
Abstract
The authors present a case of a 56-year-old man with altered mental status. Magnetic resonance imaging (MRI) of the brain revealed non-enhancing abnormalities on T2 and FLAIR imaging in the brainstem, cerebellum, and cerebrum. Immunohistochemisty demonstrated precursor T-cell lymphoblastic lymphoma. After treatment with methotrexate, he improved clinically without focal sensorimotor deficits and with improving orientation. MRI showed almost complete resolution of brainstem and cerebral lesions. To the authors’ knowledge, there are only five previous reports of primary central nervous system T-cell lymphoblastic lymphoma. Since treatable, it deserves consideration in patients with altered mental status and imaging abnormalities that include diffuse, non-enhancing changes with increased signal on T2-weighted images.
Similar content being viewed by others

Introduction
Primary central nervous system (CNS) lymphomas account for 5% of primary brain tumors, of which the vast majority are of B-cell origin [2]. Although rare, the incidence of primary CNS lymphoma appears to be increasing in both immunocompromised and immunocompetent patients, even when controlling for improving diagnostic technology [18]. Primary CNS T-cell lymphomas (TPCNSL) constitute between 3.6 and 8.5% of primary CNS lymphomas in France and Japan, respectively [2, 12]. In the largest study in the United States, 2% of primary CNS lymphomas were of T-cell origin [9]. Brainstem involvement is even more rare, occurring in 9% of TPCNSL [17, 22]. Primary CNS lymphomas are classified morphologically similar to systemic lymphomas. Primary CNS lymphomas are most commonly diffuse large-cell lymphomas, with lymphoblastic types occurring in only 2.9% [15]. To our knowledge, of the reports which describe histologic analysis, only five report the lymphoblastic type of TPCNSL (Table 1) [4, 11, 19–21]. In this report, we present a 56-year-old man with altered mental status and diffuse non-enhancing magnetic resonance imaging (MRI) abnormalities in the brainstem, cerebellum, and cerebral hemisphere with pathology findings consistent with primary brain T-cell lymphoblastic lymphoma.
Clinical details
History and Examination
The patient is a 56-year-old immunocompetent white man with a 6-week history of headaches, memory disturbance, decreased hearing, diplopia, and weakness. The patient presented to an outside hospital for increasing confusion, lethargy, and vomiting. History was significant for a recent tick bite with positive Lyme disease serology. Prior MRI had demonstrated abnormal enhancement of cranial nerves III and IV bilaterally. The presumptive diagnosis had been Lyme neuroborreliosis and the patient had undergone treatment with doxycycline and prednisone prior to presentation. Improvement was noted, however it was short-lived. Lumbar puncture was performed at the outside hospital which showed mild lymphocytic pleocytosis and oligoclonal bands with 59 white blood cells (WBC) per mm3 (normal < 5), glucose of 36 mg/dl, and protein 154 mg/dl (normal 18-58). Lyme polymerase chain reaction testing of the cerebrospinal fluid (CSF) was negative. On exam, the patient was afebrile, hypertensive 205/105, and had a nonfocal neurologic exam. There was no evidence of nuchal rigiditiy, rashes, lymphadenopathy, or joint swelling. Laboratory studies were significant for 20,700 WBC per mm3 and an erythrocyte sedimentation rate of 13 mm/h. HIV status was negative. The patient’s neurologic status subsequently worsened requiring tracheal intubation for airway protection. The patient, now unresponsive, was transferred to the Virginia Commonwealth University Health System for further evaluation and management.
Imaging
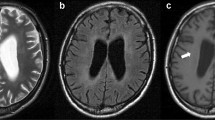
Repeat imaging was performed shortly after presentation to our hospital. Computed tomography (CT) of the brain revealed subtle low attenuation in the brainstem without evidence of hemorrhage, infarction, or mass. Brain MRI revealed widespread, nearly symmetric, non-enhancing areas with increased signal on T2 (Fig. 1a) and FLAIR (Fig. 1b) imaging in the pons, with extension to the medulla and midbrain, cerebellum, right frontal cortex, and bilateral temporal lobes, with extension to the Sylvian region and right parietal lobe. There was no mass effect or midline shift.

Axial MRI obtained before biopsy shows diffuse hyperintensity on T2-weighted images in the pons (a) and extending to the temporal lobes bilaterally (b). Preoperative FLAIR image also demonstrates the lesion in the pons and temporal lobes (c). One month postoperative and post-chemotherapy axial T2-weighted images demonstrate substantial resolution of the lesion in the pons (d) and cerebral hemispheres (e)
Operation and postoperative course
The patient underwent small right craniotomy for open biopsy of the frontal lesion and stereotactic transfrontal, transpeduncular biopsy of the pontine area of T2 signal abnormality. Pathologic examination was consistent with lymphoblastic type TPCNSL and is described below. Normal chest and abdominal CTs supported the CNS origin of the disease. Postoperatively, the patient remained markedly obtunded with impaired respiratory function, requiring tracheostomy and percutaneous endoscopic gastrostomy tube placement. During this period the patient also developed a persistent right facial nerve palsy. The patient was treated with one dose of intrathecal methotrexate, followed by five courses of high dose intravenous methotrexate with leucovorin rescue over the ensuing weeks. Subsequently, the patient improved clinically and was able to eventually transition from inpatient to outpatient neurorehabilitation. On discharge from the rehabilitation program, the patient was alert and oriented to all spheres with improved higher cognitive function and, with the exception of the right facial droop, an intact motor and sensory exam. The postoperative MRI at 1 month showed almost complete resolution of brainstem and cerebral lesions (Fig. 1c, d). The patient continued to do well until 11 months when MRI showed recurrence of disease. The patient was then treated with temozolomide but subsequently succumbed to the disease at 15 months.
Histologic examination
Brainstem biopsy specimen showed atypical lymphocytic infiltrate with prominent reactive astrocytosis and spongiform changes in the white matter (Fig. 2a, b). Subpial spread of malignant cells was noted in the cortical specimen (Fig. 2c). Immunohistochemical analysis demonstrated positive staining for the T-cell marker CD3 (Fig. 2d), CD7, CD45, CD10, CD99 (data not shown), and were weakly positive for both CD1a and CD5 (data not shown). Staining for CD4 was equivocal. CD8, CD34, and CD 68 were negative, as was staining for the B-cell marker CD20 (data not shown). Malignant cells were also positive for TdT (Fig. 2e). The specimen also exhibited a high Ki-67 labeling index (Fig. 2f). CSF analysis showed atypical lymphoid cells consistent with precursor T-cell lymphoblastic lymphoma/leukemia. Bone marrow biopsy demonstrated no evidence of malignancy.

Pathologic specimens obtained from stereotactic biopsy demonstrated diffuse lymphocytic infiltrate in the specimen from the pons (a). Spongiform changes in the white matter and malignant infiltration in Virchow-Robbins spaces were also noted. Magnification of 100× demonstrates malignant lymphocytes (b). The cortical specimen demonstrated subpial infiltration (c). The malignant lymphocytes present in the specimen from the pons stained positive for CD3 (d) and TdT (e). The specimen demonstrated a high Ki-67 labeling index(f)
Discussion and review of the literature
Lymphoblastic lymphomas are characterized by medium-sized blast cells with a high nuclear to cytoplasmic ratio, fine chromatin, and inconspicuous nucleoli [5]. As in the present case, immunophenotyping demonstrating CD3 and TdT positivity confirms the diagnosis of T-cell lymphoblastic lymphoma. TPCNSL of the lymphoblastic type is an extremely rare tumor with only five reported cases in the literature (Table 1) [4, 11, 19–21]. Of the four which provided clinical characteristics, two were supratentorial, one was leptomeningeal, and one was located in the cerebellum. In contrast, the present tumor was located in the pons with diffuse extension to brainstem, cerebellar, and cerebral structures. Taken together, these findings parallel those of the single large multicenter study of TPCNSL, which reported that the majority of these tumors are located supratentorially [22].
Additional clinical characteristics of the lymphoblastic type are similar to those of TPCNSL. The overall median age of patients with TPCNSL is 60 [22]. With the exception of one case report describing a 2-year-old female [4], the remaining cases and ours involve patients in the fifth and sixth decades [11, 19, 21]. There is a male predominance of TPCNSL [22]. Including the present report, 3/5 (60%) of the reported cases of lymphoblastic type TPCNSL are male [19, 21]. In the present report, lumbar puncture revealed an elevated CSF protein, which was true in 79% of TPCNSL [22].
One previously reported patient with leptomeningeal disease presented with altered mental status, as did the present patient [21]. The two remaining reports which described presenting symptoms demonstrated focal neurologic deficits: dysphasias and hemiparesis, which were consistent with their locations in the left frontal lobe [11, 19]. These observations underscore the diversity of presentations possible in this rare disease. Primary CNS lymphoma can be difficult to distinguish from other neurologic diseases [14]. Interestingly, there is one case report of Lyme neuroborreliosis initially misdiagnosed as CNS lymphoma, which led to delay in treatment [24]. Conversely, in the present case, the patient presented with a history of positive Lyme serology and MRI findings consistent with cranial neuritis. When the patient failed to respond to antibiotic and corticosteroid treatment, brain biopsy was performed. Since this tumor is responsive to methotrexate and other chemotherapy treatment, it is important to maintain a high index of suspicion when evaluating patients with non-focal neurologic manifestations and diffuse imaging abnormalities. Early brain biopsy should be considered to obtain a definitive diagnosis.
Imaging features of lymphoblastic TPCNSL appear to be variable. Previous reports utilized CT scanning (Table 1). One tumor demonstrated enhancement after contrast administration [11]. One tumor was hyperdense relative to gray matter and one was hypodense [4, 19]. CT imaging in the present report demonstrated subtle diffuse hypodensity in the brainstem. This variability in CT imaging is in contrast to other primary CNS lymphomas which are generally hyperdense and universally enhancing [10]. A recent series of MRI analysis of seven patients with TPCNSL reported that the majority of these tumors present radiographically as mass lesions that are hypointense on T1-weighted images and hyperintense on T2-weighted images which enhance [16]. While imaging in the present study demonstrated high T2 signal intensity, the abnormality was diffusely infiltrative with no enhancement after contrast administration.
Median survival for TPCNSL is 25 months [22]. A prior case series and literature review suggested that pathology might be predictive of survival, with a higher frequency of long-term survivors having low-grade lymphocytic TPCNSL [23]. A larger, more recent retrospective study showed no difference in survival based on low-grade versus anaplastic histologic features [22]. Of the four previous reports of lymphoblastic TPCNSL, only one patient survived for greater than 1 year [4]. The patient in the present case relapsed after 11 months and succumbed to the disease at 15 months despite salvage therapy. Taken together, these data suggest a shorter survival for patients with TPCNSL of the lymphoblastic type. Recent multicenter studies of primary CNS lymphoma, including patients with TPCNSL treated with high-dose methotrexate in combination with procarbazine and vincristine, have demonstrated prolonged survival relative to single agent therapy [1, 3, 7, 13]. Others have demonstrated improved survival in patients treated with methotrexate and osmotic blood-brain barrier disruption [6]. Preliminary analysis of three patients with TPCNSL treated with blood brain barrier disruption and intra-arterial methotrexate showed similar encouraging responses [8]. Future studies examining the affect of newer treatment regimens such as these would be difficult for TPCNSL of the lymphoblastic type due to the rarity of the disease. However, this type of study would be necessary before definitively designating this histology as a poor prognostic indicator.
Conclusions
TPCNSL of the lymphoblastic type is an extremely rare disease, with only five previously reported cases in the literature. The cases reported demonstrate that this is a diverse disease with variable clinical presentations, anatomic locations, and imaging findings. Because of the possibility of misdiagnosis and rapid progression, early biopsy appears to be indicated to obtain a definitive diagnosis and facilitate institution of appropriate chemotherapy. Nevertheless, it appears that lymphoblastic TPCNSL confers a worse prognosis than other types of TPCNSL.
References
Abrey LE, Yahalom J, DeAngelis LM (2000) Treatment for primary CNS lymphoma: the next step. J Clin Oncol 18:3144–3150
Bataille B, Delwail V, Menet E, Vandermarcq P, Ingrand P, Wager M, Guy G, Lapierre F (2000) Primary intracerebral malignant lymphoma: report of 248 cases. J Neurosurg 92:261–266
Batchelor T, Carson K, O’Neill A, Grossman SA, Alavi J, New P, Hochberg F, Priet R (2003) Treatment of primary CNS lymphoma with methotrexate and deferred radiotherapy: a report of NABTT 96–07. J Clin Oncol 21:1044–1049
Bogdahn U, Bogdahn S, Mertens HG, Dommasch D, Wodarz R, Wunsch PH, Kuhl P, Richter E (1986) Primary non-Hodgkin’s lymphomas of the CNS. Acta Neurol Scand 73:602–614
Chan JK, Banks PM, Cleary ML, Delsol G, Wolf-Peeters C, Falini B, Gatter KC, Grogan TM, Harris NL, Isaacson PG et al (1994) A proposal for classification of lymphoid neoplasms (by the International Lymphoma Study Group). Histopathology 25:517–536
Dahlborg SA, Henner WD, Crossen JR, Tableman CM, Petrillo A, Braziel R, Neuwelt EA (1996) Non-AIDS primary CNS lymphoma: first example of a durable response in a primary brain tumor using enhanced chemotherapy delivery without cognitive loss and without radiotherapy. Cancer J Sci Am 2:166–174
DeAngelis LM, Seiferheld W, Schold SC, Fisher B, Schultz CJ (2002) Combination chemotherapy and radiotherapy for primary central nervous system lymphoma: Radiation Therapy Oncology Group Study 93–10. J Clin Oncol 20:4643–4648
Doolittle ND, Neuwelt EA (2008) Primary central nervous system lymphoma of T-cell phenotype: a rare variant of Non-Hodgkin’s Lymphoma treated with cns directed therapy. Blood (ASH Annual Meeting Abstracts) 112(11): 4965
Ferreri AJ, Reni M, Pasini F, Calderoni A, Tirelli U, Pivnik A, Aondio GM, Ferrarese F, Gomez H, Ponzoni M, Borisch B, Berger F, Chassagne C, Iuzzolino P, Carbone A, Weis J, Pedrinis E, Motta T, Jouvet A, Barbui T, Cavalli F, Blay JY (2002) A multicenter study of treatment of primary CNS lymphoma. Neurology 58:1513–1520
Go JL, Lee SC, Kim PE (2006) Imaging of primary central nervous system lymphoma. Neurosurg Focus 21:E4
Grant JW, Gallagher PJ, Jones DB (1986) Primary cerebral lymphoma. A histologic and immunohistochemical study of six cases. Arch Pathol Lab Med 110:897–901
Hayabuchi N, Shibamoto Y, Onizuka Y (1999) Primary central nervous system lymphoma in Japan: a nationwide survey. Int J Radiat Oncol Biol Phys 44:265–272
Herrlinger U, Kuker W, Uhl M, Blaicher HP, Karnath HO, Kanz L, Bamberg M, Weller M (2005) NOA-03 trial of high-dose methotrexate in primary central nervous system lymphoma: final report. Ann Neurol 57:843–847
Hunt MA, Jahnke K, Murillo TP, Neuwelt EA (2006) Distinguishing primary central nervous system lymphoma from other central nervous system diseases: a neurosurgical perspective on diagnostic dilemmas and approaches. Neurosurg Focus 21:E3
Jellinger KA, Paulus W (1992) Primary central nervous system lymphomas–an update. J Cancer Res Clin Oncol 119:7–27
Kim EY, Kim SS (2005) Magnetic resonance findings of primary central nervous system T-cell lymphoma in immunocompetent patients. Acta Radiol 46:187–192
McCue MP, Sandrock AW, Lee JM, Harris NL, Hedley-Whyte ET (1993) Primary T-cell lymphoma of the brainstem. Neurology 43:377–381
Olson JE, Janney CA, Rao RD, Cerhan JR, Kurtin PJ, Schiff D, Kaplan RS, O’Neill BP (2002) The continuing increase in the incidence of primary central nervous system non-Hodgkin lymphoma: a surveillance, epidemiology, and end results analysis. Cancer 95:1504–1510
Provinciali L, Signorino M, Ceravolo G, Pasquini U (1988) Onset of primary brain T-lymphoma simulating a progressive leukoencephalopathy. Ital J Neurol Sci 9:377–381
Schiffer D, Chio A, Giordana MT, Novero D, Palestro G, Soffietti R, Vasario E (1987) Primary lymphomas of the brain: a clinico-pathologic review of 37 cases. Tumori 73:585–592
Schmitt-Graff A, Pfitzer P (1983) Cytology of the cerebrospinal fluid in primary malignant lymphomas of the central nervous system. Acta Cytol 27:267–272
Shenkier TN, Blay JY, O’Neill BP, Poortmans P, Thiel E, Jahnke K, Abrey LE, Neuwelt E, Tsang R, Batchelor T, Harris N, Ferreri AJ, Ponzoni M, O’Brien P, Rubenstein J, Connors JM (2005) Primary CNS lymphoma of T-cell origin: a descriptive analysis from the international primary CNS lymphoma collaborative group. J Clin Oncol 23:2233–2239
Villegas E, Villa S, Lopez-Guillermo A, Petit J, Ribalta T, Graus F (1997) Primary central nervous system lymphoma of T-cell origin: description of two cases and review of the literature. J Neurooncol 34:157–161
Walther EU, Seelos K, Bise K, Mayer M, Straube A (2004) Lyme neuroborreliosis mimicking primary CNS lymphoma. Eur Neurol 51:43–45
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Clark, A.J., Lee, K., Broaddus, W.C. et al. Primary brain T-cell lymphoma of the lymphoblastic type presenting as altered mental status. Acta Neurochir 152, 163–168 (2010). https://doi.org/10.1007/s00701-009-0433-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00701-009-0433-z