV samples formed a clade and had a close relationship to each other regardless where they were collected.
PiCV infection affects healthy and diseased young pigeons and is thought to be responsible for immune suppression, resulting in complex and variable syndromes. This virus affects pigeons across the world, and outbreaks may have a devastating impact on poultry farming if the virus is left unchecked. Our study provides information about the prevalence of PiCV in eastern China, reveals characteristics of PiCV genomes, and lays the foundation for further research about genetic variation and molecular evolutionary relationships in the family Circoviridae. Future research should aim to include more samples of PiCV from across its distribution area and should explore the pathogenic mechanism of PiCV and methods for culturing this virus.
References
Todd D, Weston JH, Soike D, Smyth JA (2001) Genome sequence determinations and analyses of novel circoviruses from goose and pigeon. Virology 286:354–362
Woods LW, Latimer KS, Barr BC, Niagro FD, Campagnoli RP, Nordhausen RW, Castro AE (1993) Circovirus-like infection in a pigeon. J Vet Diagn Invest 5:609–612
Woods LW, Latimer KS, Niagro FD, Riddell C, Crowley AM, Anderson ML, Daft BM, Moore JD, Campagnoli RP, Nordhausen RW (1994) A retrospective study of circovirus infection in pigeons: nine cases (1986–1993). J Vet Diagn Invest 6:156–164
Smyth JA, Carroll BP (1995) Circovirus infection in European racing pigeons. Vet Rec 136:173–174
Gough RE, Drury SE (1996) Circovirus-like particles in the bursae of young racing pigeons. Vet Rec 138:167
Soike D (1997) Circovirus infections in pigeons. Tierarztliche Praxis 25:52–54
Abadie J, Nguyen F, Groizeleau C, Amenna N, Fernandez B, Guereaud C, Guigand L, Robart P, Lefebvre B, Wyers M (2001) Pigeon circovirus infection: pathological observations and suggested pathogenesis. Avian Pathol 30:149–158
Franciosini MP, Fringuelli E, Tarhuni O, Guelfi G, Todd D, Casagrande Proietti P, Falocci N, Asdrubali G (2005) Development of a polymerase chain reaction-based in vivo method in the diagnosis of subclinical pigeon circovirus infection. Avian Dis 49:340–343
Duchatel JP, Todd D, Curry A, Smyth JA, Bustin JC, Vindevogel H (2005) New data on the transmission of pigeon circovirus. Vet Rec 157:413–415
Chen CL, Wang PX, Lee MS, Shien JH, Shien HK, Ou SJ, Chen CH, Chang PC (2006) Development of a polymerase chain reaction procedure for detection and differentiation of duck and goose circovirus. Avian Dis 50:92–95
Todd D (2000) Circoviruses: immunosuppressive threats to avian species: a review. Avian Pathol 29:373–394
Raue R, Schmidt V, Freick M, Reinhardt B, Johne R, Kamphausen L, Kaleta EF, Muller H, Krautwald-Junghanns ME (2005) A disease complex associated with pigeon circovirus infection, young pigeon disease syndrome. Avian Pathol 34:418–425
Duchatel JP, Todd D, Smyth JA, Bustin JC, Vindevogel H (2006) Observations on detection, excretion and transmission of pigeon circovirus in adult, young and embryonic pigeons. Avian Pathol 35:30–34
Mankertz A, Hattermann K, Ehlers B, Soike D (2000) Cloning and sequencing of columbid circovirus (coCV), a new circovirus from pigeons. Arch Virol 145:2469–2479
Csagola A, Lorincz M, Tombacz K, Wladar Z, Kovacs E, Tuboly T (2012) Genetic diversity of pigeon circovirus in Hungary. Virus Genes 44:75–79
Yu XP, Zhu C, Zheng XT, Mu AX, Yu HT (2009) Cloning and analysis of the complete genomes of pigeon circovirus from Zhejiang Province. Bing Du Xue Bao 25:355–361
Daum I, Finsterbusch T, Hartle S, Gobel TW, Mankertz A, Korbel R, Grund C (2009) Cloning and expression of a truncated pigeon circovirus capsid protein suitable for antibody detection in infected pigeons. Avian Pathol 38:135–141
Xiang QW, Wang X, Xie ZJ, Sun YN, Zhu YL, Wang SJ, Liu HJ, Jiang SJ (2012) ORF3 of duck circovirus: a novel protein with apoptotic activity. Vet Microbiol 159:251–256
Fauquet CM, Mayo MA (2001) The 7th ICTV report. Arch Virol 146:189–194
Phenix KV, Weston JH, Ypelaar I, Lavazza A, Smyth JA, Todd D, Wilcox GE, Raidal SR (2001) Nucleotide sequence analysis of a novel circovirus of canaries and its relationship to other members of the genus Circovirus of the family Circoviridae. J Gen Virol 82:2805–2809
Ball NW, Smyth JA, Weston JH, Borghmans BJ, Palya V, Glavits R, Ivanics E, Dan A, Todd D (2004) Diagnosis of goose circovirus infection in Hungarian geese samples using polymerase chain reaction and dot blot hybridization tests. Avian Pathol 33:51–58
Soike D, Hattermann K, Albrecht K, Segales J, Domingo M, Schmitt C, Mankertz A (2001) A diagnostic study on columbid circovirus infection. Avian Pathol 30:605–611
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739
Duchatel JP, Todd D, Willeman C, Losson B (2009) Quantification of pigeon circovirus in serum, blood, semen and different tissues of naturally infected pigeons using a real-time polymerase chain reaction. Avian Pathol 38:143–148
Zhang Z, Lu C, Wang Y, Wang S, Dai D, Chen Z, Fan H (2011) Molecular characterization and epidemiological investigation of Pigeon circovirus isolated in eastern China. J Vet Diagn Invest 23:665–672
Coletti M, Franciosini MP, Asdrubali G, Passamonti F (2000) Atrophy of the primary lymphoid organs of meat pigeons in Italy associated with circoviruslike particles in the bursa of Fabricius. Avian Dis 44:454–459
Soike D, Albrecht K, Hattermann K, Schmitt C, Mankertz A (2004) Novel circovirus in mulard ducks with developmental and feathering disorders. Vet Rec 154:792–793
Schoemaker NJ, Dorrestein GM, Latimer KS, Lumeij JT, Kik MJ, van der Hage MH, Campagnoli RP (2000) Severe leukopenia and liver necrosis in young African grey parrots (Psittacus erithacus erithacus) infected with psittacine circovirus. Avian Dis 44:470–478
Hattermann K, Soike D, Grund C, Mankertz A (2002) A method to diagnose Pigeon circovirus infection in vivo. J Virol Methods 104:55–58
Roy P, Dhillon AS, Lauerman L, Shivaprasad HL (2003) Detection of pigeon circovirus by polymerase chain reaction. Avian Dis 47:218–222
Freick M, Muller H, Raue R (2008) Rapid detection of pigeon herpesvirus, fowl adenovirus and pigeon circovirus in young racing pigeons by multiplex PCR. J Virol Methods 148:226–231
Halami MY, Nieper H, Muller H, Johne R (2008) Detection of a novel circovirus in mute swans (Cygnus olor) by using nested broad-spectrum PCR. Virus Res 132:208–212
Finsterbusch T, Steinfeldt T, Caliskan R, Mankertz A (2005) Analysis of the subcellular localization of the proteins Rep, Rep’ and Cap of porcine circovirus type 1. Virology 343:36–46
Yu XP, Zheng XT, He SC, Zhu JL, Shen QF (2005) Cloning and analysis of the complete genome of a Goose circovirus from Yongkang Zhejiang. Wei sheng wu xue bao (Acta Microbiol Sin) 45:860–864
Duchatel JP, Jauniaux T, Smyth J, Habsch I, de Bournonville M, Losson B, Todd D (2010) Effect of a commercial paratyphus vaccine on the development of pigeon circovirus infection in young pigeons (Columba livia domestica). J Avian Med Surg 24:107–114
Bassami MR, Berryman D, Wilcox GE, Raidal SR (1998) Psittacine beak and feather disease virus nucleotide sequence analysis and its relationship to porcine circovirus, plant circoviruses, and chicken anaemia virus. Virology 249:453–459
Dayaram A, Goldstien S, Zawar-Reza P, Gomez C, Harding JS, Varsani A (2013) Identification of starling circovirus in an estuarine mollusc (Amphibola crenata) in New Zealand using metagenomic approaches. Genome Announc 1:e00278–13
Fu GH, Cheng LF, Shi SH, Peng CX, Chen HM, Huang Y (2008) Genome cloning and sequence analysis of duck circovirus. Bing Du Xue Bao 24:138–143
Acknowledgments
This study was supported by the Natural Science Foundation of Jiangsu Province (BK2012083), the Industrialization Project of University of Jiangsu Province (JHT-5) and the Research Foundation of Jinling Institute of Technology (40610047).
Author information
Authors and Affiliations
Corresponding author
Additional information
Z. Zhang and W. Dai contributed equally to this work.
Rights and permissions
About this article

Cite this article
Zhang, Z., Dai, W., Wang, S. et al. Epidemiology and genetic characteristics of pigeon circovirus (PiCV) in eastern China. Arch Virol 160, 199–206 (2015). https://doi.org/10.1007/s00705-014-2255-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-014-2255-4