
Abstract
Changes of hepatic sinusoids are crucial in the pathogenesis of liver cirrhosis and portal hypertension. Liver injury leads to distinct morphological abnormalities such as loss of sinusoidal fenestration, vasoconstriction, and angiogenesis as well as molecular changes. Communication between the two key cells in this hepatic microenvironment—hepatic stellate cells (HSC) and sinusoidal endothelial cells (SEC)—has been studied for many years and several canonical pathways have been elucidated, such as decreased eNOS activity or increased PDGF and TGF-β production leading to activation and migration of HSC. In recent studies, alternative pathways of intercellular communication in liver diseases have been described such as cell-derived extracellular vesicles called exosomes, which deliver cell compounds to their target cells. Moreover, such extracellular vesicles may link injury to inflammation in alcoholic hepatitis. While inflammation leading to liver fibrosis has been studied in detail, in some circumstances pathways other than the known canonical inflammatory pathways may contribute to hepatic fibrogenesis. For example, in congestive hepatopathy, sinusoidal dilatation and fibrosis have been shown to be mediated by non-inflammatory mechanisms and associated with sinusoidal thrombi. A recently developed murine model further enables experimental studies of this disease entity. Increasing knowledge about these alternative disease pathways in liver injury, inflammation, and fibrosis may reveal possible target molecules for future therapies. This article builds upon a seminar given at the recent 3rd JSGE International Topic Conference in Sendai, Japan, and reviews the areas outlined above.
Similar content being viewed by others
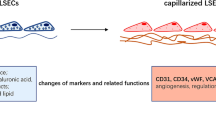

Overview of changes in hepatic sinusoids in the pathogenesis of liver cirrhosis and portal hypertension
The hepatic sinusoids with the involved hepatic sinusoidal endothelial cells (SEC), smooth muscle cells, and pericyte-like hepatic stellate cells (HSC) form an intrahepatic microcirculatory unit, where these cells are intimately associated with one another and communicate through paracrine and autocrine effects [1]. Changes in this microenvironment are crucial in the early steps of fibrogenesis and include sinusoidal remodeling, vasoconstriction, endothelial dysfunction, and angiogenesis [2]. This article provides background information pertaining to the hepatic sinusoids and then focuses on recent developments pertaining to sinusoidal pathobiology, as recently presented at the 3rd JSGE International Topic Conference in Sendai, Japan.
Sinusoidal endothelial cells
SEC have a very unique phenotype distinct from conventional endothelial cells, characterized by multiple fenestrae and the lack of a basement membrane [3–5]. Serving as a mechanical sieve, fenestration permits steric selection of transfer from sinusoidal space to hepatic parenchyma [6, 7]. In addition, SEC show constitutive expression of nitric oxide (NO) synthase, which can be upregulated by increased blood flow and shear stress [8] as well as paracrine factors from other cells such as VEGF [9, 10]. Through this mechanism known as autoregulation, hepatic sinusoids can adapt to increased intrahepatic blood flow and decrease intrahepatic pressure [8]. Further physiological functions of SEC are leukocyte diapedesis through expression of adhesion molecules, endocytosis (known as the hepatic reticuloendothelial system), and bacterial processing [5]. As the first cells interacting with portal vein components, SEC are exposed to different bacterial compounds. Bacterially derived lipopolysaccharides (LPS) bind to their receptor TLR-4 expressed on SEC, inducing injury and inflammation [11]. Interestingly, recent studies have shown that LPS can also induce fibrosis-associated angiogenesis by interacting with TLR-4, highlighting the role of SEC in the so-called gut–liver axis [12, 13].
In liver disease, the SEC phenotype changes dramatically [1, 14]. Liver injury leads to endothelial dysfunction with loss of fenestrae and deposition of a basement membrane, a process that is known as capillarization [6, 15, 16]. Besides these morphological changes, changes in paracrine and autocrine function are dramatic, too. NO production usually decreases, leading to an increasing intrahepatic pressure due to an inability to maintain intrasinusoidal autoregulation by vasodilatation [1, 17]. In addition, decreased NO production activates a contractile phenotype of HSC, inducing extracellular matrix production and migratory capacity [18, 19]. Finally, recent studies revealed the importance of angiogenesis in the process of fibrogenesis and their interdependency [12, 20]. As fibrogenesis is considered a dynamic process, adequate blood supply and metabolically highly active cells require sprouting of new vessels, as we know from tumorigenesis [21]. Angiogenic factor released by neighboring cells such as hypoxic hepatocytes (VEGF production via HIF1α) and activated HSC secreting VEGF or angiopoietin further activate SEC and promote SEC matrix invasion [20–23]. Additionally, angiogenesis is known to be induced via the LPS-TLR-4 pathway [12]. The concept of parallel development of fibrogenesis and angiogenesis has further been supported by the beneficial effects of tyrosine-kinase inhibitors such as sorafenib or sunitinib on hepatic fibrogenesis [24, 25].
Hepatic stellate cells
Most of the changes in SEC phenotype occur in crosstalk with neighboring cells, of which HSC are the most important [21]. HSC are pericyte-like cells in the space of Dissé (located between SEC and hepatocytes), and they are usually maintained in their quiescent state as fat-storing cells [26]. However, as described for SEC, HSC can undergo dramatic phenotype transformation in the context of liver injury: a decrease in NO production by SEC, which normally induces HSC quiescence, leads to HSC activation characterized by enhanced contractility, increased migratory capacity, deposition of extracellular matrix components such as fibronectin and collagen I or III, upregulation of smooth muscle alpha actin (α-SMA), and increased release of autocrine and paracrine factors. Besides this canonical NO pathway for quiescence and activation of HSC, [18, 19] several other soluble factors from surrounding cells (such as hepatocytes, Kupffer-cells, and lymphocytes) play an important role in HSC activation [26]. Interestingly, studies of different extracellular matrices have revealed that matrix stiffness itself is a key determinant of HSC activation, although involved proteins and receptors have not been identified so far [27]. Activated HSC show decreased vitamin A, increased stress (actin) bundles, prominent rough endoplasmic reticulum, and both increased PDGF receptors and α-SMA [1, 28]. The latter seems to be a key step in HSC activation—a rather complex process—which dynamically regulates behavior and function of HSC [1]. Expression of α-SMA directly correlates with activation of myofibroblasts [29]. Among paracrine and autocrine factors released by activated HSC, TGF-β is the most pro-fibrogenic by stimulating production of fibrillar and non-fibrillar matrix components [20, 26, 30]. Interestingly, in a physiological state, TGF-β has a dual role and maintains homeostasis by promoting and inhibiting downstream fibrogenic pathways via SMAD phosphorylation [31]. However, in liver injury, the TGF-β co-receptor neuropilin 1 promotes the myofibroblast phenotype by inducing distinctive downstream SMAD activation [31]. Another canonical pathway in HSC activation, which promotes motility, migration and recruitment, is PDGF ligand binding to its receptors [32–35]. Again, neuropilin 1 has been shown to be an important downstream regulator of PDGF signaling in activated HSC [32]. In summary, activated HSC with their myofibroblast phenotype are the most pro-fibrogenic cells in the liver [20].
Paracrine function and crosstalk between sinusoidal endothelial cells and hepatic stellate cells
Besides the canonical pathway of constitutive expression of NO in SEC and its role in the quiescence of HSC [1, 19], numerous paracrine and autocrine factors and SEC/HSC crosstalk mechanisms have been identified and discussed in other reviews [1, 9, 20, 21]. Therefore, the following examples of intercellular communication are used illustratively and do not claim to provide a complete overview.
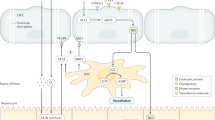
As mentioned before, NO is a key player in the hepatic sinusoid with a crucial role in maintenance of physiological SEC and HSC phenotypes. NO is synthesized by a constitutively active NO synthase and released from SEC in response to diverse external stimuli including mechanical forces such as shear stress [8, 36]. Given the paracrine effect of NO on smooth muscle cells and HSC and its autocrine effect on SEC, shear stress leads to a decrease in intrahepatic resistance as a response to increased blood flow, and maintains SEC phenotype [8]. By release of NO from SEC, the latter not only prevent HSC activation, they also promote reversion to quiescence [19]. Thus, reduced NO not only increases intrahepatic resistance, it also plays an important role in the early steps of liver fibrosis [2]. Interdependent with NO, VEGF is a key regulator of SEC phenotype maintenance [9] as it leads to formation and maintenance of fenestrae [1, 37–40]. Interestingly, VEGF, which is released from hepatocytes and HSC, can act on SEC via both an NO-independent and NO-dependent pathway [10, 41]. Of note, only the latter is critical for early steps of liver fibrosis, as reduced NO leads to capillarization and HSC activation in cirrhosis, [9] while VEGF is even upregulated in diseased liver, which can be explained by hypoxic hepatocytes (via HIF1α) and activated HSC secreting VEGF [22, 42]. Taken together, the VEGF-stimulated NO-dependent pathway is an illustrative example on how hepatocytes, SEC, and HSC communicate and how interruption of this pathway can lead to fibrogenesis (for details see Fig. 1). Besides NO signaling, PDGF and TGF-β, two profibrotic growth factors, are key players in SEC-HSC crosstalk and both promote HSC migration and recruitment [21]. Interestingly, these two pathways seem to be interdependent, as PDGFR-α has been recently shown to be an important co-receptor promoting TGF-β signaling in HSC via transcriptional and posttranscriptional regulation of TGF-β receptors [43]. In addition, signals of TGF-β and PDGF seem to converge at the level of the c-abl kinase further supporting a significant crosstalk between these two factors [44]. PDGF and TGF-β are released by SEC, Kupffer cells and HSC, therefore acting both via autocrine and paracrine mechanisms [26]. While PDGF is the most potent proliferative stimulus toward HSC, TGF-β contributes to HSC-based collagen deposition and stimulates production of matrix proteins [45]. PDGF released by capillarized SEC can act via its PDGFR-β receptor and promote an angiogenic phenotype of HSC, which further facilitates angiogenesis [46]. However, PDGF can also stimulate production of matrix proteins (such as Fibronectin) in HSC [47]. Of note, these proteins do not seem to be cell products only; they themselves regulate cell migration and angiogenesis. Fibronectin may lead to AKT activation and subsequent SEC chemotaxis by transactivation of FGFR1 independent from FGF-ligand [48]. Besides these endogenous factors involved in SEC-HSC crosstalk, therefore promoting fibrogenesis in diseased liver, exogenous factors (among which bacterial LPS is one of the best studied) seem to play a crucial role, too. TLR-4, the corresponding receptor, is expressed on every major liver cell type including HSC and SEC. LPS-TLR-4 signaling leads to TGF-β pathway activation and regulates the production of fibronectin in HSC as well as promotes fibrosis-associated angiogenesis by increasing SEC matrix invasion [12, 13]. While all those crosstalks between SEC and HSC are key in early stages of liver fibrogenesis, SEC seem to be master regulators in the process of liver regeneration, too [49]. SEC-derived angiopoetin-2 (Ang-2) has been shown to regulate liver regeneration by its downregulation in the early phase, leading to reduction of SEC-derived TGF-β and therefore promoting hepatocyte proliferation, and by recovery of Ang-2 levels during a later phase, leading to increased VEGF-2 signaling and therefore promoting angiogenesis [50].

NO pathway in HSC-SEC crosstalk. NO is produced and released from SEC in response to external stimuli such as shear stress (autoregulation) and in response to other paracrine factors such as VEGF. Released NO maintains the SEC phenotype, prevents HSC activation, and promotes reversion to quiescence. Therefore, reduction in NO is a key step in early fibrosis
New pathophysiological aspects in hepatic injury, inflammation, and fibrosis
While inflammation leading to liver fibrosis has been studied in detail, processes other than the known canonical inflammatory pathways may contribute to hepatic fibrogenesis, such as intrasinusoidal thrombosis in the pathogenesis of congestive hepatopathy. Furthermore, recent studies have revealed new pathways of intercellular communication beyond the concept of chemokines and growth factors such as the cell-derived extracellular vesicles, which may additionally link injury to inflammation in alcoholic hepatitis.
Role of hepatic sinusoids in the pathogenesis of congestive hepatopathy
Chronic hepatic congestion known as congestive hepatopathy leads to hepatic fibrosis and mainly occurs in the setting of right-sided heart failure [51]. Other possible causes for impaired hepatic venous outflow are Budd–Chiari syndrome or long-term survival after successful Fontan surgery [52]. In the latter, decreased cardiac index together with increased right atrial pressure (termed central venous hypertension), which results from direct attachment of the right atrium to the pulmonary artery, leads to liver damage and fibrosis [51]. Of note, one-third of those patients show hepatomegaly and abnormal liver function tests [53]. After 11.5 years, more than one-quarter present with liver cirrhosis [54]. However, most patients with chronic hepatic congestion remain asymptomatic and develop impaired liver function late in the disease course [51]. Laboratory findings are mainly elevated parameters of cholestasis [55, 56]. Although congestive hepatopathy has been recognized for years and has been steadily increasing due to high prevalence of chronic heart failure and improved survival of patients with complex congenital heart defects after surgery, little is known about its pathogenesis. Hypotheses were generated from retrospective ex-vivo human liver specimens, which showed variable distribution of parenchymal fibrosis correlating with fibrous obliteration of hepatic and portal veins [57, 58]. The generally accepted concept of parenchymal extinction considered congestive fibrosis to be a response to hypoxia, pressure, and hepatocellular necrosis [59]. This concept was broadened by intrahepatic venous thrombosis in recent years. Degree of thrombosis was thought to trigger degree of necrosis and consecutive fibrogenesis [58]. Of particular interest, fibrosis in congestive hepatopathy did not seem to result from inflammation, as no inflammatory infiltrates have been found in liver specimens from patients having undergone the Fontan procedure [60]. So far, these concepts have not been tested experimentally due to the non-existence of an adequate animal model. Nonetheless, evidence for the role of thrombosis in liver fibrogenesis has been increasing in recent years. While possession of Factor V Leiden mutations has been shown to be a risk factor in rapid fibrosis progression in HCV [61], low-molecular weight heparin and warfarin prevented hepatic fibrogenesis caused by carbon tetrachloride in the rat [62]. Furthermore, the influence of Factor V Leiden polymorphism on fibrosis progression has been confirmed experimentally [63].
Simonetto et al. now have developed a murine model where they surgically generated hepatic venous outflow obstruction by partially ligating the suprahepatic abdominal inferior vena cava (pIVCL) in order to reduce its diameter by 70 % [64]. Histological changes with centrilobular necrosis, vascular extravasation, and sinusoidal dilatation as well as immunostaining for α-SMA and collagen (with a centrilobular and perisinusoidal distribution) were consistent with those in human hepatic congestion. Moreover, there were no signs of inflammation involved in congestive fibrogenesis, while significant inflammation in regard to histology and expression of proinflammatory cytokines was observed in murine cirrhotic models with bile duct ligation. With this model, Simonetto et al. could show that both fibrin and mechanical vascular strain stimulated fibronectin fibril assembly by HSC and confirmed the role of thrombin in HSC activation. Of note, a 6-week course of oral warfarin did not only decrease spleen and liver mass, it also reduced intrahepatic fibrin, α-SMA (as a marker of HSC activation), and finally fibrosis, further supporting the role of intrahepatic thrombosis in fibrogenesis [64]. These findings broaden the concept of hepatic sinusoids in early stages of liver fibrosis. Furthermore, development of this murine model for congestive hepatopathy enables experimental investigations primarily focusing on the role of intrasinusoidal thrombosis and dilatation as well as anticoagulation with traditional and newer oral anticoagulants as a potential therapeutic option in liver fibrogenesis.
Exosome signaling as a new form of SEC/HSC crosstalk
Exosomes and their role for physiological secretion of proteins such as the elimination of transferrin receptors by reticulocytes have been identified more than two decades ago [65, 66]. However, in recent years exosomes have been increasingly recognized as a new way of intercellular communication by promoting protein and lipid exchange between exosome producing cells and target cells [67]. Exosomes are extracellular membrane-enclosed vesicles, produced in multivesicular bodies (MVB) and released into the extracellular space or into biological fluids by fusion of MVB with the plasma membrane [68]. They are characterized by the following criteria: diameter of 40–100 nm, density of 1.13–1.19 g/mL, cup- or deflated football-shaped morphology, and a sediment at 100,000 ×g [67]. They contain common marker proteins such as tetraspanins (e.g., CD 9, CD 10, or CD 26), endosome-associated proteins (Alix, TSG101), cytoplasmic heat shock proteins, and—most important for cell–cell interaction—cell-type specific proteins and nucleic acids [69, 70]. More than 40,000 proteins, 7500 RNA, and 1100 lipid molecules have been identified from more than 286 exosomal studies [71]. Exosome signaling seems to be promoted by interaction via unknown receptors on target cells inducing downstream signaling or via direct fusion of the exosomal membrane with the target cell membrane delivering their content into the cell [65, 69, 70, 72]. Different cells in different organs and tissues have been identified as exosome producing or target cells [73]. In the liver, those are liver epithelia (hepatocytes, cholangiocytes), natural killer T cells, HSC, and SEC [68]. Of interest, liver cells have been shown to be targets even for exosomes originating from other organs such as the intestine [74]. The current knowledge about the role of exosomes in liver diseases has been discussed previously by Masyuk et al. [68], who reviewed the involvement of exosomes in HCC, HCV, and liver inflammation, and their potential role as early diagnostic and prognostic markers from the urine or blood. Herein, we highlight the role of exosome signaling in HSC-SEC crosstalk. Hedgehog ligands are known to activate downstream pathways in endothelial cells through exosomes during embryogenesis [75]. In a recent study, Witek et al. [76] showed that those hedgehog ligands play an important role in HSC-SEC crosstalk and angiogenesis via exosome-enriched microparticles. PDGF-treated HSC and cholangiocytes release exosomes with hedgehog ligands, which induce hedgehog-dependent changes in SEC with upregulation of several genes leading to an angiogenic phenotype. Of note, bile duct ligation in mice increased the release of such exosomes, too, supporting further evidence for a key role of extracellular vesicles in liver fibrogenesis [76]. While HSC communicate with SEC via exosomes, the same seems to be true vice versa. Wang et al. [77] published a study showing that SEC-derived exosomes containing SK1 regulate HSC signaling and migration through Fibronectin-integrin-dependent exosome adherence and dynamin-2-dependent exosome internalization. In more detail, SK1 has been shown to be an SEC-derived exosome protein activated by FGF-2, which has been known to be important in the release of exosomes in liver fibrosis. Exosomes overexpressing SK1 contained more S1P, an HSC chemotactic factor, and led to HSC migration via AKT activation in the target cell. Adhesion and endocytosis have been shown to be mediated through fibronectin (on exosomes) –integrin (on HSC) interaction and dynamin-2, respectively [77]. Thereby, Wang et al. not only highlighted the potential role of exosomes in HSC-SEC crosstalk and liver fibrogenesis, they also added important knowledge about how these vesicles may interact with their target cells. In conclusion, exosomes, which have been studied for years, are considered a new means of SEC-HSC crosstalk in the pathophysiology of liver fibrosis and therefore present potential targets for future therapies (Fig. 2).

Extracellular vesicle signaling. Different ways of EV signaling have been identified. Exosomes originate from multivesicular bodies and are released at the plasma membrane. Microvesicles are larger and shed directly from the plasma membrane
From injury to inflammation in alcoholic hepatitis
Alcoholic hepatitis (AH) as a severe form of alcoholic liver disease (ALD) with high mortality due to liver or renal failure, bacterial infection, or portal hypertension can occur at any stage of ALD [78, 79]. Although ALD and AH significantly contribute to liver-related morbidity and mortality, pathomechanisms still remain elusive [80]. Nonetheless, several concepts for ethanol-induced liver damage exist: while ethanol has a direct effect on hepatocytes resulting in apoptosis through generation of acetaldehyde (via ADH) and ROS accumulation (via increased metabolism by CYP2E1), [81, 82] it can also act through indirect mechanisms. Excessive alcohol consumption disrupts the intestinal epithelial barrier and leads to an alteration of the gut flora. Increased gut permeability and dysbiosis results in higher levels of LPS binding to TLR-4 receptors on liver cells (such as HSC, SEC, and hepatocytes) which then induces liver injury and inflammation as described above [83, 84]. Although neutrophil infiltration [85] is the predominant histological feature in AH and results from hepatocyte degeneration, there is increasing evidence for the role of liver-resident macrophages called Kupffer cells, which are activated in AH by both hepatocyte injury and the LPS signaling pathway [83, 86, 87]. Of note, Kupffer cell inflammation response parallels the extent of injury and fibrosis [88]. Herein, we review on two novel pathomechanisms involved in intercellular communication leading to AH: microvesicles and high-mortality-group box 1 (HMGB1). While exosomes originate from MVB and are released at the plasma membrane by fusion of exosome and MVB membranes, microvesicles (MV) shed directly from the plasma membrane [65]. They have a size of 0.1–1 µm, contain cellular compounds such as lipids, proteins, RNA, and microRNA, and act as a vector between MV-producing cells and their target cells [89]. MV are increasingly recognized as a means of intercellular communication. In addition, numerous studies have uncovered their role in liver diseases with increased MV levels, MV formation, and decreased MV clearance [89]. While MV seem to contribute to the communication between tumor cells and between tumor cells and their environment, they seem to have a dual function in fibrosis: on one side they promote fibrinolysis by enhancing expression of MMP, [90] on the other side they promote fibrosis by increased angiogenesis [91]. In a very recent study, MV have now been identified as an important pathomechanism in early ethanol-induced damage and as a novel form of crosstalk between hepatocytes and liver-resident macrophages. Verma et al. [92] could show that ethanol results in the release of MV from hepatocytes via activation of the pro-apoptotic protein caspase 3. These MV have further been shown to stimulate macrophage activation by CD40 ligand—a member of the TNF family—and to induce inflammatory cytokines leading to liver inflammation [92].
Activation of TLR-4 downstream signaling leading to sterile inflammation is promoted by endogenous cell products such as the so-called danger-associated molecular patterns (DAMPs) [93]. HGMB1 is a protein released from the nucleus of injured cells [94], acting as an activator of TLR-4 (as LPS or DAMPs) with a role in ischemic liver injury and viral hepatitis [95, 96]. In a recent study, Seo et al. was able to show that HMGB1 translocates from the nucleus of ethanol-injured hepatocytes, resulting in higher intracellular levels and ultimately leading to increased migration of HSC and SEC [97]. Thereby, they directly link the first hit of ethanol-induced liver injury to the two key players in the early steps of liver fibrogenesis. Of note, recruitment of both HSC and SEC could be blocked by the HMGB1-antibody and siRNA, respectively, promoting new ideas for future therapies.
Summary
Recent studies have described alternative pathways of intercellular communication in liver diseases such as cell-derived extracellular vesicles, which deliver cell compounds to their target cells. Moreover, such extracellular vesicles may link injury to inflammation in AH. While inflammation leading to liver fibrosis has been studied in detail, elements other than the known canonical inflammatory pathways may contribute to hepatic fibrogenesis. In congestive hepatopathy, sinusoidal dilatation and fibrosis have been shown to be mediated by non-inflammatory mechanisms and associated with sinusoidal thrombi. A recently developed murine model further enables experimental studies of this disease entity. Increasing knowledge about these alternative disease pathways in liver injury, inflammation, and fibrosis may reveal possible target molecules for future therapies.
References
Iwakiri Y, Shah V, Rockey DC. Vascular pathobiology in chronic liver disease and cirrhosis—current status and future directions. J Hepatol. 2014;61(4):912–24.
Bosch J, Groszmann RJ, Shah VH. Evolution in the understanding of the pathophysiological basis of portal hypertension: How changes in paradigm are leading to successful new treatments. J Hepatol. 2015;62(1 Suppl):S121–30.
Arii S, Imamura M. Physiological role of sinusoidal endothelial cells and Kupffer cells and their implication in the pathogenesis of liver injury. J Hepatobiliary Pancreat Surg. 2000;7(1):40–8.
Braet F, De Zanger R, Baekeland M, Crabbé E, Van Der Smissen P, Wisse E. Structure and dynamics of the fenestrae-associated cytoskeleton of rat liver sinusoidal endothelial cells. Hepatology. 1995;21(1):180–9.
Hilscher MB, Huebert RC, Shah VH. Hepatic sinusoidal endothelial cells. In: Dufour JD, Clavien PA, editors. Signaling pathways in liver diseases. New York: Wiley; 2015. p. 73–81.
Wisse E. An electron microscopic study of the fenestrated endothelial lining of rat liver sinusoids. J Ultrastruct Res. 1970;31(1):125–50.
Burt AD, Le Bail B, Balabaud C. Morphological investigation of sinusoidal cells. Semin Liver Dis. 1993;13:21–38.
Shah V, Haddad FG, Garcia-Cardena G, Frangos JA, Mennone A, Groszmann RJ, et al. Liver sinusoidal endothelial cells are responsible for nitric oxide modulation of resistance in the hepatic sinusoids. J Clin Invest. 1997;100(11):2923–30.
DeLeve LD. Liver sinusoidal endothelial cells in hepatic fibrosis. Hepatology. 2015;61(5):1740–6.
DeLeve LD, Wang X, Hu L, McCuskey MK, McCuskey RS. Rat liver sinusoidal endothelial cell phenotype is maintained by paracrine and autocrine regulation. Am J Physiol Gastrointest Liver Physiol. 2004;287(4):G757–63.
Dauphinee SM, Karsan A. Lipopolysaccharide signaling in endothelial cells. Lab Invest. 2006;86(1):9–22.
Jagavelu K, Routray C, Shergill U, O’Hara SP, Faubion W, Shah VH. Endothelial cell toll-like receptor 4 regulates fibrosis-associated angiogenesis in the liver. Hepatology. 2010;52(2):590–601.
Zhu Q, Zou L, Jagavelu K, Simonetto DA, Huebert RC, Jiang ZD, et al. Intestinal decontamination inhibits TLR4 dependent fibronectin-mediated cross-talk between stellate cells and endothelial cells in liver fibrosis in mice. J Hepatol. 2012;56(4):893–9.
Iwakiri Y, Grisham M, Shah V. Vascular biology and pathobiology of the liver: Report of a single-topic symposium. Hepatology. 2008;47(5):1754–63.
Xu B, Broome U, Uzunel M, Nava S, Ge X, Kumagai-Braesch M, et al. Capillarization of hepatic sinusoid by liver endothelial cell-reactive autoantibodies in patients with cirrhosis and chronic hepatitis. Am J Pathol. 2003;163(4):1275–89.
Schaffner F, Poper H. Capillarization of hepatic sinusoids in man. Gastroenterology. 1963;44:239–42.
Rockey DC, Chung JJ. Reduced nitric oxide production by endothelial cells in cirrhotic rat liver: Endothelial dysfunction in portal hypertension. Gastroenterology. 1998;114(2):344–51.
Langer DA, Das A, Semela D, Kang-Decker N, Hendrickson H, Bronk SF, et al. Nitric oxide promotes caspase-independent hepatic stellate cell apoptosis through the generation of reactive oxygen species. Hepatology. 2008;47(6):1983–93.
Deleve LD, Wang X, Guo Y. Sinusoidal endothelial cells prevent rat stellate cell activation and promote reversion to quiescence. Hepatology. 2008;48(3):920–30.
Thabut D, Shah V. Intrahepatic angiogenesis and sinusoidal remodeling in chronic liver disease: New targets for the treatment of portal hypertension? J Hepatol. 2010;53(5):976–80.
Lee JS, Semela D, Iredale J, Shah VH. Sinusoidal remodeling and angiogenesis: A new function for the liver-specific pericyte? Hepatology. 2007;45(3):817–25.
Corpechot C, Barbu V, Wendum D, Kinnman N, Rey C, Poupon R, et al. Hypoxia-induced VEGF and collagen I expressions are associated with angiogenesis and fibrogenesis in experimental cirrhosis. Hepatology. 2002;35(5):1010–21.
Lee S, Chen TT, Barber CL, Jordan MC, Murdock J, Desai S, et al. Autocrine VEGF signaling is required for vascular homeostasis. Cell. 2007;130(4):691–703.
Tugues S, Fernandez-Varo G, Muñoz-Luque J, Ros J, Arroyo V, Rodés J, et al. Antiangiogenic treatment with sunitinib ameliorates inflammatory infiltrate, fibrosis, and portal pressure in cirrhotic rats. Hepatology. 2007;46(6):1919–26.
Hennenberg M, Trebicka J, Stark C, Kohistani AZ, Heller J, Sauerbruch T. Sorafenib targets dysregulated Rho kinase expression and portal hypertension in rats with secondary biliary cirrhosis. Br J Pharmacol. 2009;157(2):258–70.
Caligiuri A, Marra F. Stellate cells. In: Dufour JF, Clavien PA, editors. Signaling pathways in liver diseases. New York: Wiley; 2015. p. 34–52.
Wells RG. The role of matrix stiffness in hepatic stellate cell activation and liver fibrosis. J Clin Gastroenterol. 2005;39(4 Suppl 2):S158–61.
Kent G, Gay S, Inouye T, Bahu R, Minick OT, Popper H. Vitamin A-containing lipocytes and formation of type III collagen in liver injury. Proc Natl Acad Sci USA. 1976;73(10):3719–22.
Nakatani T, Honda E, Hayakawa S, Sato M, Satoh K, Kudo M, et al. Effects of decorin on the expression of alpha-smooth muscle actin in a human myofibroblast cell line. Mol Cell Biochem. 2008;308(1–2):201–7.
Friedman SL. Cytokines and fibrogenesis. Semin Liver Dis. 1999;19(2):129–40.
Cao Y, Szabolcs A, Dutta SK, Yaqoob U, Jagavelu K, Wang L, et al. Neuropilin-1 mediates divergent R-Smad signaling and the myofibroblast phenotype. J Biol Chem. 2010;285(41):31840–8.
Cao S, Yaqoob U, Das A, Shergill U, Jagavelu K, Huebert RC, et al. Neuropilin-1 promotes cirrhosis of the rodent and human liver by enhancing PDGF/TGF-beta signaling in hepatic stellate cells. J Clin Invest. 2010;120(7):2379–94.
Lai CC, Henningson C, DiMaio D. Bovine papillomavirus E5 protein induces oligomerization and trans-phosphorylation of the platelet-derived growth factor beta receptor. Proc Natl Acad Sci USA. 1998;95(26):15241–6.
Westermark B, Siegbahn A, Heldin CH, Claesson-Welsh L. B-type receptor for platelet-derived growth factor mediates a chemotactic response by means of ligand-induced activation of the receptor protein-tyrosine kinase. Proc Natl Acad Sci USA. 1990;87(1):128–32.
Claesson-Welsh L, Eriksson A, Morén A, Severinsson L, Ek B, Ostman A, et al. cDNA cloning and expression of a human platelet-derived growth factor (PDGF) receptor specific for B-chain-containing PDGF molecules. Mol Cell Biol. 1988;8(8):3476–86.
Michel T, Feron O. Nitric oxide synthases: Which, where, how, and why? J Clin Invest. 1997;100(9):2146–52.
Shah V, Toruner M, Haddad F, Cadelina G, Papapetropoulos A, Choo K, et al. Impaired endothelial nitric oxide synthase activity associated with enhanced caveolin binding in experimental cirrhosis in the rat. Gastroenterology. 1999;117(5):1222–8.
Funyu J, Mochida S, Inao M, Matsui A, Fujiwara K. VEGF can act as vascular permeability factor in the hepatic sinusoids through upregulation of porosity of endothelial cells. Biochem Biophys Res Commun. 2001;280(2):481–5.
Yokomori H, Oda M, Yoshimura K, Nagai T, Ogi M, Nomura M, et al. Vascular endothelial growth factor increases fenestral permeability in hepatic sinusoidal endothelial cells. Liver Int. 2003;23(6):467–75.
May D, Djonov V, Zamir G, Bala M, Safadi R, Sklair-Levy M, et al. A transgenic model for conditional induction and rescue of portal hypertension reveals a role of VEGF-mediated regulation of sinusoidal fenestrations. PLoS One. 2011;6(7):e21478.
Xie G, Wang X, Wang L, Atkinson RD, Kanel GC, Gaarde WA, et al. Role of differentiation of liver sinusoidal endothelial cells in progression and regression of hepatic fibrosis in rats. Gastroenterology. 2012;142(4):918–27 (e6).
Kwon SH, Jeong SW, Jang JY, Lee JE, Lee SH, Kim SG, et al. Cyclooxygenase-2 and vascular endothelial growth factor in chronic hepatitis, cirrhosis and hepatocellular carcinoma. Clin Mol Hepatol. 2012;18(3):287–94.
Liu C, Li J, Xiang X, Guo L, Tu K, Liu Q, et al. PDGF receptor-α promotes TGF-β signaling in hepatic stellate cells via transcriptional and posttranscriptional regulation of TGF-β receptors. Am J Physiol Gastrointest Liver Physiol. 2014;307(7):G749–59.
Daniels CE, Wilkes MC, Edens M, Kottom TJ, Murphy SJ, Limper AH, et al. Imatinib mesylate inhibits the profibrogenic activity of TGF-beta and prevents bleomycin-mediated lung fibrosis. J Clin Invest. 2004;114(9):1308–16.
Friedman SL. Liver fibrosis—from bench to bedside. J Hepatol. 2003;38(Suppl 1):S38–53.
Semela D, Das A, Langer D, Kang N, Leof E, Shah V. Platelet-derived growth factor signaling through ephrin-b2 regulates hepatic vascular structure and function. Gastroenterology. 2008;135(2):671–9.
Thabut D, Routray C, Lomberk G, Shergill U, Glaser K, Huebert R, et al. Complementary vascular and matrix regulatory pathways underlie the beneficial mechanism of action of sorafenib in liver fibrosis. Hepatology. 2011;54(2):573–85.
Zou L, Cao S, Kang N, Huebert RC, Shah VH. Fibronectin induces endothelial cell migration through β1 integrin and Src-dependent phosphorylation of fibroblast growth factor receptor-1 at tyrosines 653/654 and 766. J Biol Chem. 2012;287(10):7190–202.
Wang R, Huebert RC, Shah VH. Sinusoidal endothelial cells coordinate liver regeneration and angiogenesis via angiopoietin-2: An ode to prometheus. Gastroenterology. 2014;147(2):533–4.
Hu J, Srivastava K, Wieland M, Runge A, Mogler C, Besemfelder E, et al. Endothelial cell-derived angiopoietin-2 controls liver regeneration as a spatiotemporal rheostat. Science. 2014;343(6169):416–9.
Møller S, Bernardi M. Interactions of the heart and the liver. Eur Heart J. 2013;34(36):2804–11.
Asrani SK, Asrani NS, Freese DK, Phillips SD, Warnes CA, Heimbach J, et al. Congenital heart disease and the liver. Hepatology. 2012;56(3):1160–9.
Wu FM, Ukomadu C, Odze RD, Valente AM, Mayer JE, Earing MG. Liver disease in the patient with Fontan circulation. Congenit Heart Dis. 2011;6(3):190–201.
Baek JS, Bae EJ, Ko JS, Kim GB, Kwon BS, Lee SY, et al. Late hepatic complications after Fontan operation; non-invasive markers of hepatic fibrosis and risk factors. Heart. 2010;96(21):1750–5.
Poelzl G, Ess M, Mussner-Seeber C, Pachinger O, Frick M, Ulmer H. Liver dysfunction in chronic heart failure: Prevalence, characteristics and prognostic significance. Eur J Clin Invest. 2012;42(2):153–63.
Myers RP, Lee SS. Cirrhotic cardiomyopathy and liver transplantation. Liver Transpl. 2000;6(4 Suppl 1):S44–52.
Wanless IR, Wong F, Blendis LM, Greig P, Heathcote EJ, Levy G. Hepatic and portal vein thrombosis in cirrhosis: Possible role in development of parenchymal extinction and portal hypertension. Hepatology. 1995;21(5):1238–47.
Wanless IR, Liu JJ, Butany J. Role of thrombosis in the pathogenesis of congestive hepatic fibrosis (cardiac cirrhosis). Hepatology. 1995;21(5):1232–7.
Moschcowitz E. The morphology and pathogenesis of cardiac fibrosis of the liver. Ann Intern Med. 1952;36(4):933–55.
Kendall TJ, Stedman B, Hacking N, Haw M, Vettukattill JJ, Salmon AP, et al. Hepatic fibrosis and cirrhosis in the Fontan circulation: A detailed morphological study. J Clin Pathol. 2008;61(4):504–8.
Wright M, Goldin R, Hellier S, Knapp S, Frodsham A, Hennig B, et al. Factor V Leiden polymorphism and the rate of fibrosis development in chronic hepatitis C virus infection. Gut. 2003;52(8):1206–10.
Abe W, Ikejima K, Lang T, Okumura K, Enomoto N, Kitamura T, et al. Low molecular weight heparin prevents hepatic fibrogenesis caused by carbon tetrachloride in the rat. J Hepatol. 2007;46(2):286–94.
Anstee QM, Goldin RD, Wright M, Martinelli A, Cox R, Thursz MR. Coagulation status modulates murine hepatic fibrogenesis: Implications for the development of novel therapies. J Thromb Haemost. 2008;6(8):1336–43.
Simonetto DA, Yang HY, Yin M, de Assuncao TM, Kwon JH, Hilscher M, et al. Chronic passive venous congestion drives hepatic fibrogenesis via sinusoidal thrombosis and mechanical forces. Hepatology. 2015;61(2):648–59.
Raposo G, Stoorvogel W. Extracellular vesicles: Exosomes, microvesicles, and friends. J Cell Biol. 2013;200(4):373–83.
Pan BT, Teng K, Wu C, Adam M, Johnstone RM. Electron microscopic evidence for externalization of the transferrin receptor in vesicular form in sheep reticulocytes. J Cell Biol. 1985;101(3):942–8.
Théry C, Amigorena S, Raposo G, Clayton A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr Protoc Cell Biol. 2006;30(3.22):3.22.1–29.
Masyuk AI, Masyuk TV, Larusso NF. Exosomes in the pathogenesis, diagnostics and therapeutics of liver diseases. J Hepatol. 2013;59(3):621–5.
Vlassov AV, Magdaleno S, Setterquist R, Conrad R. Exosomes: current knowledge of their composition, biological functions, and diagnostic and therapeutic potentials. Biochim Biophys Acta. 2012;1820(7):940–8.
Bang C, Thum T. Exosomes: new players in cell–cell communication. Int J Biochem Cell Biol. 2012;44(11):2060–4.
Keerthikumar S, Chisanga D, Ariyaratne D, Al Saffar H, Anand S, Zhao K, et al. ExoCarta: a web-based compendium of 39 Exosomal Cargo. J Mol Biol. 2015. doi: 10.1016/j.jmb.2015.09.019 (Epub ahead of print).
Sun D, Zhuang X, Zhang S, Deng ZB, Grizzle W, Miller D, et al. Exosomes are endogenous nanoparticles that can deliver biological information between cells. Adv Drug Deliv Rev. 2013;65(3):342–7.
Arendt BK, Walters DK, Wu X, Tschumper RC, Jelinek DF. Multiple myeloma dell-derived microvesicles are enriched in CD147 expression and enhance tumor cell proliferation. Oncotarget. 2014;5(14):5686–99.
Deng ZB, Zhuang X, Ju S, Xiang X, Mu J, Wang Q, et al. Intestinal mucus-derived nanoparticle-mediated activation of Wnt/β-catenin signaling plays a role in induction of liver natural killer T cell anergy in mice. Hepatology. 2013;57(3):1250–61.
Vokes SA, Yatskievych TA, Heimark RL, McMahon J, McMahon AP, Antin PB, et al. Hedgehog signaling is essential for endothelial tube formation during vasculogenesis. Development. 2004;131(17):4371–80.
Witek RP, Yang L, Liu R, Jung Y, Omenetti A, Syn WK, et al. Liver cell-derived microparticles activate hedgehog signaling and alter gene expression in hepatic endothelial cells. Gastroenterology. 2009;136(1):320–30 (e2).
Wang R, Ding Q, Yaqoob U, de Assuncao TM, Verma VK, Hirsova P, et al. Exosome adherence and internalization by hepatic stellate cells triggers sphingosine 1-phosphate dependent migration. J Biol Chem. 2015;290(52):30684–96.
Singal AK, Kamath PS, Gores GJ, Shah VH. Alcoholic hepatitis: Current challenges and future directions. Clin Gastroenterol Hepatol. 2014;12(4):555–64 (quiz e31–2).
Lucey MR, Mathurin P, Morgan TR. Alcoholic hepatitis. N Engl J Med. 2009;360(26):2758–69.
Gao B, Shah VH. Combination therapy: New hope for alcoholic hepatitis? Clin Res Hepatol Gastroenterol. 2015;39(Suppl 1):S7–11.
Beier JI, McClain CJ. Mechanisms and cell signaling in alcoholic liver disease. Biol Chem. 2010;391(11):1249–64.
Wang K. Molecular mechanisms of hepatic apoptosis. Cell Death Dis. 2014;5:e996.
Szabo G. Gut–liver axis in alcoholic liver disease. Gastroenterology. 2015;148(1):30–6.
Schnabl B, Brenner DA. Interactions between the intestinal microbiome and liver diseases. Gastroenterology. 2014;146(6):1513–24.
Altamirano J, Miquel R, Katoonizadeh A, Abraldes JG, Duarte-Rojo A, Louvet A, et al. A histologic scoring system for prognosis of patients with alcoholic hepatitis. Gastroenterology. 2014;146(5):1231–9 (e1–6).
Wang HJ, Gao B, Zakhari S, Nagy LE. Inflammation in alcoholic liver disease. Annu Rev Nutr. 2012;32:343–68.
Wheeler MD, Kono H, Yin M, Nakagami M, Uesugi T, Arteel GE, et al. The role of Kupffer cell oxidant production in early ethanol-induced liver disease. Free Radic Biol Med. 2001;31(12):1544–9.
Hernandez-Gea V, Friedman SL. Pathogenesis of liver fibrosis. Annu Rev Pathol. 2011;6:425–56.
Lemoinne S, Thabut D, Housset C, Moreau R, Valla D, Boulanger CM, et al. The emerging roles of microvesicles in liver diseases. Nat Rev Gastroenterol Hepatol. 2014;11(6):350–61.
Kornek M, Popov Y, Libermann TA, Afdhal NH, Schuppan D. Human T cell microparticles circulate in blood of hepatitis patients and induce fibrolytic activation of hepatic stellate cells. Hepatology. 2011;53(1):230–42.
Povero D, Eguchi A, Niesman IR, Andronikou N, de Mollerat du Jeu X, Mulya A, et al. Lipid-induced toxicity stimulates hepatocytes to release angiogenic microparticles that require Vanin-1 for uptake by endothelial cells. Sci Signal. 2013;6(296):ra88.
Verma VK, Li H, Wang R, Hirsova P, Mushref M, Liu Y, et al. Alcohol stimulates macrophage activation through caspase dependent hepatocyte derived release of CD40L containing extracellular vesicles. J Hepatol. 2015. doi: 10.1016/j.jhep.2015.11.020 (Epub ahead of print).
Kawai T, Takeuchi O, Fujita T, Inoue J, Mühlradt PF, Sato S, et al. Lipopolysaccharide stimulates the MyD88-independent pathway and results in activation of IFN-regulatory factor 3 and the expression of a subset of lipopolysaccharide-inducible genes. J Immunol. 2001;167(10):5887–94.
Degryse B, Bonaldi T, Scaffidi P, Müller S, Resnati M, Sanvito F, et al. The high mobility group (HMG) boxes of the nuclear protein HMG1 induce chemotaxis and cytoskeleton reorganization in rat smooth muscle cells. J Cell Biol. 2001;152(6):1197–206.
Li L, Chen L, Hu L, Liu Y, Sun HY, Tang J, et al. Nuclear factor high-mobility group box1 mediating the activation of Toll-like receptor 4 signaling in hepatocytes in the early stage of nonalcoholic fatty liver disease in mice. Hepatology. 2011;54(5):1620–30.
Majumdar M, Ratho R, Chawla Y, Singh MP. High levels of circulating HMGB1 as a biomarker of acute liver failure in patients with viral hepatitis E. Liver Int. 2013;33(9):1341–8.
Seo YS, Kwon JH, Yaqoob U, Yang L, De Assuncao TM, Simonetto DA, et al. HMGB1 recruits hepatic stellate cells and liver endothelial cells to sites of ethanol-induced parenchymal cell injury. Am J Physiol Gastrointest Liver Physiol. 2013;305(11):G838–48.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Greuter, T., Shah, V.H. Hepatic sinusoids in liver injury, inflammation, and fibrosis: new pathophysiological insights. J Gastroenterol 51, 511–519 (2016). https://doi.org/10.1007/s00535-016-1190-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00535-016-1190-4