
Abstract
Smith–Magenis syndrome (SMS), a neurodevelopmental disorder characterized by dysmorphic features, intellectual disability (ID), and sleep disturbances, results from a 17p11.2 microdeletion or a mutation in the RAI1 gene. We performed exome sequencing on 6 patients with SMS-like phenotypes but without chromosomal abnormalities or RAI1 variants. We identified pathogenic de novo variants in two cases, a nonsense variant in IQSEC2 and a missense variant in the SAND domain of DEAF1, and candidate de novo missense variants in an additional two cases. One candidate variant was located in an alpha helix of Necdin (NDN), phased to the paternally inherited allele. NDN is maternally imprinted within the 15q11.2 Prader–Willi Syndrome (PWS) region. This can help clarify NDN’s role in the PWS phenotype. No definitive pathogenic gene variants were detected in the remaining SMS-like cases, but we report our findings for future comparison. This study provides information about the inheritance pattern and recurrence risk for patients with identified variants and demonstrates clinical and genetic overlap of neurodevelopmental disorders. Identification and characterization of ID-related genes that assist in development of common developmental pathways and/or gene-networks, may inform disease mechanism and treatment strategies.



Similar content being viewed by others
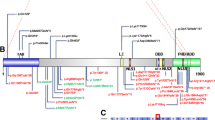
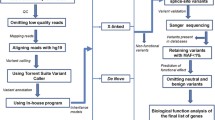

References
Alexander-Bloch AF, McDougle CJ, Ullman Z, Sweetser DA (2016) IQSEC2 and X-linked syndromal intellectual disability. Psychiatr Genet 26:101–108. doi:10.1097/ypg.0000000000000128
Beneduzzi D, Iyer AK, Trarbach EB, Silveira-Neto AP, Silveira LG, Tusset C, Yip K, Mendonca BB, Mellon PL, Latronico AC (2011) Mutational analysis of the necdin gene in patients with congenital isolated hypogonadotropic hypogonadism. Eur J Endocrinol 165:145–150. doi:10.1530/EJE-11-0199
Bentley DR, Balasubramanian S, Swerdlow HP et al (2008) Accurate whole human genome sequencing using reversible terminator chemistry. Nature 456:53–59. doi:10.1038/nature07517
Blasius TL, Cai D, Jih GT, Toret CP, Verhey KJ (2007) Two binding partners cooperate to activate the molecular motor Kinesin-1. J Cell Biol 176:11–17. doi:10.1083/jcb.200605099
Bodian DL, Solomon BD, Khromykh A, Thach DC, Iyer RK, Link K, Baker RL, Baveja R, Vockley JG, Niederhuber JE (2014) Diagnosis of an imprinted-gene syndrome by a novel bioinformatics analysis of whole-genome sequences from a family trio. Mol Genet Genomic Med 2:530–538. doi:10.1002/mgg3.107
Chen L, Jensik P, Walkiewicz M, Alaimo JT, Mullegama SV, Elsea SH (2015) Functional characterization of novel DEAF1 mutations in clinical whole-exome sequencing of intellectual disability patients and its regulation of the RAI1 gene; (Program#387). In: Presented at the 65th Annual Meeting of The American Society of Human Genetics, Baltimore, MD, October 10, 2015
Chen L, Tao Y, Song F, Yuan X, Wang J, Saffen D (2016) Evidence for genetic regulation of mRNA expression of the dosage-sensitive gene retinoic acid induced-1 (RAI1) in human brain. Sci Rep 6:19010. doi:10.1038/srep19010
Clayton-Smith J, O’Sullivan J, Daly S et al (2011) Whole-exome-sequencing identifies mutations in histone acetyltransferase gene KAT6B in individuals with the Say-Barber-Biesecker variant of Ohdo syndrome. Am J Hum Genet 89:675–681. doi:10.1016/j.ajhg.2011.10.008
Contestabile A, Sintoni S (2013) Histone acetylation in neurodevelopment. Curr Pharm Des 19:5043–5050
Deciphering Developmental Disorders Study (2015) Large-scale discovery of novel genetic causes of developmental disorders. Nature 519:223–228. doi:10.1038/nature14135
Driscoll DJ, Miller JL, Schwartz S, Cassidy SB (1993) Prader–Willi Syndrome. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH (eds) Gene Reviews (R). University of Washington, Seattle, WA
Guex N, Peitsch MC, Schwede T (2009) Automated comparative protein structure modeling with SWISS-MODEL and Swiss-PdbViewer: a historical perspective. Electrophoresis 30(Suppl 1):S162–173. doi:10.1002/elps.200900140
Iossifov I, O’Roak BJ, Sanders SJ et al (2014) The contribution of de novo coding mutations to autism spectrum disorder. Nature 515:216–221. doi:10.1038/nature13908
Jay P, Rougeulle C, Massacrier A et al (1997) The human necdin gene, NDN, is maternally imprinted and located in the Prader–Willi syndrome chromosomal region. Nat Genet 17:357–361. doi:10.1038/ng1197-357
Juyal RC, Figuera LE, Hauge X, Elsea SH, Lupski JR, Greenberg F, Baldini A, Patel PI (1996) Molecular analyses of 17p11.2 deletions in 62 Smith-Magenis syndrome patients. Am J Hum Genet 58:998–1007
Kim MK, Kang YK (1999) Positional preference of proline in alpha-helices. Protein Sci 8:1492–1499. doi:10.1110/ps.8.7.1492
Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J (2014) A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 46:310–315. doi:10.1038/ng.2892
Ku CS, Polychronakos C, Tan EK, Naidoo N, Pawitan Y, Roukos DH, Mort M, Cooper DN (2013) A new paradigm emerges from the study of de novo mutations in the context of neurodevelopmental disease. Mol Psychiatry 18:141–153. doi:10.1038/mp.2012.58
Lek M, Karczewski KJ, Minikel EV et al (2016) Analysis of protein-coding genetic variation in 60,706 humans. Nature 536:285–291. doi:10.1038/nature19057
Lopez AJ, Wood MA (2015) Role of nucleosome remodeling in neurodevelopmental and intellectual disability disorders. Front Behav Neurosci 9:100. doi:10.3389/fnbeh.2015.00100
Loviglio MN, Beck CR, White JJ et al (2016) Identification of a RAI1-associated disease network through integration of exome sequencing, transcriptomics, and 3D genomics. Genome Med 8:105. doi:10.1186/s13073-016-0359-z
McCarthy SE, Gillis J, Kramer M et al (2014) De novo mutations in schizophrenia implicate chromatin remodeling and support a genetic overlap with autism and intellectual disability. Mol Psychiatry 19:652–658. doi:10.1038/mp.2014.29
Miller DT, Adam MP, Aradhya S et al (2010) Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet 86:749–764. doi:10.1016/j.ajhg.2010.04.006
Pavone M, Caldarelli V, Khirani S et al (2015) Sleep disordered breathing in patients with Prader–Willi syndrome: a multicenter study. Pediatr Pulmonol 50:1354–1359. doi:10.1002/ppul.23177
Rauch A, Wieczorek D, Graf E et al (2012) Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. Lancet 380:1674–1682. doi:10.1002/ppul.23177
Richards S, Aziz N, Bale S et al (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med 17:405–424. doi:10.1038/gim.2015.30
Ronemus M, Iossifov I, Levy D, Wigler M (2014) The role of de novo mutations in the genetics of autism spectrum disorders. Nat Rev Genet 15:133–141. doi:10.1038/nrg3585
Schaaf CP, Gonzalez-Garay ML, Xia F et al (2013) Truncating mutations of MAGEL2 cause Prader–Willi phenotypes and autism. Nat Genet 45:1405–1408. doi:10.1038/ng.2776
Shoubridge C, Tarpey PS, Abidi F et al (2010) Mutations in the guanine nucleotide exchange factor gene IQSEC2 cause nonsyndromic intellectual disability. Nat Genet 42:486–488. doi:10.1038/ng.588
Slager RE, Newton TL, Vlangos CN, Finucane B, Elsea SH (2003) Mutations in RAI1 associated with Smith-Magenis syndrome. Nat Genet 33:466–468. doi:10.1038/ng1126
Smith ACM, Boyd KE, Elsea SH, Finucane BM, Haas-Givler B, Gropman A, Laje G, Magenis E, Potocki L. (2012) Smith-Magenis Syndrome Seattle (WA): University of Washington, Seattle; 2001 . Available from: https://www.ncbi.nlm.nih.gov/books/NBK1310/. Accessed 28 Jun 2012
Szklarczyk D, Morris JH, Cook H et al (2016) The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. doi:10.1093/nar/gkw937
Teer JK, Mullikin JC (2010) Exome sequencing: the sweet spot before whole genomes. Hum Mol Genet 19:R145–R151. doi:10.1093/hmg/ddq333
Teer JK, Green ED, Mullikin JC, Biesecker LG (2012) VarSifter: visualizing and analyzing exome-scale sequence variation data on a desktop computer. Bioinformatics 28:599–600. doi:10.1093/bioinformatics/btr711
Tennese AA, Gee CB, Wevrick R (2008) Loss of the Prader–Willi syndrome protein necdin causes defective migration, axonal outgrowth, and survival of embryonic sympathetic neurons. Dev Dyn 237:1935–1943. doi:10.1002/dvdy.21615
Tran Mau-Them F, Willems M, Albrecht B et al (2014) Expanding the phenotype of IQSEC2 mutations: truncating mutations in severe intellectual disability. Eur J Hum Genet 22:289–292. doi:10.1038/ejhg.2013.113
Vilboux T, Ciccone C, Blancato JK, Cox GF, Deshpande C, Introne WJ, Gahl WA, Smith ACM, Huizing M (2011) Molecular analysis of the retinoic acid induced 1 Gene (RAI1) in patients with suspected Smith-Magenis syndrome without the 17p11.2 deletion. PLoS One 6:14. doi:10.1371/journal.pone.0022861
Vissers LE, de Ligt J, Gilissen C et al (2010) A de novo paradigm for mental retardation. Nat Genet 42:1109–1112. doi:10.1038/ng.712
Vulto-van Silfhout AT, Rajamanickam S, Jensik PJ et al (2014) Mutations affecting the SAND domain of DEAF1 cause intellectual disability with severe speech impairment and behavioral problems. Am J Hum Genet 94:649–661. doi:10.1016/j.ajhg.2014.03.013
Waltl S (2014) Intellectual disability: novel mutations in DEAF1 cause speech impairment and behavioral problems. Clin Genet 86:507–508. doi:10.1111/cge.12475
White J, Beck CR, Harel T et al (2016) POGZ truncating alleles cause syndromic intellectual disability. Genome Med 8:3. doi:10.1186/s13073-015-0253-0
Xu B, Ionita-Laza I, Roos JL, Boone B, Woodrick S, Sun Y, Levy S, Gogos JA, Karayiorgou M (2012) De novo gene mutations highlight patterns of genetic and neural complexity in schizophrenia. Nat Genet 44:1365–1369. doi:10.1038/ng.2446
Zanella S, Watrin F, Mebarek S, Marly F, Roussel M, Gire C, Diene G, Tauber M, Muscatelli F, Hilaire G (2008) Necdin plays a role in the serotonergic modulation of the mouse respiratory network: implication for Prader–Willi syndrome. J Neurosci 28:1745–1755. doi:10.1523/JNEUROSCI.4334-1707.2008
Acknowledgements
We thank the SMS-like patients and their families for participating in this study. The authors appreciate the support of Settara C Chandrasekharappa, Ph.D., and the Genomics Core of the National Human Genome Research Institute, NIH. This study was partially supported by an NIH Bench to Bedside award (to ACM Smith) and by the Intramural Research Program of the National Human Genome Research Institute, NIH, Bethesda, Maryland, USA.
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethics standards.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

{kind=link}
Cite this article
Berger, S.I., Ciccone, C., Simon, K.L. et al. Exome analysis of Smith–Magenis-like syndrome cohort identifies de novo likely pathogenic variants. Hum Genet 136, 409–420 (2017). https://doi.org/10.1007/s00439-017-1767-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-017-1767-x