
Abstract
Nucleotide polymorphisms in a set of nuclear genes were studied in a sympatric population of pines Pinus mugo and Pinus sylvestris that includes trees classified as pure species and polycormic (multi-stemmed) individuals of potentially hybrid origin. Patterns of genetic diversity were compared between those groups of samples and to the reference allopatric populations of the species in Europe. Polymorphisms at the gene loci clearly distinguished pure parental species as measured by conventional frequency-based statistics and Bayesian assignment of samples into separate genetic clusters. Most individuals classified based on phenotypic assessments as putative hybrids were genetically very similar to P. mugo showing no existing average net divergence and genetic assignment to the same genetic cluster. On the other hand, individuals of P. sylvestris showed homogenous genetic background to the reference populations of the species from Central and Northern Europe. Ten individuals of admixed genetic composition were found in all three groups of samples; however, the majority of hybrids except one individual were identified across the samples classified as P. mugo and polycormic pines. Those trees that contained a mixture of nuclear gene haplotypes observed in the reference populations of pure species and cpDNA from P. mugo, most likely represent the first generation of hybrids. Analysis of the allelic frequency spectra and compound neutrality tests identified deviations from neutrality at several genes. This contact zone seems suitable for selection of a mapping population both in hybrid and parental species for admixture mapping to effectively search for polymorphisms that may play role in species adaptive variation and speciation.
Similar content being viewed by others

Introduction
Natural hybridisation is an important process that creates recombinants from interspecific mating between divergent parental taxa where they come into geographic contact (Arnold and Martin 2010). Hybridization occurs in roughly 10 % of animal species and 25 % of plant species and it may have various evolutionary consequences for the taxa involved (Baack and Rieseberg 2007). For instance, it may cause the swamping of the species with the smaller effective population size by gene flow from the more abundant species, integration of genetic material from one species into another through repeated back-crossing (introgression), homoploid hybrid speciation in which the new hybrid lineages become reproductively isolated from parental populations, and finally, the transfer of adaptive traits across species boundaries (Baack and Rieseberg 2007). There are well-documented examples which show that natural selection favours hybrid genotypes that may have equivalent or even higher fitness as compared to parental species due to environmental selection (Arnold et al. 2004; Minder and Widmer 2008). Even in the case of initially reduced fertility or viability of hybrids from early generations, gene flow can proceed in the populations leading to the propagation of hybrids and adaptive divergence (Gross and Rieseberg 2005).
Natural hybridisation was postulated between closely related Scots pine (Pinus sylvestris L.) and the taxa from the Pinus mugo complex including dwarf mountain pine (P. mugo T.) (Christensen 1987). Despite close phylogenetic relationships, the species are highly differentiated in phenotype (tree/shrub), geographical range (widespread/restricted) and ecology (generalist/specialist). P. sylvestris is the most widespread and economically important forest tree species in Europe and Asia, whereas P. mugo is an endemic species typical to the mountain regions of Central and Southern Europe. The present distribution of Scots pine is a result of postglacial migration from several glacial refugia (Pyhäjärvi et al. 2008). It is supposed that recolonisation created zones of secondary contacts between isolated local populations from ice-free regions which survived the last glacial maximum with populations from southern refugia. As the ranges of P. sylvestris and the taxa from the P. mugo complex overlapped in some part of their distribution, hybridisation between the species has likely contributed to high diversification observed especially within the P. mugo complex.
At present, those closely related but ecologically differentiated taxa form several contact zones in Central Europe that create unique environments for comparative studies of interspecific hybridization, introgression and the maintenance of species differences in the presence of gene flow. One of them is a sympatric population of P. sylvestris and P. mugo at the ‘Bór na Czerwonem’ peatbog in the Nowotarska Valley, Poland. This population contains a mixture of individuals that could be classified as both pure species and polycormic (multi-stemmed) trees of untypical morphology. The sympatric occurrence of phenotypically differentiated taxa in a very diverse habitat of the peatbog complexes provides a unique opportunity for genomic studies of the role of introgressive hybridization and ecological selection on the species adaptive divergence and evolution. However, nucleotide polymorphisms at nuclear genomes of individuals from contact zones of P. sylvestris and the taxa from the P. mugo complex have not been studied so far.
Here, we evaluated hybridization patterns and the role of interspecific gene flow in shaping genetic variation in a contact zone of dwarf mountain pine (P. mugo) and Scots pine (P. sylvestris). Using nucleotide sequence variation in a multilocus nuclear gene dataset and a set of the reference allopatric populations of the species, we looked at the patterns of population divergence through ecological selection and adaptation in the presence of interspecific gene exchange. Specifically, we tested for the patterns of neutral and adaptive variation at the loci and assessed the role of hybridization and selection in generating the genomic patterns of diversity in the specific peatbog habitats of the species contact zones as compared to the reference allopatric populations of the species in Europe.
Materials and methods
Sampling and DNA extraction
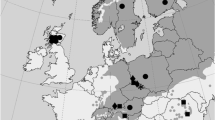
Seeds from 60 individual trees were collected from Bόr na Czerwonem reserve in Nowotarska Valley, Poland (Table 1). This stand is represented by three major phenotypic groups of individuals including (1) bushy P. mugo-like pines, (2) oligo- and polycormic (multi-stemmed) pines of untypical morphology and a height of over 2 m that cover most of the peatbog and (3) monocormic (single-stemmed) P. sylvestris-like individuals that are dominant at some central and north-eastern parts of the peatbog. Selected phenotypic traits, i.e. growth form, bark colour of the upper part of a trunk and main branches, colour and shape of needles and setting angle of conelet from the previous year were used for preliminary taxonomic classification (Christensen 1987). In total, 20 trees from each of the three groups were sampled. In the course of analyses (see below), 10 putative hybrids were found among those 60 samples from the studied contact zone and they were grouped separately in most analyses. In addition, several reference populations from the European distribution of the species were used for the comparisons of the level of nucleotide variation due to hybridization and selection in the hybrid zone vs. genetic differentiation in the allopatric populations of the species not affected by interspecific gene flow (Fig. 1; Table 1). In different analysis, the reference populations were treated separately but also the nearest allopatric populations of the pure species were grouped together to compare a similar number of samples relative to the putative hybrid zone (Table 2). In total, 194 samples were analysed from the contact zone and the reference locations (Table 2). Genomic DNA was extracted from megagametophytes from germinated seeds using DNeasy Plant Mini Kit (Qiagen).

Geographical location of the analysed hybrid population (asterisk) and the reference allopatric populations of P. mugo (filled square) and P. sylvestris (filled circle). Distribution map of Scots pine marked in grey
PCR amplification and DNA sequencing
Nucleotide diversity patterns were studied in a set of eight nuclear gene loci related to cellular metabolism, transport, signal transduction and transcription regulation (Online resource 1) (Ersoz et al. 2010). In addition, the species diagnostic cpDNA marker for P. sylvestris vs. P. mugo in trnF-trnL region (Taberlet et al. 1991) was screened in the samples. This DraI restriction enzyme PCR–RFLP marker was developed based on a single nucleotide polymorphism that leads to an undigested PCR product for P. sylvestris and a digested in one place (two bands) for P. mugo (Wachowiak et al. 2000). As cpDNA is paternally inherited in pines and transmitted by pollen, the comparative analysis of the phenotypes and composition of the chloroplast genomes in each individual may be useful to identify hybrids. PCR amplification was performed with Thermo MBS thermal cyclers and carried out in a total volume of 15 µl containing about 15 ng of haploid template DNA, 10 µM of each of dNTP, 0.2 µM of each forward and reverse primers, 0.15U of Taq DNA polymerase, 1× BSA, 1.5 mM of MgCl2 and 1× PCR buffer (BioLabs). Standard amplification procedures were used with initial denaturation at 94 °C for 3 min. followed by 35 cycles with 30 s. denaturation at 94 °C, 30 s. annealing at 60 °C for nuclear loci and 53 °C for trnF-trnL region and 1 min. 30 s. extension at 72 °C, and a final 5 min. extension at 72 °C. PCR fragments were purified using ExoI-Sap (exonuclease I, Shrimp Alkaline Phosphatase) enzymatic treatment. About 20 ng of the PCR product was used as a template in 10 μl sequencing reactions with the Big Dye Terminator DNA Sequencing Kit (Applied Biosystems, Carlsbad, CA, USA) performed by the Genomed (Warsaw, Poland) sequencing service. Multilocus haplotypes were determined by direct sequencing of haploid DNA from megagametophyte (maternally derived haploid tissue surrounding embryo, which in gymnosperms has the same genotype as the egg cell). CodonCode Aligner software ver. 3.7.1 (Codon Code Corporation, Dedham, MA, USA) was used for editing of the chromatograms, visual inspection of all polymorphic sites detected and alignment and some insertion/deletions were manually adjusted across the samples using GenDoc. The reference sequence from Pinus taeda was used for outgroup comparisons. Haplotype sequence data at the nuclear loci analyzed are deposited in GenBank (NCBI accession number: KM277840-KM277893).
Tests for interspecific gene flow
We tested for introgressive hybridization and admixture patterns in the pine species by comparing the level of nucleotide and haplotype polymorphisms, divergence and difference in the allelic frequency spectra between different groups of samples. The samples included the reference pure species populations, hybrids identified in this contact zone and the remaining groups of samples from Bόr na Czerwonem including P. mugo-like, P. sylvestris-like and oligo- and polycormic pines (Table 2). Nucleotide diversity was measured as the average number of nucleotide differences per site (π) between two sequences (Nei 1987). Multilocus estimates of population mutation parameter, theta (θ W, equal to 4N e μ, where N e is the effective population size and μ is the mutation rate per nucleotide site per generation) (Watterson 1975), were computed based on the number of total and/or silent segregating sites and the length of each locus. The number of haplotypes (N e) and haplotype diversity (H d) were computed for each gene using DnaSP v.5. The number and frequency of unique and shared haplotypes in pairwise comparisons between species were calculated with Arlequin v.3 (Excoffier et al. 2005). Locus-by-locus estimates of net divergence between groups of samples (Nei 1987), the number of shared, exclusive and fixed polymorphic sites and haplotypes for each locus were determined using SITES 1.1. Clustering analysis based on a Bayesian assignment of samples to different groups was applied to look at the relationships between samples from the contact zone and the reference populations of the species using BAPS 6.0 software (Corander and Tang 2007). In the genetic mixture analysis, each locus was input separately as a fasta file using the MLST format and ten independent runs were conducted for each K (1–30) to estimate the number of clusters for all samples combined. The codon linkage model was used, the number of iterations used to estimate admixture coefficients for the individuals was set to 100, the number of the reference individuals was set to 100 and the number of iterations used to estimate admixture coefficients for the reference individuals was 10. The number of populations was inferred from the combined maximum likelihood and the highest posterior probability estimates over all runs. The software was also used for Bayesian admixture analysis that uses genotype information for each marker to estimate admixture parameters. A relationship between groups of samples defined was further evaluated based on the mean genetic distance. The number of base differences per sequence from averaging over all sequence pairs between groups was calculated using MEGA software (Tamura et al. 2011). Genetic differentiation in pairwise comparisons between populations was measured as Wright’s fixation index (Weir and Cockerham 1984), F ST over all polymorphic sites detected and tested for significance by 1,000 permutations of the samples between populations (Excoffier et al. 2005). We also performed the analysis of the genomic composition of paternally transmitted cpDNA in samples from the contact zone of the species. In this analysis, PCR products of diagnostic trnF-trnL marker were digested with DraI restriction enzyme and scored after electrophoresis on 2 % agarose gel as species-specific to P. sylvestris (an undigested product) and species-specific to P. mugo (a digested product with two bands).
Tests for natural selection
We looked if natural selection due to local adaptation to specific peatbog environments affected genes studied in both parental species and hybrids. The loci were examined for the evidence of selection based on the analysis of the allelic frequency spectra as compared to the genetic background of the reference populations and departures from neutral expectations of polymorphisms vs. divergence at the interspecific level. Deviations from the frequency distribution spectrum expected under the standard neutral model of evolution were assessed using the frequency spectrum test and coalescence-based approaches (Tajima 1989). The distribution of Tajima’s D test statistics was investigated for each population or regional groups of populations. The significance of multilocus estimates of the test statistics was evaluated by comparison to a distribution generated by 1,000 coalescent simulations using the HKA programme. Orthologous sequences from the outgroup species were used in the Hudson-Kreitman-Aguadé (HKA) test (Jiggins et al. 2008) to look for overall departures from neutral expectations by assessing the level of multilocus polymorphism and divergence. Deviations of particular genes from the allelic and polymorphic sites frequency distribution spectra expected under the standard neutral model of evolution were investigated using two compound neutrality tests including HEW and DHEW (Zeng et al. 2007). Significance levels of the above tests were determined by carrying out 10,000 coalescent simulations based on Watterson’s estimator of theta as implemented in dh package. For neutrality test that needs a species outgroup, we used orthologous GenBank sequences of P. taeda to contrast the level of intraspecific polymorphisms with interspecific divergence that should be positively correlated for neutrally evolving loci (Hudson et al. 1987). The genetic differentiation at the loci was measured as fixation index (F ST) and its significance was evaluated by 1,000 permutations of the samples between different groups using Arlequin v.3 software (Excoffier et al. 2005).
Results
Genetic variation
About ~3.2 kbp of nuclear DNA sequence was aligned across all nuclear genes and samples providing a set of 146 polymorphic sites. Similar average total nucleotide diversity (π tot = ~0.005) and multilocus estimates of nucleotide diversity (θ tot = 0.004) were found in the group of the reference P. mugo populations, P. mugo from Bόr na Czerwonem and ten individuals of admixed ancestry classified as hybrids (Table 2). Diversity of samples classified as P. sylvestris and polycormic pines was slightly lower and similar to the reference P. sylvestris population (Table 2). In the species contact zone, no net divergence was observed between P. mugo vs. polycormic pines and divergence to the reference P. mugo populations was marginal (0.0002–0.0005) and several times lower as compared to P. sylvestris form Bόr na Czerwonem and the reference populations (0.0016–0.0018). Similarly, no divergence was found between P. sylvestris from that population and the reference samples of the species (Table 3). Hybrids showed overall slightly lower divergence to P. sylvestris than P. mugo. Out of all 132 haplotypes detected across 8 loci in all 194 samples analysed, the majority (56 %) were unique (present only once). Similar proportion of haplotypes (23–29 % out of total identified) was found in each of the group of samples defined (Table 2). Only one unique haplotype was found in the group of hybrids. The average haplotype diversity (H d) ranged between 0.43 and 0.64 (Table 2).
Population structure and differentiation
At all polymorphic sites combined, high differentiation (F ST = 0.28–0.38) was found between the reference populations of P. mugo and P. sylvestris (P < 0.01). Hybrids showed significant differentiation to all other analysed groups of samples; however, the absolute values were slightly lower in reference to Scots pine than other groups (Table 4). Samples of polycormic pines from Bόr na Czerwonem showed significant differentiation to P. sylvestris (F ST = 0.353) from that area and all the reference populations of Scots pine (F ST = 0.31–0.34). However, they showed no differentiation to P. mugo from Bόr na Czerwonem and the reference P. mugo populations (Table 4). Two genetic clusters were detected in mixture analysis using BAPS software. A close genetic similarity was found between polycormic pines from Bόr na Czerwonem, P. mugo from that area and the reference P. mugo populations that all belonged to one cluster. The other group was formed by the samples of P. sylvestris from Bόr na Czerwonem and the reference Scots pine populations (Fig. 2). No admixture was found between the reference populations of P. mugo and P. sylvestris indicating strong discriminating power of the nucleotide polymorphisms at the nuclear loci to distinguish each species. Evidence of admixture was found at ten individuals in total including four samples from the group of polycormic pines, five from P. mugo and one from P. sylvestris (Fig. 2). The group of admixed samples contained a mixture of haplotypes unique for pure P. sylvestris and P. mugo populations. The only unique allele in hybrids was found in two samples at locus Pr4-21 and it resulted from a single mutation. Based on the genetic distance analyses, the admixed samples showed closer genetic relationship with P. sylvestris (Supplementary Fig. 1). All analysed samples phenotypically characterised as peatbog forms of P. sylvestris had cpDNA haplotypes species-specific to P. sylvestris except one individual that had cpDNA of P. mugo. The same individual was also shown to have admixed origin in clustering analysis at nuclear gene loci (Fig. 2). All individuals from the other groups of samples defined including P. mugo, polycormic pines and hybrids had cpDNA haplotypes species-specific to P. mugo.

Bar plot from cluster analysis (BAPS) in the group of pines from Bόr na Czerwonem and the reference samples indicating two genetic clusters (K = 2). The grey and black colours represent proportional assignment to each cluster, the black vertical line separate the corresponding groups of populations. Evidence of admixture (P < 0.01) was found only across the samples from the sympatric population of the species from Bόr na Czerwonem including four trees from the group of samples preliminarily classified based on phenotypic traits as polycormic pines (BOR_PC), five from P. mugo-like (BOR_M) and one from P. sylvestris-like (BOR_S) individuals
Selection
An excess of singleton mutations as compared to expectations under the standard neutral model was detected by significantly negative multilocus Tajima’s D only in Scots pine from the reference populations of the species (D = −0.618 to −0.810, P < 0.05) (Table 2). At individual loci contrasting values of Tajima’s D were found at Pr4-12 with significantly negative values for P. sylvestris (D = −2.046, P < 0.01) from Bόr na Czerwonem vs. significantly positive value for P. mugo from that area (D = 1.724, P < 0.05). At Pr4-17 Tajima’s D was significantly negative in ten hybrid individuals (D = −1.667, P < 0.05). Both compound neutrality tests provided evidence on selection at locus Pr4-12 (P < 0.01) and DHEW test at Pr4-21 (P < 0.05) in P. sylvestris from Bόr na Czerwonem. Evidence on selection was also found at locus Pr4-4 in polycormic pines from that area in HEW test (P < 0.05).
In a multilocus HKA test, overall positive correlation between intraspecific polymorphism and interspecific divergence to the outgroup species at eight loci was found in all defined groups of samples including hybrids. Hybrids showed significant differentiation to P. mugo samples in the allelic frequency spectra at two loci including Pr4-5 and Pr4-19 and at one locus (Pr4-10) as compared to P. sylvestris. In the group of hybrids, alleles specific and observed only in the allopatric populations of P. sylvestris were found at eight samples at locus Pr4-5 and haplotypes specific to P. mugo at seven samples at locus Pr4-10. The remaining alleles at those two loci were common for both parental species. The group of hybrids showed no differentiation (P < 0.01) to any of the parental species at five loci (including Pr4-4, Pr4-12, Pr4-17, Pr4-21, Pr4-27). There was clear differentiation between P. mugo and polycormic pines vs. P. sylvestris at most loci (Supplementary Table 2). P. mugo and polycormic pines from Bόr na Czerwonem showed significant variation to some reference populations of P. mugo at four loci (Pr4-5, Pr4-17, Pr4-19, Pr4-27). No evidence of differentiation was found between P. sylvestris from Bόr na Czerwonem and the reference populations of the species.
Discussion
In our research, nuclear gene loci were sequenced and analysed for intra- and interspecific nucleotide variation in a panel of individuals derived from the contact zone and the allopatric reference stands of the two pine species. The aim was to evaluate the role of introgressive hybridization and selection on nucleotide diversity patterns of the analysed population. High genetic identity was observed between most samples from the group of oligo- and polycormic pines and P. mugo from Bόr na Czerwonem reserve as evident from very similar nucleotide diversity (π tot = ~0.004; θ tot = 0.004), non-existing net divergence and no significant differentiation in the allelic frequency spectra at all polymorphic sites combined and most individual loci. Those two groups also formed a uniform genetic cluster in a Bayesian mixture analysis, showed marginal divergence (0.0002–0.0006) to the reference P. mugo populations and shared higher proportion of haplotypes and SNPs as compared to the monocormic pines from that area classified as P. sylvestris. In contrast, P. sylvestris from Bόr na Czerwonem showed a high genetic similarity to the reference P. sylvestris populations. This genetic similarity of the corresponding groups of samples to the allopatric populations of the species indicates that the majority of analysed individuals from that area represent pure P. mugo and P. sylvestris samples. In the previous studies, the variety of morphological forms observed on this area was explained in biometric and biochemical studies as either the result of intensive hybridisation and introgression that changed the population into a hybrid swarm (Bobowicz 1990) or as a mixture of mostly pure pine species from the P. mugo complex and P. sylvestris, which phenotypes were influenced by specific growing conditions of the peatbog environments (Odrzykoski 2002). As the polymorphism at the genomic regions used in our study clearly distinguishes both putative parental species, our genetic data support the suggestion that exceptional morphology of some oligo- and polycormic individuals from peatbog populations may be due to environmental variation but they most likely represent P. mugo (Wachowiak et al. 2006).
However, in addition to pure species growing on this peatbog, we detected ten individuals in total that clearly result from admixture between P. mugo and P. sylvestris. The majority of hybrid individuals were identified in a group of samples classified initially based on phenotypic traits as P. mugo and/or oligo- and polycormic trees except one monocormic individual classified based on phenotypic assessments as P. sylvestris. Therefore, our preliminary phenotypic classification of the samples based on some basic biometric traits failed to distinguish hybrids. All the hybrids had cpDNA of P. mugo and they contained a mixture of nuclear gene haplotypes observed in the reference allopatric populations of both parental species. The only unique haplotype found in two hybrid trees resulted from a single point mutation. That group of hybrids showed closer genetic similarity to P. sylvestris evident from the higher number of specific P. sylvestris alleles at the loci and lower net divergence. Previous nucleotide diversity studies in pines indicated a high intragenic recombination rate (González-Martinez et al. 2006; Wachowiak et al. 2009). Considering the genetic composition of hybrids and lack of recombining genotypes, it seems that those trees most likely represent first generation hybrids with P. sylvestris as a maternal species.
Our results correspond with some previous observations. Barriers against interspecific hybridisation and no evidence of bidirectional gene flow between P. sylvestris and P. mugo were suggested in some previous research that indicated hybrid seeds derived only from P. sylvestris-like individuals pollinated with P. mugo but not from reciprocal crossings (Wachowiak et al. 2005b). Lack of hybrids resulting from hybridization between P. mugo as a maternal and P. sylvestris as a paternal tree and putative hybrid individuals from reverse crossing combinations were found based on a joint analysis of cpDNA, izozymes and phenotypic characteristics of trees (Wachowiak and Prus-Głowacki 2008). So far, the only evidence of reciprocal hybridization was found in a sympatric population of P. sylvestris and peatbog pine (Pinus uliginosa Neumann), a taxon from the P. mugo complex (Wachowiak et al. 2005a). Analyses of the genetic composition of seeds derived from hybrid trees would be useful to assess other possible hybridization and/or introgression trajectories of those individuals. However, the presence of hybrid embryos would not necessarily mean that such hybrids succeed and exist in peatbog environments, as far as we can conclude from our results. It will also be necessary to grow hybrid seedlings to look at the phenotypic variation and underlying genetic variability of morphological forms. Our results suggest that the first generation hybrids may express extreme phenotypic variability as compared to parental species.
Our study provides evidence on selection at some of the analysed loci. Natural selection can cause fixation of advantageous alleles that have a positive fitness effect and potential to speed up adaptation in new genetic background of hybrids (De Carvalho et al. 2010; Martin et al. 2006). Two loci in our hybrid dataset showed increased frequency towards alleles specific to P. sylvestris at calcium-dependent protein kinase (Pr4-5) and alleles specific to P. mugo at mys transcription factor (Pr4-10). Such increase of frequency of alleles unique to one of the parental species and not observed at other loci suggests that they are under selection in the hybrids’ genetic background and potentially increase their fitness in a peatbog environment. In the case of parental species, strong directional selection at some loci due to local adaptation in ecologically diverged peatbog environments should increase differentiation between the peatbog and the reference allopatric populations of the species as a result of selection for different alleles in different populations. In presence of no population structure within parental species observed in our dataset, significant difference in the allelic frequency spectra was found at a few loci in P. mugo. For instance, at calcium-dependent protein kinase (Pr4-5) and cytochrome P450 reductase (Pr4-17), only a subset of alleles (two in each case) was found in P. mugo samples as compared to the reference allopatric populations of the species. In contrast, no evidence of allelic frequency difference to the reference populations was found across P. sylvestris samples. However, both Scots pine samples from the hybrid zone and the reference populations showed evidence on selection at two loci including proton myo-inositol transporter and receptor protein kinase (Pr4-12 and Pr4-21) in compound neutrality tests. This departure from neutrality most likely reflects the species-wide pattern of selection at the genes in the European range that, however, cannot be directly linked to adaptive variation in peatbog environments. Our study reports a set of new genes with patterns of selection in the hybrid zone of two closely related pine species that contribute to so far a few such loci detected in pines (e.g. Eveno et al. 2008; Kujala and Savolainen 2012; Wachowiak et al. 2009).
Conclusions
Polymorphisms at the analysed genomic regions can discriminate both studied pine species. These polymorphisms could be used for tracking interspecific gene flow and evaluation of species composition in other contact zones of the species where individuals with mixed morpho-anatomical characteristics were described [e.g. in the Alps (Christensen 1987), Rila Mts. (Yurukov and Tashev 1992)]. Our study shows that the examined contact zone includes the majority of pure parental species individuals and some proportion of hybrids (~17 %). Considering the species composition and environmental gradients not optimal for either of the parental species, the investigated and potentially similar hybrid zones seem suitable to study the influence of a local habitat on natural selection at the genes involved in local adaptation of hybrids and parental species from contrasting environments. We identified several genes that may be under natural selection as evident from the pattern of nucleotide polymorphisms in the samples from the hybrid zone and the reference parental populations. Our study shows that it will be possible to select a suitable mapping population of a sufficient size both in hybrid and parental species for admixture mapping to effectively genotype and search for polymorphisms at many genomic regions that may play role in species adaptive variation and speciation.
References
Arnold ML, Martin NH (2010) Hybrid fitness across time and habitats. Trends Ecol Evol 25:530–536
Arnold ML, Bouck AC, Scott Cornman R (2004) Verne Grant and Louisiana Irises: is there anything new under the sun? New Phytol 161:143–149. doi:10.1046/j.1469-8137.2003.00856.x
Baack EJ, Rieseberg LH (2007) A genomic view of introgression and hybrid speciation. Curr Opin Genet Dev 17:513–518. doi:10.1016/j.gde.2007.09.001
Bobowicz MA (1990) Hybrids between Pinus mugo Turra × Pinus sylvestris L. from ‘‘Bόr na Czerwonem’’ reserve in Novotarska Valey [in Polish]. Wydawnictwo Naukowe UAM, Poznań
Christensen KI (1987) A morphometric study of the Pinus mugo Turra complex and its natural hybridization with P. sylvestris L. (Pinaceae). Feddes Repert 98:623–635. doi:10.1002/fedr.19870981113
Corander J, Tang J (2007) Bayesian analysis of population structure based on linked molecular information. Math Biosci 205:19–31
De Carvalho D et al (2010) Admixture facilitates adaptation from standing variation in the European aspen (Populus tremula L.), a widespread forest tree. Molec Ecol 19:1638–1650. doi:10.1111/j.1365-294X.2010.04595.x
Ersoz ES, Wright MH, González-Martínez SC, Langley CH, Neale DB (2010) Evolution of disease response genes in loblolly pine: insights from candidate genes. PLoS One 5:e14234. doi:10.1371/journal.pone.0014234
Eveno E et al (2008) Contrasting patterns of selection at Pinus pinaster Ait. Drought stress candidate genes as revealed by genetic differentiation analyses. Molec Biol Evol 25:417–437. doi:10.1093/molbev/msm272
Excoffier L, Laval G, Schneider S (2005) Arlequin ver. 3.0: an integrated software package for population genetics data analysis. Evol Bioinform Online 1:47–50
González-Martinez SC, Ersoz E, Brown GR, Wheeler NC, Neale DB (2006) DNA sequence variation and selection of tag SNPs at candidate genes for drought-stress response in Pinus taeda L. Genetics 172:1915–1926
Gross BL, Rieseberg LH (2005) The ecological genetics of homoploid hybrid speciation. J Heredity 96:241–252. doi:10.1093/jhered/esi026
Hudson RR, Kreitman M, Aguade M (1987) A test of neutral molecular evolution based on nucleotide data. Genetics 116:153–159
Jiggins CD, Salazar C, Linares M, Mavarez J (2008) Hybrid trait speciation and Heliconius butterflies. Philos Trans, Ser B 363:3047–3054. doi:10.1098/rstb.2008.0065
Kujala S, Savolainen O (2012) Sequence variation patterns along a latitudinal cline in Scots pine (Pinus sylvestris): signs of clinal adaptation? Tree Genet Genomes 8:1451–1467
Martin NH, Bouck AC, Arnold ML (2006) Detecting adaptive trait introgression between Iris fulva and I. brevicaulis in highly selective field conditions. Genetics 172:2481–2489. doi:10.1534/genetics.105.053538
Minder AM, Widmer A (2008) A population genomic analysis of species boundaries: neutral processes, adaptive divergence and introgression between two hybridizing plant species. Molec Ecol 17:1552–1563. doi:10.1111/j.1365-294X.2008.03709.x
Nei M (1987) Molecular evolutionary genetics. Columbia University Press, New York
Odrzykoski IJ (2002) Genetic variation study of Dwarf mountain pine (Pinus mugo) with the use of molecular and biochemical markers [in Polish]. Wydawnictwo Naukowe UAM, Poznań
Pyhäjärvi T, Salmela MJ, Savolainen O (2008) Colonization routes of Pinus sylvestris inferred from distribution of mitochondrial DNA variation. Tree Genet Genomes 4:247–254
Taberlet P, Gielly L, Pautou G, Bouvet J (1991) Universal primers for amplification of three non-coding regions of chloroplast DNA. Pl Molec Biol 17:1105–1109. doi:10.1007/bf00037152
Tajima F (1989) Statistical-method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123:585–595
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Molec Biol Evol 28:2731–2739. doi:10.1093/molbev/msr121
Wachowiak W, Prus-Głowacki W (2008) Hybridisation processes in sympatric populations of pines Pinus sylvestris L., P. mugo Turra and P. uliginosa Neumann. Pl Syst Evol 271:29–40
Wachowiak W, Leśniewicz K, Odrzykoski I, Augustyniak H, Prus-Głowacki W (2000) Species specific cpDNA markers useful for studies on the hybridization between Pinus mugo and P. sylvestris. Acta Soc Bot Poloniae 69:273–276
Wachowiak W, Celiński K, Prus-Głowacki W (2005a) Evidence of natural reciprocal hybridisation between Pinus uliginosa and P. sylvestris in the sympatric population of the species. Flora 200:563–568. doi:10.1016/j.flora.2005.06.007
Wachowiak W, Lewandowski A, Prus-Głowacki W (2005b) Reciprocal controlled crosses between Pinus sylvestris and P. mugo verified by a species-specific cpDNA marker. J Appl Genet 46:41–43
Wachowiak W, Odrzykoski I, Myczko Ł, Prus-Głowacki W (2006) Lack of evidence on hybrid swarm in the sympatric population of Pinus mugo and P. sylvestris. Flora 201:307–316. doi:10.1016/j.flora.2005.08.004
Wachowiak W, Balk P, Savolainen O (2009) Search for nucleotide diversity patterns of local adaptation in dehydrins and other cold-related candidate genes in Scots pine (Pinus sylvestris L.). Tree Genet Genomes 5:117–132
Watterson GA (1975) On the number of segregating sites in genetical models without recombination. Theor Popul Biol 7:256–276
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolution 38:1358–1370
Yurukov S, Tashev A (1992) Studies of natural hybrids between Scots pine (Pinus sylvestris L.) and Mountain pine (Pinus mugo Turra) in the South-east Rila Mts. Nauka za Gorata 29:39–43
Zeng K, Shi S, Wu C-I (2007) Compound tests for the detection of hitchhiking under positive selection. Molec Biol Evol 24:1898–1908
Acknowledgments
The research was financially supported by the Polish National Science Centre (Grant No. 2011/01/B/NZ8/01634). WBŻ acknowledge financial support from Polish National Science Centre (Grant No. DEC-2012/05/E/NZ9/03476). We thank Euforgen network for providing distribution map of Scots pine.
Author information
Authors and Affiliations
Corresponding author
Additional information
Handling editor: Sylvain Razafimandimbison.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Wachowiak, W., Cavers, S. & Żukowska, W.B. Interspecific gene flow and ecological selection in a pine (Pinus sp.) contact zone. Plant Syst Evol 301, 1643–1652 (2015). https://doi.org/10.1007/s00606-014-1182-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00606-014-1182-x