
Abstract
Endemic island plant species with a narrow distribution are often, but not always, linked to low genetic variation within populations and a lack of differentiation among populations. Cedrus brevifolia is a narrow endemic island tree species of Cyprus. Its range is restricted to a single forest, divided into five neighbouring sites. This study, using biparentally inherited nuclear microsatellites and paternally inherited plastid (chloroplast) microsatellites, assessed the genetic variation of C. brevifolia within its sole population and the level of genetic differentiation among formed sites. The results from both markers showed high diversity (nuclear H T = 0.70; plastid H T = 0.93), strongly suggesting that the species did not experience severe bottleneck events or extensive genetic drift. Besides, the maintenance of a high genetic diversity in C. brevifolia may suggest that it originates from a widespread congener species. Significant genetic differentiation at nuclear (G ST = 0.052) and plastid (G ST = 0.119) markers was found among the formed sites. Remarkably, the relatively high genetic differentiation found at plastid markers was comparable to values observed in two widespread congener cedar species. The genetic differentiation probably occurred due to fragmentation of a previously uniform population. This would lead to the shaping of different genetic groups (Bayesian analysis) and to significant population substructure. Furthermore, significant values observed for both isolation by distance and large-scale spatial genetic structure could indicate ineffective gene flow among sites and the early geographical isolation of the more isolated sites from the core population.
Similar content being viewed by others

Introduction
Endemic species have been of interest to population geneticists for a long time. Narrow endemic species have often been recorded to have low diversity (e.g. Loveless and Hamrick 1984; Hamrick et al. 1992; Frankham 1995; Carrió et al. 2010) as a consequence of consanguineous mating (high inbreeding) and random loss of alleles due to drift, which is more probable when the effective population size is low (Hamrick et al. 1979, 1992; Young et al. 1996; Carrió et al. 2010). Therefore, the low genetic variability of narrow endemic species is explained by a limited effective population size as a result of founder effects (e.g. Pinus torreyana—Ledig and Conkle 1983; Waters and Schaal 1991) and genetic bottlenecks (e.g. Picea omorika—Ballian et al. 2006; Narsi et al. 2008). This phenomenon of significant low genetic variation is more obvious in island endemic species than in non-endemic congener species (Frankham 1997).
However, comparisons of genetic diversity between narrow and widespread congeneric species for a series of genetic diversity estimators (i.e. percentage of polymorphic loci, mean number of alleles per locus and observed heterozygosity) have shown that the view of rare species lacking genetic variation is an overgeneralization (Gitzendanner and Soltis 2000). Although narrow species have, on average, less genetic variation than their widespread congeners, there is a wide range in values, and levels of diversity are highly correlated within a genus. Thus, some rare species exhibit reduced genetic variation, whereas others maintain levels of diversity equal to or exceeding those of widespread congeners (e.g. Abies equitrojani—Gulbaba et al. 1998; Pinus rzedowskii—Delgado et al. 1999; Abies nebrodensis—Parducci et al. 2001; Conte et al. 2004; Nothofagus alessandrii—Torres-Díaz et al. 2007). Such high levels of diversity may have resulted from the species not having been strongly affected by genetic drift and/or inbreeding processes, because their population sizes may never have been reduced below a certain threshold. Similarly, narrowly distributed species do not necessarily demonstrate high differentiation among populations (Gitzendanner and Soltis 2000; Peakall et al. 2003). Life history traits (i.e. breeding system, seed and pollen dispersal, longevity) have also been regarded as good predictors of genetic diversity within and among populations (Hamrick and Godt 1989, 1996; Carrió et al. 2010). Hence, wind-pollinated and/or wind-dispersed outcrossing species show a higher proportion of genetic diversity within populations, in combination with low levels of genetic differentiation (Hamrick and Godt 1996; Hamrick et al. 1992; Nybom 2004; Carrió et al. 2010).
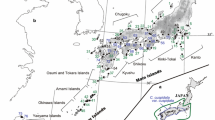
Cedrus brevifolia is a monoecious and wind-pollinated narrow endemic conifer species of the Cyprus flora, believed to be native, as its presence on the island is mentioned as early as the first millennium bc in Theophrastus (370–287 bc) (Hort 1980). “The Red Flora Data Book of Cyprus” (Tsintides et al. 2007) lists Cyprus cedar as a vulnerable species, whilst its habitat type is included in Annex I of the Directive 92/43/EEC as a priority habitat type. Despite the assumption that the species had a wider distribution in the past (Zohary 1973; Thirgood 1987), during the last three centuries its range has been confined to one non-uniform population (Fig. 1) on the mountains of the Paphos forest (Cyprus Forestry Department 2005). Nowadays, this population is patchy, and divided by the Cyprus Forest Department into five neighbouring geographic regimes (referred to as ‘sites’) separated by mountain ranges. C. brevifolia ranges from the upper limits of the meso-Mediterranean to the mid supra-Mediterranean zone (altitude 900–1,400 m above sea level). A recent inventory estimated that approximately 15,800 trees with a diameter at breast height (DBH) over 12 cm and 134,700 trees with a DBH less than 12 cm are growing in this area (Cyprus Forestry Department 2000).

Map of the locations of the 11 sampling plots within the C. brevifolia natural population. Between the main area of Tripylos (plots 1A, 1D, 1E, 1F) and the remaining plots from this subpopulation (plots 1B, 1C) there exist cohorts of C. brevifolia trees which are not obvious on the map
C. brevifolia is well-differentiated from other species of the genus based on morphological and ecophysiological traits such as short needles and slow growth, resistance to aphids, and the highest tolerance to drought in all cedar species (e.g. Vidakovic 1991; Ladjal et al. 2005). In contrast, phylogenetic studies on cedar species have shown a strong genetic relationship between C. brevifolia and its widespread congener C. libani, especially with populations from Turkey (Scaltsoyiannes 1999; Fady et al. 2000; Bou Dagher-Kharrat et al. 2007; Qiao et al. 2007). Qiao et al. (2007), analysing plastid DNA sequences, estimated the time of divergence between C. libani and C. brevifolia (molecular clock) at 7.83 ± 2.79 to 6.56 ± 1.20 million years ago. Despite this rather long divergence time and despite its situation as an isolated endemic species, C. brevifolia must not have experienced strong genetic drift. This is confirmed by the study of Bou Dagher-Kharrat et al. (2007) who, using a small C. brevifolia sample, found ample genetic diversity within Cyprus cedar, comparable to that of the other three cedar taxa.
This study is the first in-depth investigation of genetic variability and population genetic structure in the endemic and narrowly distributed C. brevifolia. Patterns of genetic variation were examined using two DNA marker types with different inheritance modes: biparentally inherited (nuclear microsatellites: Chaib et al. 2006) and paternally inherited (chloroplast microsatellites: Fady et al. 2003, who found that chloroplast DNA—referred to as “plastid”—is paternally inherited in this genus). Attempts to detect variation at maternally inherited mitochondrial DNA failed due to lack of polymorphism (Eliades 2008). The different modes of inheritance of both markers made it possible to observe different responses to evolutionary events such as bottlenecks and genetic drift (Petit and Vendramin 2007) and illustrated patterns of gene flow (Hamilton and Miller 2002) in the whole distribution area of Cyprus cedar. Two major questions were tested: (1) is the surprisingly high (for a narrow endemic island species) genetic diversity of C. brevifolia confirmed using large sample sizes and is it similarly expressed at markers of different modes of inheritance? and (2) is differentiation among the different sites occupied by the species low, as expected for a narrowly distributed and predominantly outcrossing tree species?
Materials and methods
Study location and plant material
Sampling was performed at all the identified sites, covering a sufficient part of each of the five sites (Table 1). The largest site, Tripylos, is in the centre of this area, where cedar trees occupy a nonhomogeneous area of about 398 ha (22.10 ha pure stands and 375.90 ha mixed stands; Fig. 1). In the other four sites cedar occupies smaller areas and, according to the soil conditions, occurs in pure stands (poor soil) or mixed with Pinus brutia (rich soil).
Sampling was performed throughout the entire natural distribution of C. brevifolia. In total, 11 sampling plots were sampled from the whole population, where the number of plots in each site depended on the site size. Six plots were sampled from the central site at Tripylos (plots 1A–F), two from the second largest site at Mauroi Gremoi (plots 2A, B) and one from each of the remaining sites (Fig. 1; Table 1). In each case, the sampling design covered a sufficient part of each of the five sites.
From each plot, plant material (fresh needles) was collected from 50 adult geo-referenced trees with a DBH ≥12 cm, as this is the diameter used in the most recent forest inventories for adult individuals (Cyprus Forestry Department 2000). A DBH of 12 cm corresponds to an age of about 40 (mixed stands) to 70 years (pure stands) in C. atlantica (Bertrand 2004). A total of 550 trees (50 samples per plot, Table 1) were sampled. The size and form of each plot depended on the density and distribution of the trees within them. Distances between plots ranged from 358 m (plot 1A to plot 1E) to 7,549 m (plot 4A to plot 5A).
DNA extraction and molecular markers
Total genomic DNA was extracted from 40 mg of needles, and washed with 70% ethanol. DNA extraction was performed with a DNeasy 96 plant kit following the protocol of the manufacturer (Qiagen, Hilden, Germany). The amount of DNA was checked using λ-DNA as reference on a 0.8% agarose gel and visualized by staining with ethidium bromide. The extracted DNA was stored at −20°C for later use.
Nuclear microsatellites
The six primer pairs developed by Chaib et al. (2006) for C. atlantica were tested for transferability and polymorphism in C. brevifolia. Tests were performed on 16 C. brevifolia individuals using the forward primer labelled with the fluorescent dyes 6-FAM (blue) or HEX (green). The PCR (for the primer pairs which were successfully amplified in C. brevifolia; see “Results”) was carried out in a volume of 15 μl. For primer CatXIITcC6 the PCR mix contained: 10 mM Tris-HCl (pH 9.0), 1.5 mM MgCl2, 50 mM KCl, 0.5 U Taq DNA polymerase (Qiagen), 0.2 mM of each dNTP (Qiagen), 0.6 μM of each forward and reverse primers and 2 μl (about 20 ng) of genomic DNA. For the other three primer pairs (CatITgD4, CatXITcE11 and CatXITcD12) the PCR were performed using 7.5 μl Hot Star Master Mix (Qiagen), containing: 10 mM Tris-HCl (pH 9.0), 1.5 mM MgCl2, 50 mM KCI, 0.2 mM of each dNTP, 0.75 U Taq polymerase, plus 0.6 μM of each forward and reverse primers and 2 μl (about 20 ng) of genomic DNA, in a total volume of 15 μl.
The thermal cycling for the primers CatITgD4 and CatXITcD12 consisted of denaturation at 95°C for 15 min; 1 cycle at 80°C for 4 min; 30 cycles at 94°C for 1 min, T a (annealing temperature) for 1 min (53°C and 55.5°C, respectively) and 72°C for 1 min, followed by a final extension step at 72°C for 30 min. The same procedure was followed for CatXIITcC6, but with the denaturation step lasting for 5 min, and with T a being 54°C. For CatXITcE11 a touchdown PCR was used consisting of denaturation at 95°C for 15 min; 6 cycles at 94°C for 1 min, 60°C for 1 min (about 1°C per cycle) and 72°C for 1 min, followed by 35 cycles at 94°C for 1 min, 54°C for 1 min and 72°C for 1 min; the final extension step was at 72°C for 30 min.
Plastid microsatellites (haplotypes)
The seven plastid microsatellite primer pairs (Vendramin et al. 1996) which were successfully amplified in Cedrus species (Fady et al. 2003) were tested for amplification and polymorphism in Cyprus cedar in 16 samples. The PCR mix (15 μl) contained 10 mM Tris-HCl (pH 9.0), 2.5 mM MgCl2, 50 mM KCl, 0.4 mM of each dNTPs (Qiagen), 1 U of Hot Star Taq DNA polymerase (Solis BioDyne, Tartu, Estonia), 0.5 μM of each forward and reverse primers and 2 μl (about 20 ng) of genomic DNA. The PCR programme was as described by Vendramin et al. (1996). In order to test the homology of the amplification products all fragments of different sizes were sequenced.
For both markers, PCR was carried out in a Peltier PTC-200 thermal cycler (MJ Research, Waltham, MA). PCR fragments were separated on an ABI PRISM 3100 genetic analyser, with the internal size standard GS 500 ROX (Applied Biosystems, Darmstadt, Germany). The data were scored using the software Genescan3.7 and Genotyper3.7 programs (Applied Biosystems).
Analysis of genetic diversity within plots
Nuclear microsatellites
Genetic variation of nuclear microsatellites for each of the 11 plots was assessed by calculating: the observed number of alleles (n a), allelic richness (El Mousadik and Petit 1996; Petit et al. 1998) (A R), and genetic diversity (expected heterozygosity, H E). The inbreeding coefficient (F IS) was also calculated according to Weir and Cockerham (1984), while the statistical significance of F IS was based on Bonferroni-corrected P values after 10,000 permutations. The Fstat version 2.9.3 program (Goudet 2001) was used for these analyses. The observed heterozygosity (H O) was assessed by direct counts using the program Arlequin 3.01 (Excoffier and Schneider 2005).
Plastid haplotypes
Haplotypes were identified by combining size variants at each plastid (chloroplast) locus. Genetic diversity within plots was evaluated using the program RAREFAC (Petit 2003). The following parameters were calculated: number of different haplotypes (A), number of private haplotypes (P h), genetic diversity (H E) according to Nei (1973) and haplotypic richness (H R). Haplotypic richness was estimated after rarefaction to uniform sample sizes, corresponding to the smallest population size of the dataset (El Mousadik and Petit 1996).
Analysis of genetic diversity among plots
Population structure was investigated using a Bayesian analysis implemented in the program Bayesian Analysis of Population Structure (BAPS) v.5.2 (Corander 2006). The Bayesian analysis approach treats both the number of populations and the allele frequencies in each population as random variables (Corander et al. 2004), detecting the hidden population substructure by clustering sampled populations into a panmictic group. The analysis was done for both marker types, using prior information on the plot of origin for each sampled individual. Analysis was run ten times for 105 iterations after a burn-in period of 10,000, randomly mixing the order of sampling plots in the input file. The resulting partitions were averaged based on their posterior probabilities and illustrated on a clustering graphical output.
The genetic relationships among plots were analysed by means of a cluster analysis using the neighbour-joining (NJ) algorithm (Saitou and Nei 1987) and the minimum genetic distance of Nei (1972). The NJ algorithm was preferred as it produces additive trees and does not assume identical evolutionary rates along all branches (Weising et al. 2005). Bootstrap values for the dendrogram were generated using the Populations program (Langella 2002) and 10,000 permutations over individuals for both marker types.
The overall differentiation was computed for nuclear microsatellites based on R ST (Slatkin 1995) and F ST (Weir and Cockerham 1984), using the SPAGeDI program version 1.2d (Hardy and Vekemans 2002). For plastid haplotypes, overall differentiation was computed based on: (1) differences in fragment frequencies at each locus (G ST) and (2) similarities among haplotypes (N ST) using the Permut/CpSSR program (Petit 2005). Additionally, the total diversity (H T) and the genetic diversity across plots (H S) were calculated for both markers. The significance of genetic differentiation was determined by 10,000 permutations.
The presence of phylogeographic structure at nuclear microsatellites was assessed using the SPAGeDi program version 1.2d (Hardy and Vekemans 2002) through permutations of allele sizes among alleles within a single locus (pR ST) (10,000 permutations). This analysis compares the observed R ST value (before randomisation) with the distribution of pR ST values obtained for all possible configurations of allele size permutations—or a representative subset of them—and indicates whether shifts in allele size resulting from stepwise mutations contribute to population differentiation (Hardy et al. 2003). Phylogeographic structure at plastid haplotypes was assessed by the permutation test of N ST and G ST values for significant differentiation (Pons and Petit 1996).
Population genetic structure was also examined using hierarchical analysis of molecular variance (AMOVA; Excoffier et al. 1992), with the Arlequin 3.01 program (Excoffier and Schneider 2005). Significance tests were conducted using 10,100 permutations. AMOVA was performed after grouping the plots according to their origin from the five identified sites. In addition, AMOVA was used to investigate the significance of the influence of the altitudinal range of the plots on genetic diversity.
To test the significance of isolation by distance (IBD) at both markers, a matrix of pair-wise genetic differentiation [F ST/(1−F ST) – 10,100 random permutations using the Arlequin 3.01 program] was correlated with geographical distance using the Mantel test (Mantel 1967), using a NTSYS-PC version 2.0 (9,999 random permutations). First, an analysis was done using all plots, then the most remote plots (plot 4A and plot 5A) were consecutively removed, followed by the three remaining distant plots (plots 2A and 2B, and plot 3A).
In addition, large-scale spatial genetic structure was also investigated using kinship coefficients (F ij ), which were assessed between all pairs of trees using the statistic of Loiselle et al. (1995). Further, the regression slope (b F) of kinship coefficients on log-transformed distance was computed based on the relationship between genetic similarity and geographical distance between individuals. For both analyses the statistical significance was determined using the 99% confidence interval of F ij defined after 10,000 permutations and was calculated using the SPAGeDi 1.2 program (Hardy and Vekemans 2002).
To test whether differentiation among plots resulted from genetic drift due to recent reductions in effective population size, a diversity excess (Cornuet and Luikart 1996; Luikart et al. 1998) analysis was performed using the program Bottleneck version 1.2.02 (Piry et al. 1999). Diversity excess was estimated for each plot by simulating the coalescent process (10,000 iterations), where the two-phase mutation model (since it takes into account mutational events involving several repeats; Di Rienzo et al. 1994) was applied for nuclear microsatellites and the stepwise mutation model for plastid microsatellites. To determine which plot had a significant diversity excess (H E) the Wilcoxon signed ranks test was used, which was the most appropriate for the data from this study since it can be used with few polymorphic loci and any number of individuals (Luikart et al. 1998).
Results
Characterization of marker polymorphism
Four out of the six nuclear microsatellites showed amplification (CatITgD4, CatXITcE11, CatXITcD12 and CatXIITcC6) with clear and strong bands for all samples, corresponding to 66.7% transferability from C. atlantica to C. brevifolia. For loci CatITgD4 (188–204 bp) and CatXIITcC6 (280–305 bp) alleles differed by a multiple of two nucleotides, as reported by Chaib et al. (2006) for C. atlantica. For primer CatXITcD12 (274–310 bp) the dinucleotide repeat motif was observed (as in C. atlantica—Chaib et al. 2006), but with an addition of intermitted alleles (1 bp difference) with a total frequency of 12%. These additional size variants were considered as new alleles. At locus CatXITcE11 (dinucleotide repeat motif in C. atlantica—Chaib et al. 2006), dinucleotide patterns with several gaps were observed with the first gap (between 179 and 191 bp) being the largest.
Three of the seven tested plastid microsatellite primer pairs (Pt15169, Pt63718, Pt71936) were polymorphic in Cyprus cedar, resulting in a total of 15 variants. Four variants were found for locus Pt15169 (122–125 bp), six for locus Pt63718 (94 bp, 96–100 bp) and five for locus Pt71936 (142 bp, 144–147 bp). The variants combined into 46 different haplotypes out of 120 mathematically possible combinations. From these, 31 had frequencies less than 1% in the analysed samples.
SequencingFootnote 1 the different plastid fragments obtained from the amplification confirmed the presence of microsatellite motifs (mononucleotide repeats), while their homology was confirmed by BLAST. In addition, the homology and microsatellite motif of the detected plastid fragments were confirmed by comparing the sequences with those found by Fady et al. (2008) for the same loci.
Genetic diversity within plots
Nuclear microsatellites
The total number of alleles per locus ranged from 10 (CatITgD4) to 26 (CatXITcD12), with a total of 59 alleles and an average of 14.75 alleles per locus. Six out of the 56 alleles were private for different plots (i.e. plots 1A, 1B, 1D, 2A and 5A) with allele frequencies ranging from 1 to 6.5%. Two out of the six alleles were present only in plot 5A, with a total frequency of 10.7%.
Multilocus analysis with the rarefied sample size of 45 individuals (Table 2) showed that allelic richness (A R) ranged from 6.15 (plot 1F) to 10.49 (plot 1D), with a mean value of 8.44. Gene diversity (H E) ranged from 0.61 (plot 2A) to 0.75 (plot 1D; Table 2), with a mean value of 0.67. The observed heterozygosity (H O) ranged from 0.52 (plot 2A) to 0.75 (plot 1E), with the site Exo Milos having the highest value (H O = 0.72). The mean F IS value ranged from −0.095 (plot 1B) to 0.154 (plot 2A); 99% of F IS values were not significantly different from zero (Table 2). Plot 2A was the only plot to show positive but nonsignificant F IS values for each locus, but the total F IS of this plot was significantly different from zero.
At the site level, the highest A R value (40.68) was recorded in the largest site (Tripylos, plots 1A–1F). Meanwhile, the highest H E value (0.71) was recorded in the sites Sellae tis Ellias (plot 3A) and Exo Milos (plot 5A), and the highest H O value (0.72) was recorded in the site Exo Milos (plot 3A).
Plastid haplotypes
Genetic diversity parameters estimated for haplotype data within plots are summarized in Table 3. Haplotypic richness (H R) ranged from 5.00 (plot 5A) to 22.36 (plot 3A; Table 3), with a mean value of 11.99 (rarefied sample size of 48). Mean haplotype diversity (H E) was 0.84, ranging from 0.58 (plot 4A) to 0.94 (plot 3A). In addition, the highest number of haplotypes (24) and private haplotypes (10–20% of the sample size) was recorded in plot 3A, exceeding the total number of private haplotypes in the 300 samples of Tripylos. In contrast to plot 3A, the lowest number of haplotypes (six) was recorded in plot 5A, in which no private haplotypes were detected. At the site level, a lower H R value (15.98) was recorded in Tripylos (plots 1A–1F) than in a smaller site, namely Sellae tis Ellias (plot3A, H R 22.36); the highest H E value (0.94) was also recorded in Sellae tis Ellias.
Genetic structure among plots
BAPS showed that the 11 plots were clustered into six groups for nuclear microsatellites (Fig. 2a), with plots 1A, 1B, 1F, 4A and 5A each forming a separate group and the remaining plots forming a common group. For plastid microsatellites, Bayesian analysis revealed seven groups (Fig. 2b) which corresponded to a large extent with the natural geographic subdivision of C. brevifolia. Plots from Tripylos (plots 1A–1F) divided into three groups, while both plots from Mauroi Gremoi were included in a common group. Each of the remaining three plots (plots 3A, 4A and 5A) represented a separate group. Based on this analysis of both marker types, plots 1A, 4A and 5A were each classified as a separate group.

Grouping of sampling plots according to BAPS: a based on nuclear microsatellites and b based on plastid haplotypes
For both markers the amount of genetic differentiation (Nei’s minimum genetic distance; Nei 1972) observed between plot 4A (Throni) and plot 5A (Exo Milos) was higher than among the other plots. Clustering of plots based on the NJ dendrograms using Nei’s minimum genetic distances for both markers did not reflect the geographic subdivisions or the location of plots in the field. Based on this distance plot 1B and plot 5A were grouped together with the highest bootstrap value (91%) for nuclear microsatellites; for plastid haplotypes the highest bootstrap value (73%) was found between plot 2A and plot 2B (Fig. 3). For both markers, however, low bootstrap values (less than 50%) were obtained for the other clades of the NJ trees.

NJ tree of genetic markers with matrix of genetic distance using Nei’s minimum genetic distances based on a nuclear microsatellites and b plastid haplotypes. Numbers indicate percentage bootstrap support values from 10,000 replications
The genetic differentiation among plots for nuclear microsatellites was 0.052 for the infinite allele model (F ST) and 0.055 for the stepwise mutation model (R ST). Allele size information analysis revealed that R ST was not significantly different from pR ST (P value −0.774 across all loci and plots). Nonsignificant differences (P > 0.05) were also found for haplotype differentiation measures, namely G ST (0.119) and N ST (0.105). For nuclear markers the total diversity (H T) was 0.70 and for plastid markers 0.93, while the genetic diversity across plots (H S) was 0.67 and 0.86 for nuclear and plastid markers, respectively.
The hierarchical partitioning of the genetic variation (AMOVA, Table 4) demonstrated that most variation for both markers was distributed within plots (nuclear microsatellites 94.83%, plastid haplotypes 88.56%). AMOVA showed a significant differentiation among the groups (Φ CT), which were formed according to the plot regime. In contrast, nonsignificant genetic differentiation was recorded among the three altitude groups (Φ CT). However, genetic differentiation among plots within each group (Φ SC) and within-plot differentiation (Φ ST) were significant for both groups (plot regime and altitude; Table 4).
The Mantel test showed positive linear and significant coefficient of correlation (r) among geographic and genetic distances for both markers only when the plots from the whole distribution area (sites) of Cyprus cedar were included in the analysis (r = 0.690 for nuclear microsatellites and r = 0.652 for plastid haplotypes, P < 0.01). In contrast, when the most isolated plots were removed from the analysis, the Mantel test did not show any significant correlations.
The large-scale spatial genetic structure using the kinship coefficients (F ij ) showed a continuously negative slope over space in the autocorrelogram (Fig. 4). For this analysis ten distance classes were selected in order to display an equal number of pairs of individuals in each class. In the first distance class (0–150 m) both genomes showed significant mean kinship coefficients with the plastid genome showing a higher value (F ij = 0.080) and a steeper decrease in the kinship coefficient in this class than the nuclear genome (F ij = 0.042). The plastid genome showed a significant F ij (up to 1,639 m) over a longer distance than the nuclear genome (up to about 500 m). Significant values were also observed for the regression slope (b F) analysis, where the value of b F for the plastid genome (−0.0214*) was twice that of the nuclear genome (−0.0102*).

Average kinship coefficient (F ij ) between pairs of individuals plotted against the distance at the population level for both genomes; filled symbols indicate significant deviation from zero
Finally, the bottleneck analysis according to the Wilcoxon signed ranks test for both markers indicated that none of the studied plots had significant diversity excess.
Discussion
Insights into the species’ evolutionary history
Considering that endemic C. brevifolia has been restricted to a narrow range on the highlands of Paphos forest, its level of genetic diversity is particularly interesting and contrary to other reports in the literature. The total genetic diversity (H T) of Cyprus cedar was found to be high (nuclear microsatellites 0.70, plastid haplotypes 0.93) compared to genetic diversity levels for nuclear microsatellites observed by Nybom (2004) in other endemic and narrowly distributed species (0.42 for endemic, 0.56 for narrowly distributed species). The detected nuclear diversity was in the same range as for the widespread congener C. atlantica (0.75, Chaib et al. 2006) for the same markers. With respect to plastid haplotype diversity (at the same plastid—chloroplast—loci) C. brevifolia showed the same range of diversity as its widespread congener C. atlantica (0.95 in Morocco; Terrab et al. 2006) and even higher values than its widespread congener C. libani (0.84 in Lebanon, 0.89 in Turkey, 0.86 overall; Fady et al. 2008). These results are in agreement with a phylogenetic study by Bou Dagher-Kharrat et al. (2007) who, using AFLPs, found that Cyprus cedar showed relatively higher genetic diversity than populations of the other congener cedar taxa. Remarkably, C. brevifolia showed a higher level of genetic diversity than some widespread conifer species of the Mediterranean basin, namely Pinus pinaster and Pinus halepensis (Bucci et al. 1998; Vendramin et al. 1998). Additionally, Cyprus cedar clearly exceeds the level of genetic diversity found in the widespread Pinus pinea (Vendramin et al. 2008), as well as in the narrowly distributed endemic Picea omorika (Narsi et al. 2008). This ample genetic diversity within a narrow endemic species suggests that Cyprus cedar is not characterized by a founder effect. Such an assumption could be accepted if the population is of recent origin, which is not the case for C. brevifolia (Theophrastus in Hort 1980; Qiao et al. 2007). The high genetic diversity of Cyprus cedar has possibly resulted from the absence of genetic drift (discussed below). C. brevifolia is thus another example of a narrow endemic island species with high diversity levels.
This ample genetic diversity in Cyprus cedar could be interpreted according to the argument that maintaining high genetic diversity in a narrow endemic species is due to its origin from a widespread congener species (Smith and Pham 1996). This is probably true for C. brevifolia, since earlier phylogenetic studies on cedar species found a genetic relationship between C. brevifolia and several C. libani sources from Turkey (Scaltsoyiannes 1999; Fady et al. 2000; Bou Dagher-Kharrat et al. 2007). Also, the comparatively high transferability (66.7%) of nuclear microsatellite primers from C. atlantica to C. brevifolia, compared to other studies on conifer species (González-Martínez et al. 2004; Vendramin and Hansen 2005), as well as the observation of the same polymorphic microsatellite loci in C. brevifolia and in other cedar species (Fady et al. 2003, 2008; Terrab et al. 2006), implies that cedar species (taxa) have derived from a common phylogenetic group (Scaltsoyiannes 1999; Fady et al. 2000, 2003; Qiao et al. 2007).
Another explanation for this high genetic diversity could be admixture of several C. libani sources from Turkey, a hypothesis also presented by Bou Dagher-Kharrat et al. (2007). This admixture most likely happened during the Cyprus cedar migration into the island during the Iberian connection in the Messinian (7–5 Mya). During this period the sea level dropped by 3,048 m as a result of the frying up of the Mediterranean sea (Hsü 1972). Thus, islets appeared within the Mediterranean basin (Hsü 1972), possibly contributing to flora and fauna migration from neighbouring mainland areas (i.e. Syria, Turkey) to Cyprus. The migration of animal species (e.g. elephant, hippopotamus) to the island could also have resulted in seed migration during different chronological periods. This admixture hypothesis could be reinforced by the genetic relationship between C. brevifolia from Cyprus and C. libani sources from Turkey, recorded in previous studies (Scaltsoyiannes 1999; Fady et al. 2000; Bou Dagher-Kharrat et al. 2007). The assumption that admixture is of anthropological origin should be viewed with caution, since the molecular clock (Qiao et al. 2007) places the divergence of C. brevifolia from counterpart species much earlier than the earliest confirmed site of human activity in Cyprus (10,000 bc; Mithen 2005). Further, when Theophrastus visited the island and described the species (370–287 bc), Cyprus cedar was already restricted to the mountainous area of the Paphos forest, as is the case today for the natural habitat of the species. Nevertheless, an analysis of C. libani populations covering the species distributional range and the C. brevifolia population for the same markers might support this hypothesis. Also, the assumption that C. brevifolia had a wider distribution on the island in the past should be further investigated (i.e. pollen and wood fossils).
The demographic implications of the Cyprus cedar population do not appear to suggest genetic erosion of the species. Pleistocene climate variations are known to have modified effective population sizes throughout Europe, since during these variations population bottlenecks resulted in genetic drift (Petit et al. 2003, 2008). C. brevifolia, although a narrow endemic, may have been somewhat preserved from genetic bottlenecks. Bottlenecks are assumed to have occurred during displacement and confinement of populations in Pleistocene glaciations or by raised water levels during intervening warm periods (van Staaden et al. 1996). The effective population size (N e) of Cyprus cedar in its present natural sites can thus be assumed to have never dropped below the critical threshold of 500 individuals, a sample size considered by Sherwin and Moritz (2000) as necessary to explain a heterozygosity decrease of 0.1% per generation. Additionally, even if in one of the isolated sites N e dropped below a critical size, this size would have been maintained for only a few generations. Meanwhile, the present population size of this species is large enough to counteract the effects of genetic drift. Finally, the nonsignificant inbreeding coefficient values that were generally observed are another indication that Cyprus cedar is not affected by demographic bottlenecks, as its population is at a Hardy-Weinberg equilibrium, and not significantly affected by inbreeding.
Insights into the demographic history of the C. brevifolia population
The plots of the current Cyprus cedar population were apparently divided into different genetic groups (Bayesian analysis), suggesting a substructure in the C. brevifolia population. However, the low but significant divergence among the plots for the nuclear microsatellite (G SΤ = 0.052) is in contrast to the general assumption that endemic species are characterized by high population divergence (e.g. Loveless and Hamrick 1984; Hamrick et al. 1992; Carrió et al. 2010), even for nuclear microsatellites (G ST = 0.26; Nybom 2004). In contrast, the plastid marker in C. brevifolia is characterized by significantly higher genetic differentiation (G ST = 0.119) than other cedars, comparable with values observed in its widespread congeners C. libani (G ST = 0.067; Fady et al. 2008) and C. atlantica (G ST = 0.107; Terrab et al. 2006), at the same plastid loci. AMOVA also indicated a higher percentage of differentiation for plastid haplotypes than for nuclear microsatellites. This discrepancy between the two genomes could be due to differences in effective population size (Hu and Ennos 1997; Hamilton and Miller 2002), since the effective population size of the plastid genome is half that of the nuclear genome. Thus, in non-equilibrium populations and when IBD occurs, plastid markers drift at a faster rate than nuclear markers (Hu and Ennos 1997). Meanwhile, the absence of significant phylogeographic structure, the low bootstrap values for clades in the dendrograms and the low genetic differentiation in the nuclear genome imply relatedness among the plots. This close relationship among plots could either reflect the fragmentation of an earlier, uniformly distributed population, or ample levels of gene flow between sites. Nevertheless, the significant IBD, as well as the significant differentiation among plots within groups (AMOVA), probably suggest the current existence of barriers to gene flow owing to local conditions that cannot exactly be measured. Besides, the continuously negative slope of the autocorrelogram and the negative significant regression slope imply a structure across space (e.g. Diniz-Filho and Telles 2002; Heuertz et al. 2003). The fact that the paternal plastid genome showed a clearer geographical structure (Bayesian analysis) and a significant kinship coefficient (F ij ) over longer distances than the nuclear genome appears to be an indication of reduced levels of pollen flow. Thus, the inconsistent levels of genetic differentiation between genomes could also be influenced by the different gene flow patterns (pollen and seed dispersal rates) resulting from secondary seed dispersal by animals (biotic means). The wider area of Paphos forest is also the natural habitat of the endemic mouflons (Ovis aries orientalis) which act as seed vectors.
The interpretation of fragmentation of a previously uniform population appears to be more plausible. The fragmentation might have been recent and caused by human activities (such as over-grazing before conservation laws were passed in the 20th century) or more ancient forces, caused by altitudinal and range movements amplified by the local topography during Pleistocene glaciations and interglaciations, as was typical for Euro-Mediterranean ecosystems (Petit et al. 2003). This altitudinal movement of C. brevifolia is possibly the reason for the nonsignificant differentiation between groups according to elevation (AMOVA). Additionally, the hypothesis of fragmentation would also be in accordance with the significant IBD when the most isolated sites are included in the analysis. This IBD may have resulted from different population histories, as these sites were probably separated earlier from the uniform population than the other sites. Particularly for the sites of Throni (plot 4A) and Mauroi Gremoi (plot 2A, the plot with a significant inbreeding coefficient), fragmentation could have negatively affected the maintenance of genetic variation, since genetic drift appears to have been promoted as a result of reduction in gene flow at fragmented sites.
On the contrary, the high genetic diversity of small and isolated sites (Sellae tis Ellias, plot 3A; Exo Milos, plot 5A) is in line with the argument of Young et al. (1996) that not all fragmentation events lead to reduced genetic diversity in relic populations. Based on this, the relic sites can be assumed to have had unusually high diversity before fragmentation, most likely due to middle-frequency private alleles and/or haplotypes within the site. Similarly, the maintenance of high genetic diversity has been found to be linked to the occurrence of such private alleles by Taggart et al. (1990) and Prober and Brown (1994). Additionally, the high genetic diversity in these small sites most likely implies that not enough time has elapsed since fragmentation.
In conclusion, despite the general view on endemic species (Loveless and Hamrick 1984; Hamrick et al. 1992; but see Carrió et al. 2010) and similarly to observations by Gitzendanner and Soltis (2000), C. brevifolia is both genetically diverse and genetically structured. Therefore, it constitutes an example of a narrow endemic island species with significant genetic structure. Its origin, potential contact with mainland species and population size may have been the most important components affecting the high genetic diversity observed. Additionally, this study provides further proof that the mild eastern Mediterranean climate of the late glacial maximum made it possible for mountainous conifers to conserve their high genetic diversity by maintaining large enough population sizes (Mai 1989; Fady 2005; Fady and Conord 2010).
Notes
One DNA sequence (the smallest fragment) per plastid primer pair observed in C. brevifolia was deposited in GenBank under the accession numbers FJ907514 (Pt15169), FJ907515 (Pt63718) and FJ907516 (Pt71936).
References
Ballian D, Longauer R, Mikić T, Paule L, Kajba D, Gömöry D (2006) Genetic structure of a rare European conifer, Serbian spruce (Picea omorika (Panč.) Purk.). Plant Syst Evol 260:53–63
Bertrand A (2004) Dynamic naturelle du cedre de l’Atlas (Cedrus atlantica Manetti) au Mont Ventoux. Approche spatio-temporelle et elements de modélisation. MSc Thesis, Université de Droit, Aix-Marseille III
Bou Dagher-Kharrat M, Mariette S, Lefèvre F, Fady B, Grenier-de March G, Plomion C, Savouré A (2007) Geographical diversity and genetic relationships among Cedrus species estimated by AFLP. Tree Genet Genomes 3:275–285
Bucci G, Anzidel M, Madaghiele A, Vendramin GG (1998) Detection of haplotypic variation and natural hybridization in halepensis-complex pine species using chloroplast simple sequence repeat (SSR) markers. Mol Ecol 7:1633–1643
Carrió E, Forrest AD, Güemes J, Vargas P (2010) Evaluating species nonmonophyly as a trait affecting genetic diversity: a case study of three endangered species of Antirrhinum L. (Scrophulariaceae). Plant Syst Evol 288:43–58
Chaib J, Danan S, Jouaud L, Hagen S, Lefevre F, Fady B (2006) Identification and characterization of nuclear microsatellite in Mediterranean cedar (Cedrus spp.). Mol Ecol Notes 6:840–842
Conte L, Cotti C, Schicchi R, Raimondo FM, Christofolini G (2004) Detection of ephemeral genetic sub-structure in the narrow endemic Abies nebrodensis (Lojac.) Mattei (Pinaceae) using RAPD markers. Plant Biosyst 138:279–289
Corander J (2006) BAPS: Bayesian Analysis of Population Structure manual v. 4.14. IOP PublishingWeb. http://web.abo.fi/fak/mnf/mate/jc/software/baps.html. Accessed 13 April 2011
Corander J, Waldmann P, Marttinen P, Sillanpää MJ (2004) BAPS 2: enhanced possibilities for the analysis of genetic population structure. Bioinformatic 20:2363–2369
Cornuet JM, Luikart G (1996) Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 144:2001–2014
Cyprus Forestry Department (2000) Inventory of forest vegetation and regeneration in the Trypilos nature reserve area. Sector of management and data processing service. Ministry of Agriculture, Natural Resources and Environment, Nicosia, Cyprus (in Greek).
Cyprus Forestry Department (2005) Cyprus. In: Merlo M, Croitoru L (eds) Valuing Mediterranean forests: towards total economic value. CABI Publishing, Wallingford, pp 213–228
Delgado P, Piñero D, Chaos A, Pérez-Nasser N, Alvarez-Buylla ER (1999) High population differentiation and genetic variation in the endangered Mexican pine Pinus rzedowskii (Pinaceae). Am J Bot 86:669–676
Di Rienzo A, Peterson AC, Garza JC, Valdes AM, Slatkin M, Freimer NB (1994) Mutational processes of simple-sequence repeat loci in human populations. Proc Natl Acad Sci USA 91:3166–3170
Diniz-Filho JAF, Telles MPC (2002) Spatial autocorrelation analysis and the identification of operational units for conservation in continuous populations. Conserv Biol 16:924–935
El Mousadik A, Petit RJ (1996) High level of genetic differentiation for allelic richness among populations of the argan tree [Argania spinosa (L.) Skeels] endemic to Morocco. Theor Appl Genet 92:832–839
Eliades NG (2008) Fingerprinting of genetic diversity and patterns of spatial genetic variation in the endemic tree Cedrus brevifolia (Hook f.) Henry from Cyprus: implications for its conservation. Optimus, Goettingen
Excoffier LG, Schneider S (2005) Arlequin ver. 3.0: an integrated software package for population genetics data analysis. Evol Bioinformatics Online 1:47–50
Excoffier L, Smouse P, Quaitro J (1992) Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131:479–491
Fady B (2005) Is there really more biodiversity in Mediterranean forest ecosystems? Taxon 54:905–910
Fady B, Conord C (2010) Macroecological patterns of species and genetic diversity in vascular plants of the Mediterranean basin. Divers Distrib 16:53–64
Fady B, Bariteau M, Fallour D, Giroud E, Lefèvre F (2000) Isozyme gene markers and taxonomy of Mediterranean Cedrus species. In: Panetsos K (ed) Adaptation and selection of Mediterranean Pinus and Cedrus for sustainable afforestation of marginal lands. Proceedings of the Final Conference of the EU project FAIR CT95–0097. Aristotelian University of Thessaloniki, Mytilene, pp 21–26
Fady B, Lefèvre F, Reynaud M, Vendramin GG, Bou Dagher-Kharrat M, Anzidei M, Pastorelli R, Savouré A, Bariteau M (2003) Gene flow among different taxonomic units: evidence from nuclear and cytoplasmic markers in Cedrus plantation forests. Theor Appl Genet 107:1132–1138
Fady B, Lefèvre F, Vendramin GG, Ambert A, Régnier C, Bariteau M (2008) Genetic consequences of past climate and human impact on eastern Mediterranean Cedrus libani forests. Implications for their conservation. Conserv Genet 9:85–95
Frankham R (1995) Conservation genetics. Annu Rev Genet 29:305–327
Frankham R (1997) Do island populations have less genetic variation than mainland populations? Heredity 78:311–327
Gitzendanner MA, Soltis PS (2000) Patterns of genetic variation in rare and widespread plant congeners. Am J Bot 87:783–792
González-Martínez SC, Robledo-Arnuncio JJ, Collada C, Díaz A, Williams CG, Alía R, Cervera MT (2004) Cross-amplification and sequence variation of microsatellite loci in Eurasian hard pines. Theor Appl Genet 109:103–111
Goudet J (2001) Fstat: a program to estimate and test gene diversities and fixation indices (version 2.9.3.2). University of Lausanne, Switzerland
Gulbaba AG, Velioglu E, Ozer AS, Doerksen AH, Adams WT (1998) Population genetic structure of Kazdağ Fir (Abies equitrojani Ashers. Et Sint), a narrow endemic to Turkey. Implication for in situ conservation. In: Zencirci N, Kaya Z, Anikster Y, Adams WT (eds) Proceedings of international symposium on in situ conservation of plant genetic diversity CRIFC. Ankara, Turkey, pp 271–281
Hamilton MB, Miller J (2002) Comparing relative rates of pollen and seed gene flow in the Island Model using nuclear and organelle measures of population structure. Genetics 162:1897–1909
Hamrick JL, Godt MJ (1989) Allozyme diversity in plant species. In: Brown A, Clegg M, Khaler A, Weir B (eds) Plant population genetics, breeding and genetic resources. Sinauer Press, Sunderland, pp 43–63
Hamrick JL, Godt MJW (1996) Effects of life history traits in genetic diversity in plant species. Biol Sci 351:1291–1298
Hamrick JL, Linhart YB, Mitton JB (1979) Relationships between life history characteristics and electrophoretically detectable genetic variation in plants. Annu Rev Ecol Syst 10:173–200
Hamrick JL, Godt MJW, Sherman-Broyles SL (1992) Factors influencing levels of genetic diversity in woody plant species. New Forest 6:95–124
Hardy OJ, Vekemans X (2002) SPAGeDi: a versatile computer programme to analyse spatial genetic structure at the individual or population levels. Mol Ecol Notes 2:618–620
Hardy OJ, Charbonnel N, Fréville H, Heuertz M (2003) Microsatellite allele sizes: a simple test to assess their significance on genetic differentiation. Genetics 163:1467–1482
Heuertz M, Vekemans X, Hausman JF, Palada M, Hardy OJ (2003) Estimated seed vs. pollen dispersal from spatial genetic structure in the common ash. Mol Ecol 12:2483–2495
Hort A (1980) Theophrastus: enquiry into plants, and minor works on odours and weather signs, Vols 1–2, Books 1–9. English translation, Harvard University Press, Massachusetts, pp 458–465
Hsü KJ (1972) When the Mediterranean dried up. Sci Am 277:26–36
Hu XS, Ennos RA (1997) On estimation of the ratio of pollen to seed flow among plant populations. Heredity 79:541–552
Ladjal M, Huc R, Ducrey M (2005) Drought effects on hydraulic conductivity and xylem vulnerability to embolism in diverse species and provenances of Mediterranean cedars. Tree Physiol 25:1109–1117
Langella O (2002) Populations (version 1.2.28). Centre National de la Recherche Scientifique, France. IOP PublishingWeb. http://fsffrance.org/science/biologie.en.html. Accessed 13 April 2011
Ledig FT, Conkle MT (1983) Genetic diversity and genetic structure in a narrow endemic torrey pine(Pinus torreyana Parry ex Carr). Evolution 37:79–85
Loiselle AB, Sork LV, Nason S, Graham C (1995) Spatial genetic structure of a tropical understory shrub, Psychotria officinalis (Rubiaceae). Am J Bot 82:1420–1425
Loveless M, Hamrick JL (1984) Ecological determinants of genetic structure in plant population. Annu Rev Ecol Syst 15:65–95
Luikart G, Sherwin WB, Steele BM, Allendorf FW (1998) Usefulness of molecular markers for detecting population bottlenecks via monitoring genetic change. Mol Ecol 7:963–974
Mai DH (1989) Development and regional differentiation of the European vegetation during the Tertiary. Plant Syst Evol 162:79–91
Mantel NA (1967) The detection of disease clustering and a generalized regression approach. Can J For Res 27:209–220
Mithen S (2005) After the ice: a global human history, 20,000 BC–5,000 BC. Harvard University Press, Boston, p 97
Narsi N, Bojovic S, Vendramin GG, Fady B (2008) Population genetic structure of the relic Serbian spruce, Picea omorika, inferred from plastid DNA. Plant Syst Evol 271:1–7
Nei M (1972) Genetic distance between populations. Am Nat 106:283–292
Nei M (1973) Analysis of gene diversity in subdivided populations. Proc Natl Acad Sci USA 70:3321–3323
Nybom H (2004) Comparison of different nuclear DNA markers for estimating intraspecific genetic diversity in plants. Mol Ecol 13:1143–1155
Parducci L, Szmidt AE, Ribeiro MM, Drouzas AD (2001) Taxonomic position and origin of the endemic sicilian fir Abies nebrodensis (Lojac.) Mattei based on allozyme analysis. For Genet 8:119–127
Peakall R, Ebert D, Scott LJ, Meagher PF, Offord CA (2003) Comparative genetic study confirms exceptionally low genetic variation in the ancient and endangered relictual conifer, Wollemia nobilis (Araucariacea). Mol Ecol 12:2331–2334
Petit RJ (2003) RAREFAC. IOP PublishingWeb. http://www.pierroton.inra.fr/genetics/labo/Software/Rarefac/. Accessed 13 April 2011
Petit RJ (2005) PERMUT&CpSSR. IOP PublishingWeb. http://www.pierroton.inra.fr/genetics/labo/Software/PermutCpSSR/index.html. Accessed 13 April 2011
Petit RJ, Vendramin GG (2007) Plant phylogeography based on organelle genes: an introduction. In: Weiss S, Ferrand N (eds) Phylogeography of southern European refugia: evolutionary perspectives on the origins and conservation of European biodiversity. Springer Netherlands, Dordrecht, pp 23–100
Petit RJ, El Mousadik A, Pons O (1998) Identifying populations for conservation on the basis of genetic markers. Conserv Biol 12:844–855
Petit RJ, Aguinagalde I, de Beaulieu JL, Bittkau C, Brewer S, Cheddadi R et al (2003) Glacial refugia: hotspots but not melting pots of genetic diversity. Science 300:1563–1565
Petit RJ, Hu FS, Dick CW (2008) Forests of the past: a window to future changes. Science 320:1450–1452
Piry S, Luikart G, Cornuet JM (1999) Bottleneck: a computer program for detecting recent reductions in the effective population size using allele frequency data. J Hered 90:248–249
Pons O, Petit RJ (1996) Measuring and testing genetic differentiation with ordered versus unordered alleles. Genetics 144:1237–1245
Prober SM, Brown AHD (1994) Conservation of the grassy white box woodlands: population genetics and fragmentation of Eucalyptus albens. Conserv Biol 8:1003–1013
Qiao CY, Ran JH LY, Wang XQ (2007) Phylogeny and biogeography of Cedrus (Pinaceae) inferred from sequences of seven paternal chloroplast and maternal mitochondrial DNA regions. Ann Bot 100:573–580
Saitou N, Nei M (1987) The neighbour joining method: a new method for reconstructing phylogenetic tree. Mol Biol Evol 4:406–425
Scaltsoyiannes A (1999) Allozyme differentiation and phylogeny of cedar species. Silvae Genet 48:61–68
Sherwin WB, Moritz C (2000) Managing and monitoring genetic erosion. In: Young AG, Clarke GM (eds) Genetics, demography and viability of fragmented populations. Cambridge University Press, Cambridge, pp 9–34
Slatkin M (1995) A measure of population subdivision based on microsatellite allele frequencies. Genetics 139:457–462
Smith JF, Pham TV (1996) Genetic diversity of the narrow endemic Allium aaseae (Alliaceae). Am J Bot 83:717–726
Taggart JB, McNally SF, Sharp PM (1990) Genetic variability and differentiation among founder populations of the pitcher plant (Sarracenia purpurea L.) in Ireland. Heredity 64:177–183
Terrab A, Paun O, Talavera S, Tremetsberger K, Arista M, Stuessy TF (2006) Genetic diversity and population structure in natural populations of Moroccan Atlas cedar (Cedrus atlantica; Pinaceae) determined with cpSSR markers. Am J Bot 93:1274–1280
Thirgood JV (1987) Cyprus: a chronicle of its forest, land and people. University of British Columbia Press, Vancouver
Torres-Díaz C, Ruiz E, González F, Fuentes G, Cavieres L (2007) Genetic diversity in Nothofagus alessandrii (Fagaceae), an endangered endemic tree species of the coastal Maulino forest of Central Chile. Ann Bot 100:75–82
Tsintides T, Christodoulou CS, Delipetrou P, Georghiou K (2007) The red data book of the flora of Cyprus. Cyprus Forest Association, Nicosia, p 149
van Staaden MJ, Michener GR, Chesser RK (1996) Spatial analysis of microgeographic genetic structure in Richardson’s ground squirrels. Can J Zool 74:1187–1195
Vendramin GG, Hansen OK (2005) Molecular markers for characterizing diversity in forest trees. In: Geburek T, Turok J (eds) Conservation and management of forest genetic resources in Europe. Arbora Publishers, Zvolen, pp 335–368
Vendramin GG, Lelli L, Rossi P, Morgante M (1996) A set of primers for the amplification of 20 chloroplast microsatellites in Pinaceae. Mol Ecol 5:595–598
Vendramin GG, Anzidei M, Madaghiele A, Bucci G (1998) Distribution of genetic diversity in Pinus pinaster Ait. as revealed by chloroplast microsatellites. Theor Appl Genet 97:456–463
Vendramin GG, Fady B, González-Martínez SC, Hu FS, Scotti I, Sebastiani F, Soto Á, Petit RJ (2008) Genetically depauperate but widespread: the case of an emblematic Mediterranean pine. Evolution 62:680–688
Vidakovic M (1991) Conifers. In: Brekalo B (ed) Morphology and variation. Graficki Zavod, Hrvatske, pp 129–136
Waters ER, Schaal BA (1991) No variation is detected in the chloroplast genome of Pinus torreyana. Can J For Res 21:1832–1835
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolution 38:1358–1370
Weising K, Nybon H, Wolff K, Kahl G (2005) DNA fingerprinting in plants: principles, methods, and applications, 2nd edn. Tayor & Francis, Boca Raton
Young A, Boyle T, Brown T (1996) The population genetic consequences of habitat fragmentation for plants. Trends Ecol Evol 11:413–418
Zohary M (1973) Geobotanical foundations of the Middle East, vol 2. Gustav Fischer/Swets and Zeitlinger, Stuttgart/Amsterdam
Acknowledgments
The authors would like to thank all the technical assistants at the section of Forest Genetics and Forest Tree Breeding, University Göttingen, Germany, for their technical support in the laboratory work. Many thanks also to Assistant Professor Aristotelis C. Papageorgiou (Democritus University of Thrace) for his continuous support. Warm gratitude is extended to the Cyprus Forest Department (CFD) and the Cyprus Geological Survey (CGS) for providing the basic data for the map design, as well as to Mr. Didier Betored (INRA, Avignon) for his help with the map layout. Also, many thanks to the CFD for valuable help in sampling. Last but not least, the authors thank Dr. Pablo Vargas (associate editor of the journal PSE) and the two anonymous reviewers for their insightful comments on a previous version of the manuscript. The writing of the manuscript was made possible by financial support to the first author from the Mobility Center of the European Network of Excellence EVOLTREE (EVOLution of TREEs as drivers of terrestrial biodiversity, http://www.evoltree.org/) while visiting INRA Avignon.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Eliades, NG.H., Gailing, O., Leinemann, L. et al. High genetic diversity and significant population structure in Cedrus brevifolia Henry, a narrow endemic Mediterranean tree from Cyprus. Plant Syst Evol 294, 185–198 (2011). https://doi.org/10.1007/s00606-011-0453-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00606-011-0453-z