
Abstract
Schwannomatosis is characterized by the predisposition to develop multiple schwannomas and, less commonly, meningiomas. Despite the clinical overlap with neurofibromatosis type 2 (NF2), schwannomatosis is not caused by germline NF2 gene mutations. Instead, germline mutations of either the SMARCB1 or LZTR1 tumour suppressor genes have been identified in 86% of familial and 40% of sporadic schwannomatosis patients. In contrast to patients with rhabdoid tumours, which are due to complete loss-of-function SMARCB1 mutations, individuals with schwannomatosis harbour predominantly hypomorphic SMARCB1 mutations which give rise to the synthesis of mutant proteins with residual function that do not cause rhabdoid tumours. Although biallelic mutations of SMARCB1 or LZTR1 have been detected in the tumours of patients with schwannomatosis, the classical two-hit model of tumorigenesis is insufficient to account for schwannoma growth, since NF2 is also frequently inactivated in these tumours. Consequently, tumorigenesis in schwannomatosis must involve the mutation of at least two different tumour suppressor genes, an occurrence frequently mediated by loss of heterozygosity of large parts of chromosome 22q harbouring not only SMARCB1 and LZTR1 but also NF2. Thus, schwannomatosis is paradigmatic for a tumour predisposition syndrome caused by the concomitant mutational inactivation of two or more tumour suppressor genes. This review provides an overview of current models of tumorigenesis and mutational patterns underlying schwannomatosis that will ultimately help to explain the complex clinical presentation of this rare disease.
Similar content being viewed by others


Introduction
Schwannomatosis (MIM #162091) is a rare disorder with an estimated incidence of 1/40,000–1/70,000 (Koontz et al. 2013) that is characterized by the occurrence of multiple schwannomas and, much less commonly, meningiomas. In patients with schwannomatosis, schwannomas commonly affect the spine (74%) and peripheral nerves (89%), whereas cranial nerve schwannomas (mostly trigeminal) are uncommon (8%) (Merker et al. 2012). In one-third of patients with schwannomatosis, the tumours are anatomically limited to a single limb or several contiguous segments of the spine or one half of the body (MacCollin et al. 1996; Merker et al. 2015). The most common symptom reported by schwannomatosis patients is chronic pain which may be either local or diffuse (MacCollin et al. 2005; Merker et al. 2015).
Considerable overlap has been noted between schwannomatosis and NF2 (MIM #101000) in terms of the occurrence of the associated types of tumour, but both diseases are regarded as separate clinical entities (MacCollin et al. 1996, 2003; Evans et al. 1997; reviewed by Blakeley and Plotkin 2016). Despite this clinical overlap, there are several important differences between schwannomatosis and NF2 in relation to the frequency of specific tumour types and the occurrence of certain clinical symptoms (see Table 1 and references therein). Intradermal schwannomas, ependymomas, cataract, and retinal abnormalities are all observed in patients with NF2 but are not associated with schwannomatosis. Furthermore, bilateral vestibular schwannomas, the hallmark feature of NF2, have not been reported in patients with schwannomatosis. However, unilateral vestibular schwannomas may occur in association with schwannomatosis and hence cannot be used as an exclusion criterion to distinguish between schwannomatosis and NF2 (Smith et al. 2012a, 2015, 2016; Wu et al. 2015; Mehta et al. 2016).
The majority of patients with schwannomatosis are sporadic, whereas 13–25% are familial cases (Evans et al. 1997; Antinheimo et al. 2000; MacCollin et al. 2005; Merker et al. 2012). A combination of linkage analysis in affected families and mutation screening of the NF2 gene in schwannomas indicated that schwannomatosis is not due to germline mutations in the NF2 gene (Jacoby et al. 1997; Kaufman et al. 2003; MacCollin et al. 2003). However, instead of constitutional (germline) NF2 mutations, independent somatic mutations affecting both NF2 alleles are frequently found in schwannomas of patients with schwannomatosis (Jacoby et al. 1997; Kaufman et al. 2003; Boyd et al. 2008; Hadfield et al. 2008; Sestini et al. 2008; Hutter et al. 2014; Paganini et al. 2015a; Piotrowski et al. 2014; Smith et al. 2015, 2016).
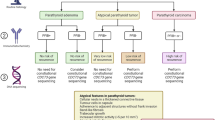
So far, two schwannomatosis predisposition genes have been identified, SMARCB1 and LZTR1 (Hulsebos et al. 2007; Sestini et al. 2008; Hadfield et al. 2008; Smith et al. 2012b; Hutter et al. 2014; Piotrowski et al. 2014; Smith et al. 2015). Further schwannomatosis predisposition genes may well exist, but they still remain to be discovered. The clinical overlap between schwannomatosis and NF2 renders differential diagnosis somewhat difficult, particularly in sporadic and mosaic cases with multiple schwannomas but without bilateral vestibular schwannomas and detectable germline NF2 gene mutations. However, comprehensive mutation testing of LZTR1, SMARCB1, and NF2 using DNA derived from blood and different tumour samples of the patient is the method of choice to distinguish between the two conditions (Castellanos et al. 2015; Smith et al. 2016). The diagnosis of schwannomatosis is predicated upon the molecular and/or clinical diagnostic criteria according to Plotkin et al. (2013) and Ostrow et al. (2016) (Fig. 1). In what follows, we review current knowledge of the mutational patterns of the known schwannomatosis predisposing genes, models of tumorigenesis, and the genotype/phenotype relationship.

Diagnostic criteria for schwannomatosis according to Ostrow et al. (2016) and Plotkin et al. (2013) based upon the criteria formulated by MacCollin et al. (2005) which predated our ability to perform molecular testing for schwannomatosis and did not consider the possibility of multiple meningiomas. a According to the findings of Castellanos et al. (2015), the deletions of 22q causing the LOH in ≥ 2 tumours should have different breakpoints for these deletions to be considered as independent events. The analysis of the extent of the LOH is necessary to exclude a large 22q deletion as the first-hit mutation (that would be identical in different tumours) which would be indicative of mosaic NF2. If an identical SMARCB1 mutation is detected in different tumours of a patient, SMARCB1-associated schwannomatosis may be diagnosed. LZTR1-associated schwannomatosis may be present, if an identical LZTR1 mutation is detected in different tumours of a patient. b High-quality MRI should include a detailed study of the internal auditory canal with slices no more than 3 mm thick. c Schwannomatosis should be considered as a possible diagnosis if two or more nonintradermal tumours are present, even if none has been pathologically confirmed to be a schwannoma; the occurrence of chronic pain in association with the tumour(s) increases the likelihood of schwannomatosis (Plotkin et al. 2013). d Smith et al. (2016) identified five patients, with unilateral vestibular schwannomas and at least two nonvestibular, nonintradermal schwannomas, who met the diagnostic criteria for NF2 but had germline LZTR1 mutations instead of germline NF2 mutations. 22q LOH: loss of heterozygosity on the long arm of chromosome 22
SMARCB1 germline mutations in patients with schwannomatosis
Linkage analysis with microsatellite markers performed in families affected with schwannomatosis served to exclude the NF2 gene as a germline-transmissible schwannomatosis predisposition gene but nevertheless suggested that such a gene could be located within an 8.48-Mb region centromeric to NF2, between markers D22S420 and D22S1148 in the vicinity of D22S1174 on chromosome 22 (MacCollin et al. 2003) (Fig. 2). This region includes SMARCB1 (also termed hSNF5 or INI1) which encompasses nine exons encoding a subunit of the human SWI/SNF chromatin-remodeling complex (reviewed by Kalimuthu and Chetty 2016). SMARCB1 appears not to be essential for the assembly of the remodeling complex (Doan et al. 2004) but is involved in targeting the SWI/SNF complex to gene promoters (Kuwahara et al. 2013). SMARCB1 also participates in a number of protein–protein interactions involving transcription factors, such as c-MYC and GLI1 (Cheng et al. 1999; Jagani et al. 2010; Stojanova et al. 2016). Biallelic SMARCB1 inactivation has been detected in a multitude of different tumour types and at high frequency in rhabdoid tumours (reviewed by Roberts and Biegel, 2009; Hollmann and Hornick 2011; Masliah-Planchon et al. 2015). Using a candidate gene approach, Hulsebos et al. (2007) investigated whether SMARCB1 might be the anticipated schwannomatosis predisposition gene. They subsequently found a germline mutation of SMARCB1 in exon 1 (c.34C > T) that was predicted to result in premature translational termination at protein position p.Gln12* in a 22-year-old female patient and her affected father. In one schwannoma from the father, Hulsebos et al. (2007) detected an additional somatic truncating SMARCB1 mutation (c.544C > T; p.Gln182*). In a second schwannoma from the same individual, the partial loss of the SMARCB1 wild-type allele was observed. These findings constituted good prima facie evidence that SMARCB1 functions as a tumour suppressor gene and that mutations in this gene predispose to schwannomatosis. Since this initial report, further studies have confirmed this conclusion, since germline SMARCB1 mutations have been identified in schwannomatosis patients from different cohorts (Supp. Table S1) (Boyd et al. 2008; Hadfield et al. 2008; Sestini et al. 2008; Rousseau et al. 2011; Smith et al. 2012b, 2014; Asai et al. 2015). These studies imply that germline SMARCB1 mutations are present in at least 48% of familial and 9.8% of sporadic schwannomatosis cases (Supp. Table S2).

Partial ideogram of chromosome 22 indicating the location of the LZTR1, SMARCB1, and NF2 genes and the microsatellite markers D22S420 (GenBank accession number Z23643.1), D22S1174 (GenBank acc. No. Z51327.1), and D22S1148 (GenBank acc. No. Z52647.1). The nucleotide numbering is given according to hg19. Linkage analysis provided the original evidence that the schwannomatosis predisposition genes are located within the ~8.5-Mb region between markers D22S420 and D22S1148 (MacCollin et al. 2003). The centromeric direction is on the left side, the telomeric direction is on the right side of the schema
Meningiomas in patients with schwannomatosis and germline SMARCB1 mutations
Biallelic inactivation of SMARCB1 has been observed in both schwannomas and meningiomas of patients with schwannomatosis (Boyd et al. 2008; Sestini et al. 2008; Hadfield et al. 2008, 2010a; Smith et al. 2012b). Meningiomas occur in 5% of schwannomatosis patients (Merker et al. 2012) and appear to be located predominantly in the cerebral falx (van den Munckhof et al. 2012). Three different families have been identified in which some members harboured germline SMARCB1 mutations and exhibited multiple schwannomas and meningiomas (Bacci et al. 2010; Christiaans et al. 2011; van den Munckhof et al. 2012; Melean et al. 2012) (Supp. Table S3). However, not all mutation carriers in these families had meningiomas, indicative of the variable expression of meningiomas in patients with germline SMARCB1 mutations. Furthermore, SMARCB1 germline mutations have not been found in patients with multiple meningiomas in the absence of schwannomatosis (Hadfield et al. 2010b). Multiple meningiomas are very rare and usually occur in the context of NF2 (reviewed by Smith 2015). Remarkably, germline mutations in SMARCE1, another component of the SWI/SNF chromatin-remodeling complex, have been identified in patients with familial multiple spinal meningiomas without NF2 (Smith et al. 2013).
Coffin–Siris syndrome, schwannomatosis, and SMARCB1 germline mutation
Recently, a patient with Coffin–Siris syndrome (MIM# 135900) and schwannomatosis has been reported to carry a germline missense mutation in exon 9 of SMARCB1 (c.1121G > A; p.Arg374Gln) (Gossai et al. 2015). The patient exhibited intellectual disability, hypotonia, microcephaly, coarse face, hypoplasia of the digits, general hirsutism, multiple schwannomas, as well as bilateral cataracts and bilateral cranial nerve schwannomas which are most unusual in the context of schwannomatosis. Patients with Coffin–Siris syndrome and germline SMARCB1 mutations have been previously reported, but none have exhibited schwannomas (Tsurusaki et al. 2012).
Mutational pattern in SMARCB1-positive schwannomas as compared with rhabdoid tumours
The recognition that mutations in the SMARCB1 gene predispose to benign schwannoma, which usually become symptomatic during adulthood, came as something of a surprise, since this gene was originally discovered in the context of its involvement in the development of atypical teratoid/rhabdoid tumours (Versteege et al. 1998; Sévenet et al. 1999a). These highly malignant tumours develop most commonly in the central nervous system in very young children who frequently die as a consequence of the malignancy before the age of 3 (Hilden et al. 2004; Lau et al. 2015). A few patients have been reported to have survived the initial tumour for up to 26 years, albeit with multiple recurrences (reviewed by Takahashi-Fujigasaki et al. 2012). Rhabdoid tumours may also develop in the kidney and less frequently, in extrarenal tissues (reviewed by Oda and Tsuneyoshi 2006), and SMARCB1 mutations have been identified in both renal and extrarenal rhabdoid tumours (Biegel et al. 2002; Savla et al. 2000; Kordes et al. 2010). Germline mutations of SMARCB1 occur in approximately one-third of patients with rhabdoid tumours (Biegel et al. 1999; Sévenet et al. 1999b; Bourdeaut et al. 2011; Eaton et al. 2011). Although most of the germline SMARCB1 mutations causing rhabdoid tumours occur de novo, familial cases have also been reported, with several affected members harbouring constitutional SMARCB1 mutations and malignant rhabdoid tumours but never developing schwannomas (Sévenet et al. 1999b; Taylor et al. 2000; Ammerlaan et al. 2008). This condition is known as rhabdoid tumour predisposition syndrome 1 (RTPS1: MIM#609322). In some cases, RTPS1 is caused by gonadal mosaicism for an SMARCB1 mutation (Sévenet et al. 1999b; Bruggers et al. 2011; Lee et al. 2002; Janson et al. 2006; Eaton et al. 2011; Gigante et al. 2016). Tumorigenesis in RTPS1 is driven by somatically acquired second-hit mutations in the wild-type SMARCB1 allele. Biallelic inactivation of SMARCB1 through the acquisition of two somatic mutations has been observed in the rhabdoid tumours of patients without germline mutations. Hence, SMARCB1 plays the role of a classic tumour suppressor gene in rhabdoid tumours according to the Knudson two-hit model (Versteege et al. 1998; Biegel et al. 1999; Uno et al. 2002; Jackson et al. 2009; Kordes et al. 2010; Bourdeaut et al. 2011).
In contrast to the highly malignant rhabdoid tumours, schwannomas are generally benign and very rarely transform to malignant tumours (reviewed by Carter et al. 2012). Differences in the position and type of germline SMARCB1 mutations have been observed in patients with schwannomatosis compared to those with rhabdoid tumours. Schwannomatosis-associated SMARCB1 mutations are preferentially located either at the 5′or 3′ end of the gene (Hulsebos et al. 2007; Hadfield et al. 2008; Rousseau et al. 2011; Smith et al. 2012b, 2014). Several recurrent SMARCB1 mutations have been identified in patients with schwannomatosis; the most common of these is the c.*82C > T mutation located in the 3′UTR (Supp. Table S4). By contrast, intragenic germline SMARCB1 mutations observed in patients with rhabdoid tumour tend to be located in the central part of the gene (Smith et al. 2014). In addition to this position effect, the mutational spectra differ, with SMARCB1 mutations in schwannomatosis patients being predominantly nontruncating, including missense and splice-site mutations as well as in-frame deletions, that lead to the production of stable transcripts (Smith et al. 2012c). By contrast, almost all germline mutations of SMARCB1 in patients with rhabdoid tumour are either protein-truncating or alternatively whole-gene or multi-exon deletions (Bourdeaut et al. 2011; Eaton et al. 2011; Smith et al. 2014; reviewed by Biegel et al. 2014). These findings are suggestive of a genotype/phenotype correlation: loss-of-function mutations occur in patients with rhabdoid tumours, whereas schwannomatosis-associated germline SMARCB1 mutations are predominantly hypomorphic (Smith et al. 2012c, 2014).
In accord with this postulate are the findings of Hulsebos et al. (2014a). These authors analysed four schwannomatosis-associated SMARCB1 mutations that were located in the 5′ region of the gene and were predicted to introduce a premature translational termination codon (PTC). Two of them, c.30delC (p.Phe10Leufs*6) and c.38delA (p.Lys13Serfs*3), may be predicted to cause frameshifts generating a PTC starting at nucleotide 44. The other two SMARCB1 mutations, c.34C > T (p.Gln12*) and c.46A > T (p.Lys16*), directly generate PTCs at their respective positions. Although transcripts containing these PTCs may reasonably be expected to be degraded by nonsense-mediated mRNA decay (NMD), stable transcripts were detected by Hulsebos et al. (2014a) in schwannoma tissue obtained from two patients harbouring either c.34C > T or c.30delC. Furthermore, Western blot analysis using frozen tumour tissue from the individual with c.30delC indicated the occurrence of a shortened protein owing to downstream translational reinitiation. This finding was confirmed by transient overexpression of a c.34C > T-containing expression vector in a cell line without endogenous SMARCB1 protein and subsequent detection of the shortened SMARCB1 protein by Western blotting. Similar overexpression experiments indicated that the mutations c.30delC, c.38delA, and c.46A > T also lead to truncated SMARCB1 proteins which may well be partially functional (Hulsebos et al. 2014a).
Many of the missense and splice-site mutations as well as in-frame deletions detected in schwannomatosis patients fail to alter SMARCB1 transcript stability, and hence, the mutant alleles are likely to encode at least partially functional proteins, as has been concluded from cyclin D1 repression assays (Smith et al. 2012c). Loss of SMARCB1 in rhabdoid tumours leads to upregulation of cyclin D1 and cell cycle progression. A cyclin D1 repression assay has shown that mutant SMARCB1 proteins, derived from expression plasmids harbouring the same missense and splice-site mutations noted in patients with schwannomatosis, were capable of suppressing cyclin D1 activity in a similar manner to the wild-type SMARCB1 protein (Smith et al. 2012c). Taken together, these observations suggest that in schwannomatosis, germline SMARCB1 mutations encode proteins with some residual functionality (Smith et al. 2012c; Hulsebos et al. 2014a). By contrast, SMARCB1 mutations in rhabdoid tumours and patients with RTPS1 are almost invariably either nonsense mutations or frameshifts, even complete gene deletions, and hence are associated with a loss-of-function (Kordes et al. 2010; Bourdeaut et al. 2011; reviewed by Biegel et al. 2014). Consequently, SMARCB1 expression is completely absent in rhabdoid tumours (Hoot et al. 2004; Judkins et al. 2004; Haberler et al. 2006; Sigauke et al. 2006; Kordes et al. 2010; reviewed by Margol and Judkins 2014).
Mosaic SMARCB1 expression in schwannomatosis-associated schwannomas
Schwannomas in patients with germline SMARCB1 mutations have been reported to exhibit a mosaic SMARCB1 protein expression pattern by immunohistochemical staining of tumour sections with the monoclonal BAF47 antibody specific for the C-terminal part of SMARCB1 raised against amino acids 257–359 (Hulsebos et al. 2007; Patil et al. 2008; Smith et al. 2012c). This mosaic pattern results from mixed immuno-positive and -negative nuclei, consistent with the expression of SMARCB1 in a subset of tumour cells. Considerable inter- and intra-tumoral variability has been observed with regard to the number of nuclei that exhibit SMARCB1 expression (Patil et al. 2008). The mosaic expression pattern may be explicable in terms of a subset of schwannoma cells retaining the wild-type SMARCB1 allele, thereby enabling the synthesis of sufficient SMARCB1 protein to give rise to positive immunostaining of the respective nuclei. In other types of tumour cell, however, the loss of the SMARCB1 wild-type allele leads to complete loss of protein expression. Although this scenario may account for some schwannomas, it cannot explain the mosaic SMARCB1 immunostaining observed in the majority of these tumours. This may be concluded from the observation that the loss of the wild-type SMARCB1 allele is readily detectable by loss of heterozygosity (LOH) analysis using polymorphic markers, suggesting that this loss affects a large proportion of tumour cells. Consequently, the mosaic SMARCB1 expression is most likely related to the hypomorphic nature of the mutations in schwannomatosis patients that encode stable mRNA transcripts giving rise to detectable amounts of SMARCB1 protein. Since, in schwannomas of patients with schwannomatosis, the wild-type SMARCB1 allele is often lost by deletion or monosomy 22, the SMARCB1 protein detected in schwannoma cells must be encoded by the mutant allele. The inability to detect mutant proteins in all tumour cells by immunostaining is likely to be a consequence of the instability of mutant SMARCB1 proteins (Hulsebos et al. 2014a). This instability results in immunologically nonreactive SMARCB1 protein degradation products in a proportion of the schwannoma cells. Since this degradation is likely to be a random process, some cells may still express detectable amounts of SMARCB1 protein resulting in a mosaic expression pattern when analysing schwannoma tissue sections (Hulsebos et al. 2014a, 2016).
Recently, an N-terminal region of SMARCB1 has been identified that encodes a winged helix DNA-binding domain (Allen et al. 2015). This domain is deleted from the shortened SMARCB1 proteins encoded by the transcripts harbouring exon 1 truncating mutations investigated by Hulsebos et al. (2014a). Furthermore, N-terminally located SMARCB1 missense mutations are predicted to destabilize the encoded protein and interfere with DNA-binding (Allen et al. 2015). Despite the reduced stability and impaired DNA-binding capacity of proteins encoded by transcripts harbouring mutations located within the N-terminal half of SMARCB1, it would appear that these proteins retain sufficient residual function to prevent the occurrence of highly malignant rhabdoid tumours during the early childhood in patients with complete loss-of-function SMARCB1 mutations.
Malignancy in patients with SMARCB1 germline mutations and schwannomatosis
Three families have been reported with germline SMARCB1 mutation carriers presenting either with rhabdoid tumours or schwannomatosis (Swensen et al. 2009; Eaton et al. 2011; Sredni and Tomita 2015). The SMARCB1 mutations identified in these families were predicted to introduce premature termination codons leading to the production of unstable transcripts that would then undergo nonsense-mediated mRNA decay (Supp. Table S5). Somatic mosaicism for the SMARCB1 mutation in the family members affected only by schwannomas cannot explain the absence of rhabdoid tumours in these patients. The existence of adult mutation carriers in these families without rhabdoid tumours is intriguing and suggests that the risk of rhabdoid tumour development is time-dependent in the sense that there may be a specific developmental time window during which the progenitor cells of rhabdoid tumours are vulnerable to SMARCB1 protein loss (Boyd et al. 2008; Biegel et al. 2014). If the cells manage to progress through this critical time period without experiencing complete SMARCB1 loss, then the individual harbouring them may not develop rhabdoid tumours despite the presence of the germline SMARCB1 mutation. This hypothesis is consistent with the peak incidence of rhabdoid tumours at 6 months of age in patients with germline SMARCB1 mutations. Furthermore, the risk of developing a rhabdoid tumour decreases dramatically after 3 years of age (Eaton et al. 2011).
Swensen et al. (2009) investigated a four-generation family with members affected either by schwannomatosis or rhabdoid tumour (Supp. Table S5). Two schwannomas from different affected members of this family were biopsied and classified as epithelioid schwannomas. This histological subtype is very rare among schwannomas detected in patients with schwannomatosis (Hart et al. 2016). Furthermore, both schwannomas were negative for SMARCB1 immunostaining, indicative of a complete loss of the SMARCB1 protein (Swensen et al. 2009). This is in stark contrast to the schwannomatosis-associated tumours which usually exhibit mosaic SMARCB1 expression (Hulsebos et al. 2007; Patil et al. 2008; Smith et al. 2012c). Taken together, these observations suggest that schwannomas observed in this family (and probably also in other families with rhabdoid tumour and schwannomatosis) may have a different biology compared with classical schwannomatosis-associated schwannomas.
In line with this postulate are the findings of Carter et al. (2012) who reported a patient with a germline SMARCB1 frameshift mutation in exon 3 and several schwannomas which were classified as ‘neuroblastoma-like’ (Supp. Table S5). Neuroblastoma-like schwannomas are extremely rare; only one other case of a patient possibly affected by schwannomatosis and a neuroblastoma-like schwannoma has been reported to date (Sulhyan et al. 2015). The female propositus described by Carter et al. (2012) had three children who carried the SMARCB1 mutation and two of them suffered from rhabdoid tumour. The propositus developed an epithelioid malignant peripheral nerve sheath tumour (MPNST) with rhabdoid features arising from a preexisting schwannoma. However, the malignant transformation of schwannomas is extremely rare and has been observed in only a few cases (Woodruff et al. 1994; Nayler et al. 1996; McMenamin and Fletcher 2001).
The above notwithstanding, malignant schwannomas were reported in two patients with familial schwannomatosis (Gonzalvo et al. 2011). However, it is unknown whether they carried SMARCB1 germline mutations. Furthermore, MPNSTs were also noted in two members of a family with schwannomatosis and two co-occurring SMARCB1 alterations, a missense mutation in exon 7 (c.864C > G; p.Asn288Lys) and a splice-site mutation located 12-bp upstream of exon 9 (c.1032-12C > G) which is predicted to lead to the insertion of 11 bp of intronic sequence in the mutant transcript (Hadfield et al. 2008; Evans et al. 2012; Smith et al. 2012b). This insertion would be predicted to introduce a frameshift that would result in the introduction of a novel stop codon (p. Arg373fsX379*). These findings raised some concern about an increased MPNST risk in schwannomatosis patients, but neither MPNSTs nor rhabdoid tumours were observed in a cohort of 87 patients with schwannomatosis, including 11 patients from seven families (Merker et al. 2012). However, it is not known how many of the patients investigated by Merker et al. (2012) carried SMARCB1 germline mutations. Further studies are necessary to assess the MPNST risk for patients with SMARCB1 mutation-positive schwannomatosis.
A risk of malignancy may exist in those schwannomatosis patients with SMARCB1 germline mutations that are less likely to be hypomorphic. Paganini et al. (2015a) reported a patient with schwannomatosis and a uterine leiomyosarcoma. The patient had a germline SMARCB1 mutation c.1118G > A involving the last nucleotide of exon 8, disrupting the donor splice site of intron 8. RNA analysis indicated that this mutation leads to the insertion of intron 8 sequences in the transcript that would be predicted to result in a frameshift. The mutant RNA has been shown to encode a C-terminally elongated protein exhibiting a different amino acid sequence and a new stop codon after an additional 48 amino acids. LOH analysis indicated that the mutant SMARCB1 allele was retained in two schwannomas and the leiomyosarcoma of the patient, whereas the wild-type allele was lost, indicative of biallelic SMARCB1 inactivation in both types of tumour (Paganini et al. 2015a). Substantiating concern about an increased risk of malignancy in a subset of patients with SMARCB1-positive schwannomatosis, Hulsebos et al. (2016) reported a type 1 papillary renal cell carcinoma (pRCC1) in a schwannomatosis patient with a germline duplication of SMARCB1 exon 7 (c.796-2246_9861 +5250dup7686) resulting in a premature stop codon (p.Glu330*). The chromosome 22 carrying the mutant SMARCB1 allele was retained in the pRCC1 and in schwannomas from this patient, whereas the wild-type SMARCB1 allele and one NF2 copy were lost by deletion. Immunohistochemical staining revealed the complete loss of SMARCB1 expression in the pRCC1 (Hulsebos et al. 2016) as observed in malignant rhabdoid tumours with biallelic SMARCB1 inactivation.
The leiomyosarcoma and the pRCC1 observed in two patients with SMARCB1 mutation-positive schwannomatosis indicate that malignancy may, indeed, occur in a subset of patients. However, the risk is probably dependent upon the SMARCB1 mutation type and may be decreased in patients with hypomorphic SMARCB1 mutations.
Models of tumorigenesis in SMARCB1-associated schwannomatosis
The classical two-hit model of tumorigenesis (Knudson 1971) does not seem to pertain in the tumours of patients with SMARCB1 germline mutations, at least in the sense that this model would require biallelic SMARCB1 inactivation to be sufficient for tumour initiation or growth. This is concluded from the observation of frequent somatic, tumour-specific NF2 mutations, and the loss of the second NF2 allele in schwannomas from patients with germline SMARCB1 mutations (Boyd et al. 2008; Sestini et al. 2008; Hadfield et al. 2008, 2010a). These findings hint at a significantly higher level of complexity than that required by the basic Knudson model. The loss of the second NF2 allele is frequently caused by the complete loss of chromosome 22 or large portions of the long arm of chromosome 22 (22q), including the second (wild-type) allele of SMARCB1 (Hadfield et al. 2010a). This pattern of mutational events points to a 4-hit/3-step model of tumorigenesis in patients with SMARCB1-positive schwannomatosis (Fig. 3a): The first hit (and step) represents the germline SMARCB1 mutation, whereas the second step involves LOH of 22q that removes the wild-type SMARCB1 allele and one of the two NF2 alleles. The third step is the somatic mutation of the remaining NF2 allele located on the chromosome harbouring the germline SMARCB1 mutation which is retained in the tumour (Boyd et al. 2008; Hadfield et al. 2008; Sestini et al. 2008). This 4-hit/3-step model of tumorigenesis probably also holds true for other benign tumours observed in patients with SMARCB1-positive schwannomatosis, since biallelic inactivation of SMARCB1 and NF2 has been observed in a meningioma and leiomyoma of patients with germline SMARCB1 mutations (van den Munckhof et al. 2012; Hulsebos et al. 2014b).

Models of tumorigenesis in schwannomatosis. a Four-hit/3-step model in patients with an heterozygous germline SMARCB1 mutation (first hit and step). The second step includes loss of heterozygosity (LOH) of 22q which removes the wild-type SMARCB1 allele and one of the two NF2 alleles. The third step is the somatic mutation of the other NF2 allele located on the chromosome harbouring the germline SMARCB1 mutation. b Four-hit/3-step model of tumorigenesis in patients with an heterozygous germline LZTR1 mutation (first hit and step). c Five-hit/3-step model of tumorigenesis in schwannomatosis. The LOH event which removes one wild-type LZTR1 allele and one copy of NF2 automatically leads to the loss of one SMARCB1 allele, which represents the fifth mutational hit
LOH of 22q in tumours from patients with germline SMARCB1 mutations does not seem to be mediated by mitotic recombination, a mechanism frequently causing LOH of other tumour suppressor genes (Makishima and Maciejewski 2011; Garcia-Linares et al. 2011; Stewart et al. 2012). In eight tumours from patients with germline SMARCB1 mutations, LOH of 22q was found to be caused exclusively by whole chromosome loss or large deletions within 22q, but not by mitotic recombination (Hadfield et al. 2010a). Copy-number neutral LOH of the NF2 locus indicative of mitotic recombination was also not observed in 17 schwannomas from patients with germline SMARCB1 mutations analysed by Piotrowski et al. (2014). By contrast, mitotic recombination accounts for 19% of the LOH events in NF2-associated schwannomas (14 of 72 schwannomas analysed) and 23% (5/22) of schwannomas from schwannomatosis patients without germline SMARCB1 mutations (Hadfield et al. 2010a). The observation that LOH in schwannomas of patients with SMARCB1 germline mutations is not mediated by mitotic recombination supports the 4-hit/3-step model of tumorigenesis from the standpoint of maximum parsimony (Fig. 3a). Although mitotic recombination would lead to biallelic inactivation of SMARCB1 by reduplication of the mutant SMARCB1 allele, the wild-type NF2 allele would be preserved. Two additional independent mutational events would then be required to inactivate both NF2 alleles. Hence, mitotic recombination would make four steps of mutation necessary to inactivate both alleles of SMARCB1 and NF2 instead of three as suggested by the model of tumorigenesis for SMARCB1-positive schwannomatosis depicted in Fig. 3a.
Although the 4-hit/3-step model accounts for the majority of the tumours in the cases of SMARCB1 mutation-positive schwannomatosis, biallelic inactivation of the NF2 gene is not observed in all schwannomas (Boyd et al. 2008; Hadfield et al. 2008, 2010a; Sestini et al. 2008; Hulsebos et al. 2007, 2016). It is estimated that at least 19% of schwannomas of patients with germline SMARCB1 mutations exhibit only mono-allelic NF2 inactivation (Supp. Table S6) (Boyd et al. 2008; Hadfield et al. 2008, 2010a; Sestini et al. 2008). In this subgroup of tumours, the inactivation of only one NF2 allele may be sufficient to promote the proliferation of the cells with biallelic SMARCB1 inactivation. Since the LOH events in SMARCB1 mutation-positive schwannoma usually include large parts of 22q (Hadfield et al. 2010a), other tumour suppressor genes located on 22q will also be haploinsufficient and this may contribute to schwannoma growth. One of these suspected tumour suppressor genes is likely to be LZTR1.
Germline mutations in LZTR1
Germline SMARCB1 mutations account for 48% of familial and 9.8% of sporadic schwannomatosis cases (Supp. Table S2) indicative of probable locus heterogeneity and the existence of additional schwannomatosis predisposition genes. Piotrowski et al. (2014) analysed 3.72 Mb of highly conserved DNA sequence within different regions of 22q, including parts of the previously defined linkage interval postulated to harbour schwannomatosis predisposition genes (MacCollin et al. 2003) and the region of the CABIN1 gene which has been suggested to be important in schwannomatosis (Buckley et al. 2005). The 3.72 Mb of highly conserved sequence were analysed by targeted next-generation sequencing which indicated germline mutations within in the LZTR1 (leucine zipper-like transcriptional regulator 1) gene in patients with schwannomatosis but lacking SMARCB1 mutations (Piotrowski et al. 2014). LZTR1 is located 2.8 Mb centromeric to SMARCB1 and 8.7 Mb centromeric to NF2 (Fig. 2), and is included in the 1.5–3-Mb region which is deleted in individuals with the DiGeorge syndrome. The protein encoded by LZTR1 contains Kelch- and BTB/POZ-domains and localizes to the Golgi complex (Nacak et al. 2006). It has also been shown to be an adaptor of the cullin 3-containing E3 ubiquitin ligase complex (Frattini et al. 2013). Somatic mutations in LZTR1 occur in 22% of glioblastomas (Frattini et al. 2013) and in several other cancers according to the Catalogue of Somatic Mutations in Cancer (COSMIC) database (Forbes et al. 2008). The biallelic inactivation of LZTR1 in glioblastomas, and the observation that LZTR1 inactivation drives self-renewal and growth of glioma spheres, are indicative of LZTR1 acting as a classic tumour suppressor gene (Frattini et al. 2013).
Piotrowski et al. (2014) identified LZTR1 germline mutations in 16 of 20 unrelated schwannomatosis patients (80%). Among these, 20 patients were 6 familial schwannomatosis cases and LZTR1 mutations were found in all of them. Eleven of the twenty patients analysed were confirmed sporadic cases and eight of these (73%) had LZTR1 mutations (Piotrowski et al. 2014). Such a high rate of LZTR1 mutation in SMARCB1 mutation-negative schwannomatosis patients was not, however, observed in subsequent studies (Hutter et al. 2014; Paganini et al. 2015b; Smith et al. 2015). In these latter studies, only 22–30% of sporadic cases and 38% of familial cases exhibited LZTR1 mutations (Supp. Table S7). This difference in the proportion of LZTR1 mutation-positive schwannomatosis cases detected may be due to the adoption of different selection criteria for the patients being analysed. Piotrowski et al. (2014) exclusively studied patients with a molecularly confirmed diagnosis of schwannomatosis. This implies that two different somatic NF2 mutations were identified in two tumours of a given patient as well as the loss of chromosome 22q encompassing the wild-type NF2 allele. By contrast, the studies with lower LZTR1 mutation detection rates mostly focussed upon patients diagnosed with schwannomatosis on the basis of clinical criteria according to Plotkin et al. (2013) and who were negative for SMARCB1 mutations. Consequently, the studies reporting lower LZTR1 mutation detection rates included a more heterogeneous group of patients whose schwannomas may not have been characterized by biallelic NF2 inactivation. Nevertheless, these studies serve to confirm that LZTR1 is a major schwannomatosis predisposition gene.
Schwannomatosis-associated LZTR1 mutations were found in nearly all exons; thus, no positional preference of mutations was observed (Hutter et al. 2014; Piotrowski et al. 2014; Paganini et al. 2015a; Smith et al. 2015). In contrast to this situation, SMARCB1 mutations causing schwannomatosis are predominantly located at the 5′ or 3′ end of the gene (Smith et al. 2014). Of the 59 LZTR1 mutations identified to date, at least 28 (47%) were protein-truncating, whilst all of the 23 missense mutations were predicted to be deleterious (Supp. Table S8). These findings suggest that the majority of schwannomatosis-associated LZTR1 mutations are not hypomorphic. Somatic loss of the wild-type LZTR1 allele is predicted to lead to biallelic loss of LZTR1 in tumours of germline mutation carriers. However, the type of the LZTR1 mutation would appear to influence LZTR1 protein expression in tumours. Schwannomas from patients with nonsense or frameshift LZTR1 mutations do not exhibit LZTR1 protein immunostaining, whereas patients carrying splicing or missense variants showed reduced immunostaining (Paganini et al. 2015b). By contrast, schwannomas from LZTR1-unrelated schwannomatosis patients, as well as patients with NF2, showed diffuse but positive LZTR1 immunostaining. Whether this variability has any effect on schwannoma growth or location is unclear but clearly warrants further investigation.
LZTR1 mutations in unaffected carriers
Ten unrelated and clinically unaffected LZTR1 mutation carriers have been identified who had relatives harbouring the same mutation and were affected by schwannomatosis (Supp. Table S8). Eight of these ten apparently nonpenetrant LZTR1 mutations were protein-truncating and highly likely to be deleterious. Even though not all clinically unaffected LZTR1 mutation carriers have been investigated by MRI [precluding the unambiguous exclusion of the occurrence of minor lesions, such as intrafascicular microlesions described by Farschtschi et al. (2016)], many of these individuals are of advanced age and some clinical symptoms should already have become apparent. The reason for the incomplete penetrance of these LZTR1 mutations is currently unclear, but it may be that somatic mosaicism for the LZTR1 mutations accounts for the absence of clinical symptoms in the unaffected mutation carriers who passed on the mutations to the affected family members. Although the respective LZTR1 mutations were detected in high proportions of peripheral blood cells of the unaffected family members, this should not be held to imply that numerous Schwann cell progenitor cells also harbour the mutation. We postulate that the LZTR1 mutations in the unaffected probands are of post-zygotic origin and are present in a minority of Schwann cell precursors. The haematopoietic progenitor cells carrying the LZTR1 mutation may possess a selective growth advantage, giving rise to a high proportion of mutation-positive blood cells. This situation would be reminiscent of patients with neurofibromatosis type 1 (NF1) and somatic mosaicism for a large deletion in the NF1 gene region spanning 1.2 Mb. The patients have a high proportion of blood cells carrying the NF1 deletion (>90%); a much lower proportion of cells with the NF1 deletion has, however, been detected in the fibroblasts and urine of these patients (Kehrer-Sawatzki and Cooper, 2008; Roehl et al. 2012). Somatic mosaicism due to a post-zygotic mutation and selective growth advantage of LZTR1 haploinsufficient blood cells may, therefore, be responsible for the apparently incomplete penetrance of some LZTR1 mutations. However, it is possible that the LZTR1 mutation in the unaffected family members is, indeed, constitutional and that the lack of clinical symptoms manifested by these LZTR1 mutation carriers may be caused by as yet unidentified modifying genes.
LZTR1 germline mutations and associated tumour spectrum
So far, patients with schwannomatosis and LZTR1 germline mutations have not been reported to exhibit meningiomas, in contrast to patients with SMARCB1 germline mutations (Hutter et al. 2014; Piotrowski et al. 2015; Paganini et al. 2015b; Smith et al. 2015, 2016). These findings suggest that LZTR1 mutations do not predispose to meningiomas. However, unilateral vestibular schwannomas can occur in patients with LZTR1 mutation-positive schwannomatosis. Recently, Smith et al. (2016) demonstrated that 5 of 70 patients (7%) presenting with a unilateral vestibular schwannoma and at least two nonintradermal, nonvestibular schwannomas have germline LZTR1 mutations; hence, these individuals have schwannomatosis rather than NF2 even, although they would fulfil current diagnostic criteria for NF2. These findings further evidence the overlap between schwannomatosis and NF2.
In contrast to LZTR1-associated schwannomatosis, SMARCB1 mutations are probably less likely to predispose to unilateral vestibular schwannomas. So far, only one solitary case of a unilateral vestibular schwannoma in a putative SMARCB1-associated schwannomatosis family has been reported (Wu et al. 2015). However, this report remains inconclusive, since, although the SMARCB1 mutation was identified in a schwannoma of family member with a unilateral schwannoma, this mutation was not confirmed to be present in the germline of this patient or in any other affected family member. Further studies are, therefore, needed to investigate a possible association between unilateral vestibular schwannomas and germline SMARCB1 mutations.
Segmental schwannomatosis
In approximately one-third of schwannomatosis patients, tumours are anatomically restricted to a single limb or a few adjacent spinal segments, thereby manifesting what appears to be a segmental form of the disease (MacCollin et al. 2005; Merker et al. 2012). Two patients have been identified with clinically symptomatic schwannomas restricted to one limb, suggestive of somatic mosaicism; however, germline LZTR1 mutations were identified in these patients (Farschtschi et al. 2016). Furthermore, microstructural magnetic resonance neurography indicated the presence of small peripheral nerve lesions (‘microlesions’) in clinically unaffected limbs of these two patients. Three additional patients were included in this study and these individuals also exhibited symptomatic schwannomas that were restricted to one limb and fascicular microlesions along the nerves of other extremities. Germline mutations in LZTR1, SMARCB1, and NF2 were not observed in these patients. However, the analysis of a schwannoma from each of these patients indicated a somatic NF2 gene mutation which is not unusual, since somatic NF2 mutations are frequently observed in schwannomas. The occurrence of asymptomatic peripheral nerve microlesions in multiple body parts in these five patients with segmental occurrence of clinically symptomatic schwannomas suggests that the disease manifestation is more widespread and not simply restricted to a single body segment (Farschtschi et al. 2016).
Models of tumorigenesis in LZTR1 mutation-positive schwannomatosis
In most schwannomas from patients harbouring LZTR1 germline mutations, the chromosome 22 is retained that harbours the germline LZTR1 mutation and an NF2 allele with a somatically acquired tumour-specific mutation. The other copy of chromosome 22 is lost or partially deleted, including the wild-type alleles of LZTR1 and NF2 (Piotrowski et al. 2014; Paganini et al. 2015b; Smith et al. 2015) (Fig. 3b). Consequently, the mutational mechanism would appear to be similar to the 4-hit/3-step model of tumorigenesis observed in schwannomas of patients with germline SMARCB1 mutations (Fig. 3a). However, since SMARCB1 is located between LZTR1 and NF2, both of which are lost by deletion or monosomy 22q, it is inevitable that one of the two SMARCB1 alleles is also lost. Hence, in practice, tumorigenesis in LZTR1-associated schwannomatosis should follow a 5-hit/3-step model (Fig. 3c). Moreover, schwannomas of patients with germline SMARCB1 mutations may follow the 5-hit/3-step model if the chromosomal region on chromosome 22 exhibiting LOH also includes LZTR1. Since LZTR1 is located proximal to SMARCB1 and NF2, the loss of one LZTR1 allele is dependent upon the extent of the region showing LOH. It is, however, unknown if the loss of one LZTR1 allele contributes to tumour growth. A mutation of the second LZTR1 allele would be necessary for the biallelic inactivation of this tumour suppressor gene. It remains to be investigated if schwannomas following either the 4-hit or the 5-hit model of tumorigenesis would exhibit differences in growth rate, proliferation index, or location.
The 3-step model of tumorigenesis appears to apply to the majority of LZTR1 mutation-positive schwannomas. However, not all schwannomas from patients with LZTR1 germline mutations exhibit this pattern of mutational events similar to that observed in some schwannomas of patients with germline SMARCB1 mutations. Instead of the biallelic NF2 inactivation implied by this model, mono-allelic NF2 inactivation was observed in 10 of the 28 (37%) schwannomas from patients with germline LZTR1 mutations analysed by Paganini et al. (2015b). In eight of these ten schwannomas, 22q LOH was detected, but an intragenic NF2 gene mutation could not be found, whereas two of ten schwannomas harboured an intragenic NF2 mutation, while heterozygosity of 22q markers was retained (Paganini et al. 2015b). Mono-allelic NF2 inactivation was detected in 3 of 11 (27%) schwannomas from LZTR1 mutation-positive patients studied by Smith et al. (2015). However, it cannot be excluded that biallelic NF2 inactivation was not detected in these studies owing to the limited resolution of the methodology employed. Further analysis would be necessary to establish whether alternative mechanisms, such as epigenetic silencing of NF2 gene expression, could be involved in the biallelic NF2 inactivation in these tumours. Indeed, hypermethylation of the NF2 promoter region has been shown to be frequent in schwannomas and could, therefore, represent an alternative mechanism of NF2 inactivation (Kino et al. 2001; Gonzalez-Gomez et al. 2003).
Predisposition to bilateral vestibular schwannomas in patients with NF2 but not schwannomatosis
NF2 mutations are detected in schwannomas irrespective of whether they occur sporadically or in the context of schwannomatosis or NF2 (Jacoby et al. 1996; Mohyuddin et al. 2002; Hadfield et al. 2010a). However, the timing of origin of the underlying mutations is different which may well impact upon the spectrum of tumours observed, in particular bilateral vestibular schwannomas (BVS). In patients with NF2, the germline NF2 gene mutation is clearly going to be present during the early embryonic development and subsets of Schwann cell precursors may be especially vulnerable to NF2 haploinsufficiency during a specific developmental time period. This could account for the high frequency (>90%) of BVS in patients with germline NF2 mutations. By contrast, in patients with sporadically occurring schwannomas and in patients with schwannomatosis, the somatic NF2 mutations occur rather later during development and neither group of patients exhibits BVS. Furthermore, the histology and growth pattern of vestibular schwannomas (VS) in NF2 are different from that of sporadic VS. VS in NF2 are multifocal, appearing “like a bunch of grapes” around the vestibular nerve (Stivaros et al. 2015). Remarkably, these multifocal tumours exhibit the same first-hit NF2 mutation but different, foci-specific second-hit NF2 mutations, indicative of the polyclonality of these tumours (Mohyuddin et al. 2002; Dewan et al. 2015). NF2-associated VS grow at multiple sites along the eighth cranial nerve and these tumour foci later merge into one tumour mass with a multi-lobulated histological pattern (Stivaros et al. 2015). By contrast, sporadic VS are usually single tumours arising from the vestibular nerve at the porus acusticus. NF2-associated VS are often more aggressive and difficult to treat than sporadic VS because of their growth pattern and the involvement of nerves. To date, seven cases of unilateral VS have been reported in patients with schwannomatosis (Smith et al. 2012a, 2015, 2016; Wu et al. 2015; Mehta et al. 2016), but neither their histology nor their growth pattern has been described.
Involvement of SMARCB1 in NF2-associated schwannomas
The main focus of this review is schwannomatosis, but the considerable clinical overlap between NF2 and schwannomatosis, as well as the shared mutational mechanisms (such as LOH of 22q), render it likely that a similar set of genes is involved in both conditions.
NF2-associated and sporadically occurring schwannomas are known to be caused by biallelic inactivation of the NF2 gene (Stemmer-Rachamimov et al. 1997; reviewed by Evans 2009). LOH of large parts of 22q is observed in 67% of NF2-associated and 56% of sporadic schwannomas (Hadfield et al. 2010a). However, copy-number neutral LOH mediated by mitotic recombination is relatively frequent, observed in 19% of NF2-associated schwannomas and in 6% of sporadic schwannomas. Nevertheless, LOH causing copy-number loss in 22q represents the most frequent second-hit mutation observed in ~50% of all NF2-associated schwannomas (Hadfield et al. 2010a) and 69% of sporadic schwannomas (Agnihotri et al. 2016). Since the LOH frequently involves large portions of chromosome 22q (Mantripragada et al. 2003; Warren et al. 2003), it is likely to be associated with the concomitant loss of several tumour suppressor genes. It has been suggested that more than two mutations are necessary for vestibular schwannoma development in NF2 patients (Woods et al. 2003). Indeed, LOH involving large parts of 22q is a key mutational step to mediate the concurrent loss of NF2, SMARCB1 and eventually also LZTR1. It should be noted that 83% of all NF2-associated schwannomas exhibit a mosaic SMARCB1 protein expression pattern resulting from intermixed tumour cells with and without SMARCB1 expression (Patil et al. 2008). This mosaic pattern has also been observed in 93% of schwannomas from patients with familial schwannomatosis and 55% of schwannomas from patients with sporadic schwannomatosis (Patil et al. 2008). However, only 5% of sporadically occurring schwannomas in patients without NF2 or schwannomatosis exhibit mosaic SMARCB1 expression. These findings suggest the frequent involvement of SMARCB1 in the pathogenesis of NF2 and schwannomatosis-associated tumours but not in sporadic schwannomas. Remarkably, the loss of LZTR1 immunostaining was not observed in seven vestibular schwannomas from seven unrelated NF2 patients (Paganini et al. 2015b).
Somatic mosaicism in schwannomatosis and unknown schwannomatosis predisposition genes
Mutation screening performed with blood-derived DNA from patients with schwannomatosis suggests that 38% of familial cases are caused by LZTR1 mutations and 48% by mutations in SMARCB1. Thus, in 14% of familial schwannomatosis cases, predisposing germline mutations have not been identified as yet. In sporadic schwannomatosis, 30% of the cases are caused by germline LZTR1 mutations and 10% by germline SMARCB1 mutations (Supp. Tables S2 and S7) (Boyd et al. 2008; Hadfield et al. 2008; Sestini et al. 2008; Rousseau et al. 2011; Smith et al. 2012b, 2014, 2015; Hutter et al. 2014; Paganini et al. 2015b). According to these assessments, the causative mutational events remain unknown in 60% of all sporadic schwannomatosis patients (Fig. 4). It is possible that a proportion of patients without intragenic germline SMARCB1 or LZTR1 mutations may harbour mutations in remote-acting regulatory regions or possess epimutations that would silence these genes, but this has not so far been investigated. Alternatively, somatic mosaicism for SMARCB1 or LZTR1 mutations could be responsible for the relatively high proportion of sporadic schwannomatosis patients without detectable germline SMARCB1 and LZTR1 mutations. However, no detectable mosaicism was found in six patients without SMARCB1 and LZTR1 mutations in their blood, as determined by analysing multiple schwannomas from these patients. In these tumours, neither SMARCB1 nor LZTR1 mutations were detected. Instead, tumour-specific somatic NF2 mutations were identified which are known to be frequent in schwannomatosis (Paganini et al. 2015b; Smith et al. 2015). Mosaicism for somatic LZTR1 or SMARCB1 mutations in patients with schwannomatosis has not so far been reported and only one case of germline (gonadal) mosaicism for a SMARCB1 mutation has been identified (Hulsebos et al. 2010). Taken together, we surmise that it is unlikely that unidentified somatic mosaicism for SMARCB1 or LZTR1 mutations would account for the high number of sporadic schwannomatosis cases without identified mutations.

Estimation of the relative proportions of familial and sporadic patients with germline mutations in LZTR1 or SMARCB1 among patients who fulfil the clinical diagnostic criteria for schwannomatosis. These estimates are derived from studies of individuals diagnosed with schwannomatosis according to clinical diagnostic criteria without preselection for those patients who have been shown to harbour different somatic NF2 gene mutations in at least two different schwannomas (Boyd et al. 2008; Hadfield et al. 2008; Sestini et al. 2008; Rousseau et al. 2011; Hutter et al. 2014; Smith et al. 2012b, 2014, 2015)
On the other hand, somatic mosaicism for an NF2 gene mutation may be much more frequent in patients considered to have schwannomatosis, but who do not carry germline SMARCB1 or LZTR1 mutations as suggested by Widemann et al. (2014). Indeed, some NF2 patients with somatic mosaicism for an NF2 gene mutation fulfil the diagnostic criteria for schwannomatosis (Plotkin et al. 2013). Further, somatic mosaicism in NF2 is not rare, since it is detected in 33% of sporadic NF2 cases with bilateral vestibular schwannomas and in up to 60% of patients with unilateral vestibular schwannoma (Moyhuddin et al. 2003; Evans et al. 2007). In patients with mosaic NF2, the ‘first-hit’ NF2 gene mutation is often present at a low level in blood cells and is sometimes only detectable in tumour tissue but not in blood cells (Evans et al. 1998, 2007; Kluwe and Mautner 1998; Kluwe et al. 2003; Paganini et al. 2014; Spyra et al. 2015; Smith et al. 2016). To distinguish between mosaic NF2 and schwannomatosis, it is necessary to perform NF2 mutation testing in more than one tumour from a given patient who does not carry a germline mutation in SMARCB1 or LZTR1 as determined by blood cell analysis. Mosaic NF2 would be confirmed if the same NF2 mutation (first-hit) is observed in two independent tumours from a given patient in addition to different tumour-specific (second-hit) mutations of the other NF2 allele. This strategy has been successfully pursued by Castellanos et al. (2015) who investigated a female patient with several painful schwannomas confined to one limb. In the schwannomas, but not the blood cells of this patient, Castellanos et al. detected a large deletion encompassing not only the NF2 gene but also large parts of chromosome 22 telomeric to NF2. Neither SMARCB1 nor LZTR1 was included within the bounds of this deletion, since these genes are located centromeric to NF2. Importantly, the deletion exhibited the same breakpoint in both schwannomas from this patient, indicating that the deletion represented the first-hit mutation. By contrast, two different intragenic NF2 gene mutations were identified which were specific to each tumour and hence represented the second-hit mutations. Consequently, the genetic diagnosis of this patient was mosaic NF2 rather than schwannomatosis. The study of Castellanos et al. (2015) demonstrates how important it is to distinguish between first-hit and second-hit mutations by the meticulous analysis of deletion breakpoints and extent of LOH to arrive at a precise diagnosis by means of molecular genetic testing.
Even if it is assumed that a certain proportion of unexplained schwannomatosis cases are caused by somatic mosaicism for an NF2 gene mutation, a subset of these unexplained cases may well be caused by mutations in a gene or genes that still remain to be identified. This subset should comprise at least 27% of all unexplained sporadic schwannomatosis cases, as concluded from the analysis of Piotrowski et al. (2014). To minimize the confounding influence of patients with somatic mosaicism for NF2 gene mutations among patients with suspected schwannomatosis, Piotrowski et al. (2014) included in their analysis of SMARCB1 mutation-negative patients only those individuals who exhibited different somatic NF2 gene mutations and LOH of chromosome 22q in at least two different tumours analysed. They were able to detect LZTR1 mutations in all six familial schwannomatosis cases analysed and in 73% of sporadic cases who were SMARCB1 mutation-negative. Consequently, 27% of sporadic schwannomatosis patients who do not exhibit germline NF2, SMARCB1, and LZTR1 mutations, but who do display tumour-specific biallelic NF2 inactivation, could, in principle, be caused by mutations in hitherto unidentified gene(s). Since Piotrowski et al. (2014) analysed a preselected group of patients and did not include those patients who, although fulfilling the diagnostic criteria for schwannomatosis, lacked biallelic NF2 inactivation in their tumours, the proportion of unexplained sporadic schwannomatosis cases caused by mutations in other as yet unidentified genes could be even higher. Mutations in these hitherto unidentified genes may also account for familial cases, since Paganini et al. (2015b) investigated six cases with familial schwannomatosis, who were negative for both SMARCB1 and LZTR1 mutations. It is possible that some of the unexplained cases are caused by gross rearrangements, multi-exon deletions, or duplications of SMARCB1 or LZTR1, but these lesions are usually much rarer than intragenic mutations and they have not been detected by any of the MLPA or SNP array analyses so far performed (Hadfield et al. 2010a; Smith et al. 2015). Taken together, we conclude that there may well be further schwannomatosis predisposition genes that still remain to be identified.
One possible candidate has been put forward by Zhang et al. (2014) who identified a heterozygous missense mutation, c.622G > C, p.Asp208His, in the COQ6 gene on chromosome 14q24.3 segregating with the disease in a family with schwannomatosis. The affected family members did not appear to harbour germline mutations in SMARCB1, LZTR1, or NF2; nor were somatic mutations of these genes detected in two schwannomas from two family members. Furthermore, immunohistochemical staining indicated normal protein expression levels of SMARCB1, LZTR1, and NF2 in both schwannomas. The latter finding is unusual, since mosaic SMARCB1 expression has been observed in 93% of schwannomas derived from patients with familial schwannomatosis (Patil et al. 2008). However, Zhang et al. (2014) did not identify biallelic inactivation of COQ6 gene in schwannomas of this family. Although the deleterious effects of the COQ6 missense mutation were validated by its lack of complementation in a coq6-deficient yeast mutant, there was no evidence for either a dominant-negative effect or a toxic gain of function of the missense COQ6 variant detected; hence, the mechanism of tumorigenesis in this schwannomatosis family remains unexplained. As opined by Trevisson et al. (2015), further studies are necessary to ascertain whether COQ6 might play a role in the etiology of schwannomatosis.
SMARCB1, LZTR1, and NF2 clearly function as classical tumour suppressor genes, and hence, it is not unreasonable to suppose that other schwannomatosis predisposition genes might also act in an onco-suppressive manner. Although it cannot be excluded that another schwannomatosis predisposition gene is located elsewhere in the genome, it is tempting to speculate that it would be located on chromosome 22, because in schwannomas, this chromosome is frequently affected by LOH. Consequently, the underlying model of tumorigenesis in these unexplained cases could also include three mutational steps similar to the above-mentioned model that accounts for SMARCB1 and LZTR1 mutation-positive tumours. Pinto et al. (2014) performed a detailed annotation of the chromosome 22-encoded proteome and identified protein products encoded by 367 genes. Among them were proteins encoded by 22 cancer-associated genes—these may represent good candidates for further schwannomatosis predisposition genes. Finally, a number of other candidate genes may be found among the genes shown by Agnihotri et al. (2016) to be somatically mutated in sporadic schwannomas (see below).
The comprehensive genome-wide analysis of the mutational landscape of somatic mutations in schwannomas of patients with schwannomatosis has not so far been performed but would improve our understanding of the tumorigenic process in schwannomatosis. Such an analysis could also help to identify altered cellular pathways that would reveal the involvement of yet unidentified schwannomatosis predisposition genes. In a recent study, Agnihotri et al. (2016) performed whole-exome sequencing of 13 cranial and 13 spinal schwannomas occurring as sporadic tumours in patients without NF2 or schwannomatosis. No germline mutations or deletions were detected for NF2, LZTR1, SMARCB1, or SMARCE1. However, these authors identified 441 somatic single-nucleotide variants located in the exomes of these 26 schwannoma samples, corresponding to 0.16 mutations per coding megabase. This number of mutations is quite low, and comparable to other low-mutation-rate tumours, such as Ewing and rhabdoid sarcomas (Lawrence et al. 2013). Agnihotri et al. (2016) observed somatic NF2 mutations and/or 22q loss in 96/125 samples (77%). Somatic LZTR1 mutations were identified in 2/26 schwannomas (8%), but somatic SMARCB1 mutations were not observed. Recurrent but low-frequency mutations were identified in eight different genes, and Agnihotri et al. validated their findings by targeted sequencing of these genes in an additional 99 sporadic schwannomas. They detected mutations in DDR1, encoding a receptor tyrosine kinase activated in lung cancer and other tumour types (Ambrogio et al. 2016), in 14/125 (11%) of the schwannomas. Other recurrently mutated genes were TSC1 (mutated in 9% of tumours), CAST (8%), ALPK2 (8%), TSC2 (7%), and TAB 3 (3%). Furthermore, 29% (37/125) of all schwannomas harboured inactivating mutations in either ARID1A or ARID1B, which along with SMARCB1, encode proteins of the SWI/SNF chromatin-remodeling complex (Nie et al. 2000). Agnihotri et al. (2016) also discovered a somatic recurrent in-frame fusion involving SH3PXD2A and HTRA1, arising through a balanced translocation on chromosome 10q in 12/25 (10%) of schwannomas analysed by RNA-sequencing. Expression of the SH3PXD2A-HTRA1 fusion in Schwann cells and a schwannoma cell line resulted in increased cell proliferation, invasive growth, and in vivo tumorigenesis. Whether this somatic fusion also contributes to tumorigenesis in the context of schwannomatosis remains to be investigated.
Conclusion and perspective
Germline mutations in either SMARCB1 or LZTR1 predispose to the development of multiple schwannomas in patients with schwannomatosis. In addition, the NF2 gene is involved in schwannomatosis-associated tumorigenesis, since it is frequently inactivated in schwannomas.
However, determining the complete mutational spectrum of all three genes, LZTR1, SMARCB1, and NF2, by comprehensive mutation testing as suggested in Fig. 5, has not so far been attempted. Such comprehensive testing would help to classify schwannomas in terms of the number of mutational hits and the biallelic or mono-allelic inactivation of these tumour suppressor genes (TSGs). A genetic classification of schwannomas might correlate with tumour growth patterns, physical location, and response to therapy.

Comprehensive mutation analysis of all three genes, LZTR1, SMARCB1, and NF2, in patients with schwannomatosis should be performed to identify the complete mutational spectra and the number of mutational hits that affect these genes. This comprehensive testing may help to classify the tumours according to their mutation-profile. The mutation analysis should also include methods, such as next-generation sequencing, which are well suited to detect somatic mosaicism with mutant cells present in low proportions. This approach should identify tumour heterogeneity and help to distinguish between mosaic NF2 and schwannomatosis, since some NF2 patients with somatic mosaicism for an NF2 gene mutation fulfil the diagnostic criteria for schwannomatosis (Plotkin et al. 2013)
From the data so far available, it is already clear that at least two and sometimes three TSGs are inactivated in schwannomas from patients with schwannomatosis, an event which is frequently mediated by LOH, including large portions of 22q. The possible involvement of further TSGs in predisposition to schwannomatosis would serve to render the mutational model even more complex.
Recurrent cancer-associated deletions involving several linked TSGs have also been observed on chromosomes 7q, 8p, and 9p in different types of tumour (Krimpenfort et al. 2007; Asou et al. 2009; Solimini et al. 2012; Xue et al. 2012; Kotini et al. 2015). The concomitant loss of several TSGs as a consequence of a single mutational event, such as a gross chromosomal deletion, may disclose an interactive or cooperative effect in the sense that the biallelic inactivation and haploinsufficiency of several TSGs collectively promote tumorigenesis of the affected cells, since several signalling pathways and other cellular processes are concurrently disturbed. Schwannomatosis serves as a paradigm for such cooperative tumorigenic effects mediated by the concomitant loss of several linked TSGs.
References
Agnihotri S, Jalali S, Wilson MR, Danesh A, Li M, Klironomos G, Krieger JR, Mansouri A, Khan O, Mamatjan Y, Landon-Brace N, Tung T, Dowar M, Li T, Bruce JP, Burrell KE, Tonge PD, Alamsahebpour A, Krischek B, Agarwalla PK, Bi WL, Dunn IF, Beroukhim R, Fehlings MG, Bril V, Pagnotta SM, Iavarone A, Pugh TJ, Aldape KD, Zadeh G (2016) The genomic landscape of schwannoma. Nat Genet 48:1339–1348. doi:10.1038/ng.3688
Allen MD, Freund SM, Zinzalla G, Bycroft M (2015) The SWI/SNF subunit INI1 contains an N-terminal winged helix DNA binding domain that is a target for mutations in schwannomatosis. Structure 23:1344–1349. doi:10.1016/j.str.2015.04.021
Ambrogio C, Gómez-López G, Falcone M, Vidal A, Nadal E, Crosetto N, Blasco RB, Fernández-Marcos PJ, Sánchez-Céspedes M, Ren X, Wang Z, Ding K, Hidalgo M, Serrano M, Villanueva A, Santamaría D, Barbacid M (2016) Combined inhibition of DDR1 and Notch signaling is a therapeutic strategy for KRAS-driven lung adenocarcinoma. Nat Med 22:270–277. doi:10.1038/nm.4041
Ammerlaan AC, Ararou A, Houben MP, Baas F, Tijssen CC, Teepen JL, Wesseling P, Hulsebos TJ (2008) Long-term survival and transmission of INI1-mutation via nonpenetrant males in a family with rhabdoid tumour predisposition syndrome. Br J Cancer 98:474–479. doi:10.1038/sj.bjc.6604156
Antinheimo J, Sankila R, Carpén O, Pukkala E, Sainio M, Jääskeläinen J (2000) Population-based analysis of sporadic and type 2 neurofibromatosis-associated meningiomas and schwannomas. Neurology 54:71–76
Asai K, Tani S, Mineharu Y, Tsurusaki Y, Imai Y, Agawa Y, Iwaki K, Matsumoto N, Sakai N (2015) Familial schwannomatosis with a germline mutation of SMARCB1 in Japan. Brain Tumor Pathol 32:216–220. doi:10.1007/s10014-015-0213-9
Asou H, Matsui H, Ozaki Y, Nagamachi A, Nakamura M, Aki D, Inaba T (2009) Identification of a common microdeletion cluster in 7q21.3 subband among patients with myeloid leukemia and myelodysplastic syndrome. Biochem Biophys Res Commun 383:245–251. doi:10.1016/j.bbrc.2009.04.004
Bacci C, Sestini R, Provenzano A, Paganini I, Mancini I, Porfirio B, Vivarelli R, Genuardi M, Papi L (2010) Schwannomatosis associated with multiple meningiomas due to a familial SMARCB1 mutation. Neurogenetics 11:73–80. doi:10.1007/s10048-009-0204-2
Biegel JA, Zhou JY, Rorke LB, Stenstrom C, Wainwright LM, Fogelgren B (1999) Germ-line and acquired mutations of INI1 in atypical teratoid and rhabdoid tumors. Cancer Res 59:74–79
Biegel JA, Tan L, Zhang F, Wainwright L, Russo P, Rorke LB (2002) Alterations of the hSNF5/INI1 gene in central nervous system atypical teratoid/rhabdoid tumors and renal and extrarenal rhabdoid tumors. Clin Cancer Res 8:3461–3467
Biegel JA, Busse TM, Weissman BE (2014) SWI/SNF chromatin remodeling complexes and cancer. Am J Med Genet C Semin Med Genet 166C:350–366. doi:10.1002/ajmg.c.31410
Blakeley JO, Plotkin SR (2016) Therapeutic advances for the tumors associated with neurofibromatosis type 1, type 2, and schwannomatosis. Neuro Oncol 18:624–638. doi:10.1093/neuonc/nov200
Bosch MM, Boltshauser E, Harpes P, Landau K (2006) Ophthalmologic findings and long-term course in patients with neurofibromatosis type 2. Am J Ophthalmol 141:1068–1077. doi:10.1016/j.ajo.2005.12.042
Bourdeaut F, Lequin D, Brugières L, Reynaud S, Dufour C, Doz F, André N, Stephan JL, Pérel Y, Oberlin O, Orbach D, Bergeron C, Rialland X, Fréneaux P, Ranchere D, Figarella-Branger D, Audry G, Puget S, Evans DG, Pinas JC, Capra V, Mosseri V, Coupier I, Gauthier-Villars M, Pierron G, Delattre O (2011) Frequent hSNF5/INI1 germline mutations in patients with rhabdoid tumor. Clin Cancer Res 17:31–38. doi:10.1158/1078-0432.CCR-10-1795
Boyd C, Smith MJ, Kluwe L, Balogh A, MacCollin M, Plotkin SR (2008) Alterations in the SMARCB1 (INI1) tumor suppressor gene in familial schwannomatosis. Clin Genet 74:358–366. doi:10.1111/j.1399-0004.2008.01060.x
Bruggers CS, Bleyl SB, Pysher T, Barnette P, Afify Z, Walker M, Biegel JA (2011) Clinicopathologic comparison of familial versus sporadic atypical teratoid/rhabdoid tumors (AT/RT) of the central nervous system. Pediatr Blood Cancer 56:1026–1031. doi:10.1002/pbc.22757
Buckley PG, Mantripragada KK, Díaz de Ståhl T, Piotrowski A, Hansson CM, Kiss H, Vetrie D, Ernberg IT, Nordenskjöld M, Bolund L, Sainio M, Rouleau GA, Niimura M, Wallace AJ, Evans DG, Grigelionis G, Menzel U, Dumanski JP (2005) Identification of genetic aberrations on chromosome 22 outside the NF2 locus in schwannomatosis and neurofibromatosis type 2. Hum Mutat 26:540–549. doi:10.1002/humu.20255
Carter JM, O’Hara C, Dundas G, Gilchrist D, Collins MS, Eaton K, Judkins AR, Biegel JA, Folpe AL (2012) Epithelioid malignant peripheral nerve sheath tumor arising in a schwannoma, in a patient with “neuroblastoma-like” schwannomatosis and a novel germline SMARCB1 mutation. Am J Surg Pathol 36:154–160. doi:10.1097/PAS.0b013e3182380802
Castellanos E, Bielsa I, Carrato C, Rosas I, Solanes A, Hostalot C, Amilibia E, Prades J, Roca-Ribas F, Lázaro C, Blanco I, Serra E; NF2 Multidisciplinary Clinics HUGTiP-ICO-IMPPC (2015) Segmental neurofibromatosis type 2: discriminating two hit from four hit in a patient presenting multiple schwannomas confined to one limb. BMC Med Genomics 8:2. doi:10.1186/s12920-015-0076-2
Cheng SW, Davies KP, Yung E, Beltran RJ, Yu J, Kalpana GV (1999) c-MYC interacts with INI1/hSNF5 and requires the SWI/SNF complex for transactivation function. Nat Genet 22:102–105. doi:10.1038/8811
Christiaans I, Kenter SB, Brink HC, van Os TA, Baas F, van den Munckhof P, Kidd AM, Hulsebos TJ (2011) Germline SMARCB1 mutation and somatic NF2 mutations in familial multiple meningiomas. J Med Genet 48:93–97. doi:10.1136/jmg.2010.082420
Dewan R, Pemov A, Kim HJ, Morgan KL, Vasquez RA, Chittiboina P, Wang X, Chandrasekharappa SC, Ray-Chaudhury A, Butman JA, Stewart DR, Asthagiri AR (2015) Evidence of polyclonality in neurofibromatosis type 2-associated multilobulated vestibular schwannomas. Neuro Oncol 17:566–573. doi:10.1093/neuonc/nou317
Doan DN, Veal TM, Yan Z, Wang W, Jones SN, Imbalzano AN (2004) Loss of the INI1 tumor suppressor does not impair the expression of multiple BRG1-dependent genes or the assembly of SWI/SNF enzymes. Oncogene 23:3462–3473. doi:10.1038/sj.onc.1207472
Dow G, Biggs N, Evans G, Gillespie J, Ramsden R, King A (2005) Spinal tumors in neurofibromatosis type 2. Is emerging knowledge of genotype predictive of natural history? J Neurosurg Spine 2:574–579. doi:10.3171/spi.2005.2.5.0574
Eaton KW, Tooke LS, Wainwright LM, Judkins AR, Biegel JA (2011) Spectrum of SMARCB1/INI1 mutations in familial and sporadic rhabdoid tumors. Pediatr Blood Cancer 56:7–15. doi:10.1002/pbc.22831
Evans DG (2009) Neurofibromatosis type 2 (NF2): a clinical and molecular review. Orphanet J Rare Dis 4:16. doi:10.1186/1750-1172-4-16
Evans DG, Huson SM, Donnai D, Neary W, Blair V, Newton V, Harris R (1992) A clinical study of type 2 neurofibromatosis. Q J Med 84:603–618
Evans DG, Mason S, Huson SM, Ponder M, Harding AE, Strachan T (1997) Spinal and cutaneous schwannomatosis is a variant form of type 2 neurofibromatosis: a clinical and molecular study. J Neurol Neurosurg Psychiatry 62:361–366
Evans DG, Wallace AJ, Wu CL, Trueman L, Ramsden RT, Strachan T (1998) Somatic mosaicism: a common cause of classic disease in tumor-prone syndromes? Lessons from type 2 neurofibromatosis. Am J Hum Genet 63:727–736. doi:10.1086/512074
Evans DG, Lye R, Neary W, Black G, Strachan T, Wallace A, Ramsden RT (1999) Probability of bilateral disease in people presenting with a unilateral vestibular schwannoma. J Neurol Neurosurg Psychiatry 66:764–767
Evans DG, Baser ME, O’Reilly B, Rowe J, Gleeson M, Saeed S, King A, Huson SM, Kerr R, Thomas N, Irving R, MacFarlane R, Ferner R, McLeod R, Moffat D, Ramsden R (2005) Management of the patient and family with neurofibromatosis 2: a consensus conference statement. Br J Neurosurg 19:5–12. doi:10.1080/02688690500081206
Evans DG, Ramsden RT, Shenton A, Gokhale C, Bowers NL, Huson SM, Pichert G, Wallace A (2007) Mosaicism in neurofibromatosis type 2: an update of risk based on uni/bilaterality of vestibular schwannoma at presentation and sensitive mutation analysis including multiple ligation-dependent probe amplification. J Med Genet 44:424–428. doi:10.1136/jmg.2006.047753
Evans DG, Ramsden RT, Shenton A, Gokhale C, Bowers N, Huson SM, Wallace AJ (2008) What are the implications in individuals with unilateral vestibular schwannoma and other neurogenic tumors? J Neurosurg 108:92–96. doi:10.3171/JNS/2008/108/01/0092
Evans DG, Huson SM, Birch JM (2012) Malignant peripheral nerve sheath tumours in inherited disease. Clin Sarcoma Res 2:17. doi:10.1186/2045-3329-2-17
Farschtschi S, Mautner VF, Pham M, Nguyen R, Kehrer-Sawatzki H, Hutter S, Friedrich RE, Schulz A, Morrison H, Jones DT, Bendszus M, Bäumer P (2016) Multifocal nerve lesions and LZTR1 germline mutations in segmental schwannomatosis. Ann Neurol 80:625–628. doi:10.1002/ana.24753
Fisher LM, Doherty JK, Lev MH, Slattery WH 3rd (2007) Distribution of nonvestibular cranial nerve schwannomas in neurofibromatosis 2. Otol Neurotol 28:1083–1090. doi:10.1097/MAO.0b013e31815a8411
Forbes SA, Bhamra G, Bamford S, Dawson E, Kok C, Clements J, Menzies A, Teague JW, Futreal PA, Stratton MR (2008) The Catalogue of Somatic Mutations in Cancer (COSMIC). Curr Protoc Hum Genet Chapter 10:Unit 10.11. doi:10.1002/0471142905.hg1011s57
Frattini V, Trifonov V, Chan JM, Castano A, Lia M, Abate F, Keir ST, Ji AX, Zoppoli P, Niola F, Danussi C, Dolgalev I, Porrati P, Pellegatta S, Heguy A, Gupta G, Pisapia DJ, Canoll P, Bruce JN, McLendon RE, Yan H, Aldape K, Finocchiaro G, Mikkelsen T, Privé GG, Bigner DD, Lasorella A, Rabadan R, Iavarone A (2013) The integrated landscape of driver genomic alterations in glioblastoma. Nat Genet 45:1141–1149. doi:10.1038/ng.2734
Garcia-Linares C, Fernández-Rodríguez J, Terribas E, Mercadé J, Pros E, Benito L, Benavente Y, Capellà G, Ravella A, Blanco I, Kehrer-Sawatzki H, Lázaro C, Serra E (2011) Dissecting loss of heterozygosity (LOH) in neurofibromatosis type 1-associated neurofibromas: importance of copy neutral LOH. Hum Mutat 32:78–90. doi:10.1002/humu.21387
Gigante L, Paganini I, Frontali M, Ciabattoni S, Sangiuolo FC, Papi L (2016) Rhabdoid tumor predisposition syndrome caused by SMARCB1 constitutional deletion: prenatal detection of new case of recurrence in siblings due to gonadal mosaicism. Fam Cancer 15:123–126. doi:10.1007/s10689-015-9836-6
Gonzalez-Gomez P, Bello MJ, Alonso ME, Lomas J, Arjona D, Campos JM, Vaquero J, Isla A, Lassaletta L, Gutierrez M, Sarasa JL, Rey JA (2003) CpG island methylation in sporadic and neurofibromatis type 2-associated schwannomas. Clin Cancer Res 9:5601–5606
Gonzalvo A, Fowler A, Cook RJ, Little NS, Wheeler H, McDonald KL, Biggs MT (2011) Schwannomatosis, sporadic schwannomatosis, and familial schwannomatosis: a surgical series with long-term follow-up. J Neurosurg 114:756–762. doi:10.3171/2010.8.JNS091900
Gossai N, Biegel JA, Messiaen L, Berry SA, Moertel CL (2015) Report of a patient with a constitutional missense mutation in SMARCB1, Coffin-Siris phenotype, and schwannomatosis. Am J Med Genet A 167A:3186–3191. doi:10.1002/ajmg.a.37356
Haberler C, Laggner U, Slavc I, Czech T, Ambros IM, Ambros PF, Budka H, Hainfellner JA (2006) Immunohistochemical analysis of INI1 protein in malignant pediatric CNS tumors: lack of INI1 in atypical teratoid/rhabdoid tumors and in a fraction of primitive neuroectodermal tumors without rhabdoid phenotype. Am J Surg Pathol 30:1462–1468. doi:10.1097/01.pas.0000213329.71745.ef
Hadfield KD, Newman WG, Bowers NL, Wallace A, Bolger C, Colley A, McCann E, Trump D, Prescott T, Evans DG (2008) Molecular characterisation of SMARCB1 and NF2 in familial and sporadic schwannomatosis. J Med Genet 45:332–339. doi:10.1136/jmg.2007.056499
Hadfield KD, Smith MJ, Urquhart JE, Wallace AJ, Bowers NL, King AT, Rutherford SA, Trump D, Newman WG, Evans DG (2010a) Rates of loss of heterozygosity and mitotic recombination in NF2 schwannomas, sporadic vestibular schwannomas and schwannomatosis schwannomas. Oncogene 29:6216–6221. doi:10.1038/onc.2010.363
Hadfield KD, Smith MJ, Trump D, Newman WG, Evans DG (2010b) SMARCB1 mutations are not a common cause of multiple meningiomas. J Med Genet 47:567–568. doi:10.1136/jmg.2009.075721
Hart J, Gardner JM, Edgar M, Weiss SW (2016) Epithelioid schwannomas: an analysis of 58 cases including atypical variants. Am J Surg Pathol 40:704–713. doi:10.1097/PAS.0000000000000589
Hollmann TJ, Hornick JL (2011) INI1-deficient tumors: diagnostic features and molecular genetics. Am J Surg Pathol 35:e47–e63. doi:10.1097/PAS.0b013e31822b325b
Hoot AC, Russo P, Judkins AR, Perlman EJ, Biegel JA (2004) Immunohistochemical analysis of hSNF5/INI1 distinguishes renal and extra-renal malignant rhabdoid tumors from other pediatric soft tissue tumors. Am J Surg Pathol 28:1485–1491
Hulsebos TJ, Plomp AS, Wolterman RA, Robanus-Maandag EC, Baas F, Wesseling P (2007) Germline mutation of INI1/SMARCB1 in familial schwannomatosis. Am J Hum Genet 80:805–810. doi:10.1086/513207
Hulsebos TJ, Kenter SB, Jakobs ME, Baas F, Chong B, Delatycki MB (2010) SMARCB1/INI1 maternal germ line mosaicism in schwannomatosis. Clin Genet 77:86–91. doi:10.1111/j.1399-0004.2009.01249
Hulsebos TJ, Kenter S, Verhagen WI, Baas F, Flucke U, Wesseling P (2014a) Premature termination of SMARCB1 translation may be followed by reinitiation in schwannomatosis-associated schwannomas, but results in absence of SMARCB1 expression in rhabdoid tumors. Acta Neuropathol 128:439–448. doi:10.1007/s00401-014-1281-3
Hulsebos TJ, Kenter S, Siebers-Renelt U, Hans V, Wesseling P, Flucke U (2014b) SMARCB1 involvement in the development of leiomyoma in a patient with schwannomatosis. Am J Surg Pathol 38:421–425. doi:10.1097/PAS.0000000000000110
Hulsebos TJ, Kenter S, Baas F, Nannenberg EA, Bleeker FE, van Minkelen R, van den Ouweland AM, Wesseling P, Flucke U (2016) Type 1 papillary renal cell carcinoma in a patient with schwannomatosis: mosaic versus loss of SMARCB1 expression in respectively schwannoma and renal tumor cells. Genes Chromosomes Cancer 55:350–354. doi:10.1002/gcc.22338
Hutter S, Piro RM, Reuss DE, Hovestadt V, Sahm F, Farschtschi S, Kehrer-Sawatzki H, Wolf S, Lichter P, von Deimling A, Schuhmann MU, Pfister SM, Jones DT, Mautner VF (2014) Whole exome sequencing reveals that the majority of schwannomatosis cases remain unexplained after excluding SMARCB1 and LZTR1 germline variants. Acta Neuropathol 128:449–452. doi:10.1007/s00401-014-1311-1
Jackson EM, Sievert AJ, Gai X, Hakonarson H, Judkins AR, Tooke L, Perin JC, Xie H, Shaikh TH, Biegel JA (2009) Genomic analysis using high-density single nucleotide polymorphism-based oligonucleotide arrays and multiplex ligation-dependent probe amplification provides a comprehensive analysis of INI1/SMARCB1 in malignant rhabdoid tumors. Clin Cancer Res 15:1923–1930. doi:10.1158/1078-0432.CCR-08-2091
Jacoby LB, MacCollin M, Barone R, Ramesh V, Gusella JF (1996) Frequency and distribution of NF2 mutations in schwannomas. Genes Chromosomes Cancer 17:45–55. doi:10.1002/(SICI)1098-2264(199609)17:1<45:AID-GCC7>3.0.CO;2-2
Jacoby LB, Jones D, Davis K, Kronn D, Short MP, Gusella J, MacCollin M (1997) Molecular analysis of the NF2 tumor-suppressor gene in schwannomatosis. Am J Hum Genet 61:1293–1302. doi:10.1086/301633
Jagani Z, Mora-Blanco EL, Sansam CG, McKenna ES, Wilson B, Chen D, Klekota J, Tamayo P, Nguyen PT, Tolstorukov M, Park PJ, Cho YJ, Hsiao K, Buonamici S, Pomeroy SL, Mesirov JP, Ruffner H, Bouwmeester T, Luchansky SJ, Murtie J, Kelleher JF, Warmuth M, Sellers WR, Roberts CW, Dorsch M (2010) Loss of the tumor suppressor Snf5 leads to aberrant activation of the Hedgehog-Gli pathway. Nat Med 16:1429–1433. doi:10.1038/nm.2251
Janson K, Nedzi LA, David O, Schorin M, Walsh JW, Bhattacharjee M, Pridjian G, Tan L, Judkins AR, Biegel JA (2006) Predisposition to atypical teratoid/rhabdoid tumor due to an inherited INI1 mutation. Pediatr Blood Cancer 47:279–284. doi:10.1002/pbc.20622
Judkins AR, Mauger J, Ht A, Rorke LB, Biegel JA (2004) Immunohistochemical analysis of hSNF5/INI1 in pediatric CNS neoplasms. Am J Surg Pathol 28:644–650
Kalimuthu SN, Chetty R (2016) Gene of the month: sMARCB1. J Clin Pathol 69:484–489. doi:10.1136/jclinpath-2016-203650
Kaufman DL, Heinrich BS, Willett C, Perry A, Finseth F, Sobel RA, MacCollin M (2003) Somatic instability of the NF2 gene in schwannomatosis. Arch Neurol 60:1317–1320. doi:10.1001/archneur.60.9.1317
Kehrer-Sawatzki H, Cooper DN (2008) Mosaicism in sporadic neurofibromatosis type 1: variations on a theme common to other hereditary cancer syndromes? J Med Genet 45:622–631. doi:10.1136/jmg.2008.059329
Kino T, Takeshima H, Nakao M, Nishi T, Yamamoto K, Kimura T, Saito Y, Kochi M, Kuratsu J, Saya H, Ushio Y (2001) Identification of the cis-acting region in the NF2 gene promoter as a potential target for mutation and methylation-dependent silencing in schwannoma. Genes Cells 6:441–454
Kluwe L, Mautner VF (1998) Mosaicism in sporadic neurofibromatosis 2 patients. Hum Mol Genet 7:2051–2055
Kluwe L, Mautner V, Heinrich B, Dezube R, Jacoby LB, Friedrich RE, MacCollin M (2003) Molecular study of frequency of mosaicism in neurofibromatosis 2 patients with bilateral vestibular schwannomas. J Med Genet 40:109–114
Knudson AG Jr (1971) Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci USA 68:820–823
Koontz NA, Wiens AL, Agarwal A, Hingtgen CM, Emerson RE, Mosier KM (2013) Schwannomatosis: the overlooked neurofibromatosis? Am J Roentgenol 200:W646–W653. doi:10.2214/AJR.12.8577
Kordes U, Gesk S, Frühwald MC, Graf N, Leuschner I, Hasselblatt M, Jeibmann A, Oyen F, Peters O, Pietsch T, Siebert R, Schneppenheim R (2010) Clinical and molecular features in patients with atypical teratoid rhabdoid tumor or malignant rhabdoid tumor. Genes Chromosomes Cancer 49:176–181. doi:10.1002/gcc.20729
Kotini AG, Chang CJ, Boussaad I, Delrow JJ, Dolezal EK, Nagulapally AB, Perna F, Fishbein GA, Klimek VM, Hawkins RD, Huangfu D, Murry CE, Graubert T, Nimer SD, Papapetrou EP (2015) Functional analysis of a chromosomal deletion associated with myelodysplastic syndromes using isogenic human induced pluripotent stem cells. Nat Biotechnol 33:646–655. doi:10.1038/nbt.3178
Krimpenfort P, Ijpenberg A, Song JY, van der Valk M, Nawijn M, Zevenhoven J, Berns A (2007) p15Ink4b is a critical tumour suppressor in the absence of p16Ink4a. Nature 448:943–946. doi:10.1038/nature06084
Kuwahara Y, Wei D, Durand J, Weissman BE (2013) SNF5 reexpression in malignant rhabdoid tumors regulates transcription of target genes by recruitment of SWI/SNF complexes and RNAPII to the transcription start site of their promoters. Mol Cancer Res 11:251–260. doi:10.1158/1541-7786.MCR-12-0390
Lau CS, Mahendraraj K, Chamberlain RS (2015) Atypical teratoid rhabdoid tumors: a population-based clinical outcomes study involving 174 patients from the Surveillance, Epidemiology, and End Results database (1973-2010). Cancer Manag Res 7:301–309. doi:10.2147/CMAR.S88561
Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH, Roberts SA, Kiezun A, Hammerman PS, McKenna A, Drier Y, Zou L, Ramos AH, Pugh TJ, Stransky N, Helman E, Kim J, Sougnez C, Ambrogio L, Nickerson E, Shefler E, Cortés ML, Auclair D, Saksena G, Voet D, Noble M, DiCara D, Lin P, Lichtenstein L, Heiman DI, Fennell T, Imielinski M, Hernandez B, Hodis E, Baca S, Dulak AM, Lohr J, Landau DA, Wu CJ, Melendez-Zajgla J, Hidalgo-Miranda A, Koren A, McCarroll SA, Mora J, Lee RS, Crompton B, Onofrio R, Parkin M, Winckler W, Ardlie K, Gabriel SB, Roberts CW, Biegel JA, Stegmaier K, Bass AJ, Garraway LA, Meyerson M, Golub TR, Gordenin DA, Sunyaev S, Lander ES, Getz G (2013) Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 499:214–218. doi:10.1038/nature12213
Lee HY, Yoon CS, Sevenet N, Rajalingam V, Delattre O, Walford NQ (2002) Rhabdoid tumor of the kidney is a component of the rhabdoid predisposition syndrome. Pediatr Dev Pathol 5:395–399. doi:10.1007/s10024-001-0259-z
Li P, Zhao F, Zhang J, Wang Z, Wang X, Wang B, Yang Z, Yang J, Gao Z, Liu P (2016) Clinical features of spinal schwannomas in 65 patients with schwannomatosis compared with 831 with solitary schwannomas and 102 with neurofibromatosis Type 2: a retrospective study at a single institution. J Neurosurg Spine 24:145–154. doi:10.3171/2015.3.SPINE141145
MacCollin M, Woodfin W, Kronn D, Short MP (1996) Schwannomatosis: a clinical and pathologic study. Neurology 46:1072–1079
MacCollin M, Willett C, Heinrich B, Jacoby LB, Acierno JS Jr, Perry A, Louis DN (2003) Familial schwannomatosis: exclusion of the NF2 locus as the germline event. Neurology 60:1968–1974
MacCollin M, Chiocca EA, Evans DG, Friedman JM, Horvitz R, Jaramillo D, Lev M, Mautner VF, Niimura M, Plotkin SR, Sang CN, Stemmer-Rachamimov A, Roach ES (2005) Diagnostic criteria for schwannomatosis. Neurology 64:1838–1845. doi:10.1212/01.WNL.0000163982.78900.AD
Makishima H, Maciejewski JP (2011) Pathogenesis and consequences of uniparental disomy in cancer. Clin Cancer Res 17:3913–3923. doi:10.1158/1078-0432.CCR-10-2900
Mantripragada KK, Buckley PG, Benetkiewicz M, De Bustos C, Hirvelä C, Jarbo C, Bruder CE, Wensman H, Mathiesen T, Nyberg G, Papi L, Collins VP, Ichimura K, Evans G, Dumanski JP (2003) High-resolution profiling of an 11 Mb segment of human chromosome 22 in sporadic schwannoma using array-CGH. Int J Oncol 22:615–622
Margol AS, Judkins AR (2014) Pathology and diagnosis of SMARCB1-deficient tumors. Cancer Genet 207:358–364. doi:10.1016/j.cancergen.2014.07.004
Masliah-Planchon J, Bièche I, Guinebretière JM, Bourdeaut F, Delattre O (2015) SWI/SNF chromatin remodeling and human malignancies. Annu Rev Pathol 10:145–171. doi:10.1146/annurev-pathol-012414-040445
Mautner VF, Tatagiba M, Lindenau M, Fünsterer C, Pulst SM, Baser ME, Kluwe L, Zanella FE (1995) Spinal tumors in patients with neurofibromatosis type 2: MR imaging study of frequency, multiplicity, and variety. Am J Roentgenol 165:951–955
Mautner VF, Lindenau M, Baser ME, Hazim W, Tatagiba M, Haase W, Samii M, Wais R, Pulst SM (1996) The neuroimaging and clinical spectrum of neurofibromatosis 2. Neurosurgery 38:880–885
Mautner VF, Lindenau M, Baser ME, Kluwe L, Gottschalk J (1997) Skin abnormalities in neurofibromatosis 2. Arch Dermatol 133:1539–1543
McMenamin ME, Fletcher CD (2001) Expanding the spectrum of malignant change in schwannomas: epithelioid malignant change, epithelioid malignant peripheral nerve sheath tumor, and epithelioid angiosarcoma: a study of 17 cases. Am J Surg Pathol 25:13–25
Mehta GU, Feldman MJ, Wang H, Ding D, Chittiboina P (2016) Unilateral vestibular schwannoma in a patient with schwannomatosis in the absence of LZTR1 mutation. J Neurosurg 5:1–3. doi:10.3171/2015.11.JNS151766
Melean G, Velasco A, Hernández-Imaz E, Rodríguez-Álvarez FJ, Martín Y, Valero A, Hernández-Chico C (2012) RNA-based analysis of two SMARCB1 mutations associated with familial schwannomatosis with meningiomas. Neurogenetics 13:267–274. doi:10.1007/s10048-012-0335-8
Merker VL, Esparza S, Smith MJ, Stemmer-Rachamimov A, Plotkin SR (2012) Clinical features of schwannomatosis: a retrospective analysis of 87 patients. Oncologist 17:1317–1322. doi:10.1634/theoncologist.2012-0162
Mohyuddin A, Neary WJ, Wallace A, Wu CL, Purcell S, Reid H, Ramsden RT, Read A, Black G, Evans DG (2002) Molecular genetic analysis of the NF2 gene in young patients with unilateral vestibular schwannomas. J Med Genet 39:315–322
Moyhuddin A, Baser ME, Watson C, Purcell S, Ramsden RT, Heiberg A, Wallace AJ, Evans DG (2003) Somatic mosaicism in neurofibromatosis 2: prevalence and risk of disease transmission to offspring. J Med Genet 40:459–463
Nacak TG, Leptien K, Fellner D, Augustin HG, Kroll J (2006) The BTB-kelch protein LZTR-1 is a novel Golgi protein that is degraded upon induction of apoptosis. J Biol Chem 281:5065–5071. doi:10.1074/jbc.M509073200
Nayler SJ, Leiman G, Omar T, Cooper K (1996) Malignant transformation in a schwannoma. Histopathology 29:189–192
Nie Z, Xue Y, Yang D, Zhou S, Deroo BJ, Archer TK, Wang W (2000) A specificity and targeting subunit of a human SWI/SNF family-related chromatin-remodeling complex. Mol Cell Biol 20:8879–8888
Oda Y, Tsuneyoshi M (2006) Extrarenal rhabdoid tumors of soft tissue: clinicopathological and molecular genetic review and distinction from other soft-tissue sarcomas with rhabdoid features. Pathol Int 56:287–295. doi:10.1111/j.1440-1827.2006.01962.x
Ostrow KL, Bergner AL, Blakeley J, Evans DG, Ferner R, Friedman JM, Harris GJ, Jordan JT, Korf B, Langmead S, Leschziner G, Mautner V, Merker VL, Papi L, Plotkin SR, Slopis JM, Smith MJ, Stemmer-Rachamimov A, Yohay K, Belzberg AJ (2016) Creation of an international registry to support discovery in schwannomatosis. Am J Med Genet A. Oct 19. doi:10.1002/ajmg.a.38024 [Epub ahead of print]
Paganini I, Mancini I, Baroncelli M, Arena G, Gensini F, Papi L, Sestini R (2014) Application of COLD-PCR for improved detection of NF2 mosaic mutations. J Mol Diagn 16:393–399. doi:10.1016/j.jmoldx.2014.02.007
Paganini I, Sestini R, Cacciatore M, Capone GL, Candita L, Paolello C, Sbaraglia M, Dei Tos AP, Rossi S, Papi L (2015a) Broadening the spectrum of SMARCB1-associated malignant tumors: a case of uterine leiomyosarcoma in a patient with schwannomatosis. Hum Pathol 46:1226–1231. doi:10.1016/j.humpath.2015.04.008
Paganini I, Chang VY, Capone GL, Vitte J, Benelli M, Barbetti L, Sestini R, Trevisson E, Hulsebos TJ, Giovannini M, Nelson SF, Papi L (2015b) Expanding the mutational spectrum of LZTR1 in schwannomatosis. Eur J Hum Genet 23:963–968. doi:10.1038/ejhg.2014.220
Parry DM, Eldridge R, Kaiser-Kupfer MI, Bouzas EA, Pikus A, Patronas N (1994) Neurofibromatosis 2 (NF2): clinical characteristics of 63 affected individuals and clinical evidence for heterogeneity. Am J Med Genet 52:450–461
Patil S, Perry A, Maccollin M, Dong S, Betensky RA, Yeh TH, Gutmann DH, Stemmer-Rachamimov AO (2008) Immunohistochemical analysis supports a role for INI1/SMARCB1 in hereditary forms of schwannomas, but not in solitary, sporadic schwannomas. Brain Pathol 18:517–519. doi:10.1111/j.1750-3639.2008.00155.x
Patronas NJ, Courcoutsakis N, Bromley CM, Katzman GL, MacCollin M, Parry DM (2001) Intramedullary and spinal canal tumors in patients with neurofibromatosis 2: MR imaging findings and correlation with genotype. Radiology 218:434–442
Pinto SM, Manda SS, Kim MS, Taylor K, Selvan LD, Balakrishnan L, Subbannayya T, Yan F, Prasad TS, Gowda H, Lee C, Hancock WS, Pandey A (2014) Functional annotation of proteome encoded by human chromosome 22. J Proteome Res 13:2749–2760. doi:10.1021/pr401169d
Piotrowski A, Xie J, Liu YF, Poplawski AB, Gomes AR, Madanecki P, Fu C, Crowley MR, Crossman DK, Armstrong L, Babovic-Vuksanovic D, Bergner A, Blakeley JO, Blumenthal AL, Daniels MS, Feit H, Gardner K, Hurst S, Kobelka C, Lee C, Nagy R, Rauen KA, Slopis JM, Suwannarat P, Westman JA, Zanko A, Korf BR, Messiaen LM (2014) Germline loss-of-function mutations in LZTR1 predispose to an inherited disorder of multiple schwannomas. Nat Genet 46:182–187. doi:10.1038/ng.2855
Plotkin SR, O’Donnell CC, Curry WT, Bove CM, MacCollin M, Nunes FP (2011) Spinal ependymomas in neurofibromatosis Type 2: a retrospective analysis of 55 patients. J Neurosurg Spine 14:543–547. doi:10.3171/2010.11.SPINE10350
Plotkin SR, Blakeley JO, Evans DG, Hanemann CO, Hulsebos TJ, Hunter-Schaedle K, Kalpana GV, Korf B, Messiaen L, Papi L, Ratner N, Sherman LS, Smith MJ, Stemmer-Rachamimov AO, Vitte J, Giovannini M (2013) Update from the 2011 international schwannomatosis workshop: from genetics to diagnostic criteria. Am J Med Genet A 161A:405–416. doi:10.1002/ajmg.a.35760
Ragge NK, Baser ME, Klein J, Nechiporuk A, Sainz J, Pulst SM, Riccardi VM (1995) Ocular abnormalities in neurofibromatosis 2. Am J Ophthalmol 120:634–641
Ragge NK, Baser ME, Riccardi VM, Falk RE (1997) The ocular presentation of neurofibromatosis 2. Eye 11:12–18
Rennie AT, Side L, Kerr RS, Anslow P, Pretorius P (2008) Intramedullary tumours in patients with neurofibromatosis type 2: MRI features associated with a favourable prognosis. Clin Radiol 63:193–200. doi:10.1016/j.crad.2007.08.003
Roberts CW, Biegel JA (2009) The role of SMARCB1/INI1 in development of rhabdoid tumor. Cancer Biol Ther 8:412–416
Roehl AC, Mussotter T, Cooper DN, Kluwe L, Wimmer K, Högel J, Zetzmann M, Vogt J, Mautner VF, Kehrer-Sawatzki H (2012) Tissue-specific differences in the proportion of mosaic large NF1 deletions are suggestive of a selective growth advantage of hematopoietic del(±) stem cells. Hum Mutat 33:541–550. doi:10.1002/humu.22013
Rousseau G, Noguchi T, Bourdon V, Sobol H, Olschwang S (2011) SMARCB1/INI1 germline mutations contribute to 10% of sporadic schwannomatosis. BMC Neurol 11:9. doi:10.1186/1471-2377-11-9
Savla J, Chen TT, Schneider NR, Timmons CF, Delattre O, Tomlinson GE (2000) Mutations of the hSNF5/INI1 gene in renal rhabdoid tumors with second primary brain tumors. J Natl Cancer Inst 92:648–650
Sestini R, Bacci C, Provenzano A, Genuardi M, Papi L (2008) Evidence of a four-hit mechanism involving SMARCB1 and NF2 in schwannomatosis associated schwannomas. Hum Mutat 29:227–231. doi:10.1002/humu
Sévenet N, Lellouch-Tubiana A, Schofield D, Hoang-Xuan K, Gessler M, Birnbaum D, Jeanpierre C, Jouvet A, Delattre O (1999a) Spectrum of hSNF5/INI1 somatic mutations in human cancer and genotype-phenotype correlations. Hum Mol Genet 8:2359–2368
Sévenet N, Sheridan E, Amram D, Schneider P, Handgretinger R, Delattre O (1999b) Constitutional mutations of the hSNF5/INI1 gene predispose to a variety of cancers. Am J Hum Genet 65:1342–1348. doi:10.1086/302639
Sigauke E, Rakheja D, Maddox DL, Hladik CL, White CL, Timmons CF, Raisanen J (2006) Absence of expression of SMARCB1/INI1 in malignant rhabdoid tumors of the central nervous system, kidneys and soft tissue: an immunohistochemical study with implications for diagnosis. Mod Pathol 19:717–725. doi:10.1038/modpathol.3800581
Smith MJ (2015) Germline and somatic mutations in meningiomas. Cancer Genet 208:107–114. doi:10.1016/j.cancergen.2015.02.003
Smith MJ, Kulkarni A, Rustad C, Bowers NL, Wallace AJ, Holder SE, Heiberg A, Ramsden RT, Evans DG (2012a) Vestibular schwannomas occur in schwannomatosis and should not be considered an exclusion criterion for clinical diagnosis. Am J Med Genet Part A 158A:215–219. doi:10.1002/ajmg.a.34376
Smith MJ, Wallace AJ, Bowers NL, Rustad CF, Woods CG, Leschziner GD, Ferner RE, Evans DG (2012b) Frequency of SMARCB1 mutations in familial and sporadic schwannomatosis. Neurogenetics 13:141–145. doi:10.1007/s10048-012-0319-8
Smith MJ, Walker JA, Shen Y, Stemmer-Rachamimov A, Gusella JF, Plotkin SR (2012c) Expression of SMARCB1 (INI1) mutations in familial schwannomatosis. Hum Mol Genet 21:5239–5245. doi:10.1093/hmg/dds370
Smith MJ, O’Sullivan J, Bhaskar SS, Hadfield KD, Poke G, Caird J, Sharif S, Eccles D, Fitzpatrick D, Rawluk D, du Plessis D, Newman WG, Evans DG (2013) Loss-of-function mutations in SMARCE1 cause an inherited disorder of multiple spinal meningiomas. Nat Genet 45:295–298. doi:10.1038/ng.2552
Smith MJ, Wallace AJ, Bowers NL, Eaton H, Evans DG (2014) SMARCB1 mutations in schwannomatosis and genotype correlations with rhabdoid tumors. Cancer Genet 207:373–378. doi:10.1016/j.cancergen.2014.04.001
Smith MJ, Isidor B, Beetz C, Williams SG, Bhaskar SS, Richer W, O’Sullivan J, Anderson B, Daly SB, Urquhart JE, Fryer A, Rustad CF, Mills SJ, Samii A, du Plessis D, Halliday D, Barbarot S, Bourdeaut F, Newman WG, Evans DG (2015) Mutations in LZTR1 add to the complex heterogeneity of schwannomatosis. Neurology 84:141–147. doi:10.1212/WNL.0000000000001129
Smith MJ, Bowers NL, Bulman M, Gokhale C, Wallace AJ, King AT, Lloyd SK, Rutherford SA, Hammerbeck-Ward CL, Freeman SR, Evans DG (2016) Revisiting neurofibromatosis type 2 diagnostic criteria to exclude LZTR1-related schwannomatosis. Neurology Nov 16. doi:10.1212/WNL.0000000000003418
Solimini NL, Xu Q, Mermel CH, Liang AC, Schlabach MR, Luo J, Burrows AE, Anselmo AN, Bredemeyer AL, Li MZ, Beroukhim R, Meyerson M, Elledge SJ (2012) Recurrent hemizygous deletions in cancers may optimize proliferative potential. Science 337:104–109. doi:10.1126/science.1219580
Spyra M, Otto B, Schön G, Kehrer-Sawatzki H, Mautner VF (2015) Determination of the mutant allele frequency in patients with neurofibromatosis type 2 and somatic mosaicism by means of deep sequencing. Genes Chromosomes Cancer 54:482–488. doi:10.1002/gcc.22259
Sredni ST, Tomita T (2015) Rhabdoid tumor predisposition syndrome. Pediatr Dev Pathol 18:49–58. doi:10.2350/14-07-1531-MISC.1
Stemmer-Rachamimov AO, Xu L, Gonzalez-Agosti C, Burwick JA, Pinney D, Beauchamp R, Jacoby LB, Gusella JF, Ramesh V, Louis DN (1997) Universal absence of merlin, but not other ERM family members, in schwannomas. Am J Pathol 151:1649–1654
Stewart DR, Pemov A, Van Loo P, Beert E, Brems H, Sciot R, Claes K, Pak E, Dutra A, Lee CC, Legius E (2012) Mitotic recombination of chromosome arm 17q as a cause of loss of heterozygosity of NF1 in neurofibromatosis type 1-associated glomus tumors. Genes Chromosomes Cancer 51:429–437. doi:10.1002/gcc.21928
Stivaros SM, Stemmer-Rachamimov AO, Alston R, Plotkin SR, Nadol JB, Quesnel A, O’Malley J, Whitfield GA, McCabe MG, Freeman SR, Lloyd SK, Wright NB, Kilday JP, Kamaly-Asl ID, Mills SJ, Rutherford SA, King AT, Evans DG (2015) Multiple synchronous sites of origin of vestibular schwannomas in neurofibromatosis Type 2. J Med Genet 52:557–562. doi:10.1136/jmedgenet-2015-103050
Stojanova A, Tu WB, Ponzielli R, Kotlyar M, Chan PK, Boutros PC, Khosravi F, Jurisica I, Raught B, Penn LZ (2016) MYC interaction with the tumor suppressive SWI/SNF complex member INI1 regulates transcription and cellular transformation. Cell Cycle 15:1693–1705. doi:10.1080/15384101.2016.1146836
Sulhyan KR, Deshmukh BD, Gosavi AV, Ramteerthakar NA (2015) Neuroblastoma-like schwannoma in a case of schwannomatosis: report of a rare case. Int J Health Sci (Qassim). 9:478–481
Swensen JJ, Keyser J, Coffin CM, Biegel JA, Viskochil DH, Williams MS (2009) Familial occurrence of schwannomas and malignant rhabdoid tumour associated with a duplication in SMARCB1. J Med Genet 46:68–72. doi:10.1136/jmg.2008.060152
Takahashi-Fujigasaki J, Matumoto M, Kan I, Oka H, Yasue M (2012) Atypical teratoid/rhabdoid tumor with 26-year overall survival: case report. J Neurosurg Pediatr 9:400–405. doi:10.3171/2012.1.PEDS11350
Taylor MD, Gokgoz N, Andrulis IL, Mainprize TG, Drake JM, Rutka JT (2000) Familial posterior fossa brain tumors of infancy secondary to germline mutation of the hSNF5 gene. Am J Hum Genet 66:1403–1406. doi:10.1086/302833
Trevisson E, Clementi M, Salviati L (2015) Is there a link between COQ6 and schwannomatosis? Genet Med 17:312–313. doi:10.1038/gim.2014.211
Tsurusaki Y, Okamoto N, Ohashi H, Kosho T, Imai Y, Hibi-Ko Y, Kaname T, Naritomi K, Kawame H, Wakui K, Fukushima Y, Homma T, Kato M, Hiraki Y, Yamagata T, Yano S, Mizuno S, Sakazume S, Ishii T, Nagai T, Shiina M, Ogata K, Ohta T, Niikawa N, Miyatake S, Okada I, Mizuguchi T, Doi H, Saitsu H, Miyake N, Matsumoto N (2012) Mutations affecting components of the SWI/SNF complex cause Coffin-Siris syndrome. Nat Genet 44:376-378. doi:10.1038/ng.2219
Uno K, Takita J, Yokomori K, Tanaka Y, Ohta S, Shimada H, Gilles FH, Sugita K, Abe S, Sako M, Hashizume K, Hayashi Y (2002) Aberrations of the hSNF5/INI1 gene are restricted to malignant rhabdoid tumors or atypical teratoid/rhabdoid tumors in pediatric solid tumors. Genes Chromosomes Cancer 34:33–41
van den Munckhof P, Christiaans I, Kenter SB, Baas F, Hulsebos TJ (2012) Germline SMARCB1 mutation predisposes to multiple meningiomas and schwannomas with preferential location of cranial meningiomas at the falx cerebri. Neurogenetics 13:1–7. doi:10.1007/s10048-011-0300-y
Versteege I, Sévenet N, Lange J, Rousseau-Merck MF, Ambros P, Handgretinger R, Aurias A, Delattre O (1998) Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature 394:203–206. doi:10.1038/28212
Warren C, James LA, Ramsden RT, Wallace A, Baser ME, Varley JM, Evans DG (2003) Identification of recurrent regions of chromosome loss and gain in vestibular schwannomas using comparative genomic hybridisation. J Med Genet 40:802–806
Widemann BC, Acosta MT, Ammoun S, Belzberg AJ, Bernards A, Blakeley J, Bretscher A, Cichowski K, Clapp DW, Dombi E, Evans GD, Ferner R, Fernandez-Valle C, Fisher MJ, Giovannini M, Gutmann DH, Hanemann CO, Hennigan R, Huson S, Ingram D, Kissil J, Korf BR, Legius E, Packer RJ, McClatchey AI, McCormick F, North K, Pehrsson M, Plotkin SR, Ramesh V, Ratner N, Schirmer S, Sherman L, Schorry E, Stevenson D, Stewart DR, Ullrich N, Bakker AC, Morrison H (2014) CTF meeting 2012: translation of the basic understanding of the biology and genetics of NF1, NF2, and schwannomatosis toward the development of effective therapies. Am J Med Genet A 164A:563–578. doi:10.1002/ajmg.a.36312
Woodruff JM, Selig AM, Crowley K, Allen PW (1994) Schwannoma (neurilemoma) with malignant transformation. A rare, distinctive peripheral nerve tumor. Am J Surg Pathol 18:882–895
Woods R, Friedman JM, Evans DG, Baser ME, Joe H (2003) Exploring the “two-hit hypothesis” in NF2: tests of two-hit and three-hit models of vestibular schwannoma development. Genet Epidemiol 24:265–272. doi:10.1002/gepi.10238
Wu J, Kong M, Bi Q (2015) Identification of a novel germline SMARCB1 nonsense mutation in a family manifesting both schwannomatosis and unilateral vestibular schwannoma. J Neurooncol 125:439–441. doi:10.1007/s11060-015-1918-7
Xue W, Kitzing T, Roessler S, Zuber J, Krasnitz A, Schultz N, Revill K, Weissmueller S, Rappaport AR, Simon J, Zhang J, Luo W, Hicks J, Zender L, Wang XW, Powers S, Wigler M, Lowe SW (2012) A cluster of cooperating tumor-suppressor gene candidates in chromosomal deletions. Proc Natl Acad Sci USA 109:8212–8217. doi:10.1073/pnas.1206062109
Zhang K, Lin JW, Wang J, Wu X, Gao H, Hsieh YC, Hwu P, Liu YR, Su L, Chiou HY, Wang D, Yuan YC, Whang-Peng J, Chiu WT, Yen Y (2014) A germline missense mutation in COQ6 is associated with susceptibility to familial schwannomatosis. Genet Med 16:787–792. doi:10.1038/gim.2014.39
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors are unaware of any conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Kehrer-Sawatzki, H., Farschtschi, S., Mautner, VF. et al. The molecular pathogenesis of schwannomatosis, a paradigm for the co-involvement of multiple tumour suppressor genes in tumorigenesis. Hum Genet 136, 129–148 (2017). https://doi.org/10.1007/s00439-016-1753-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-016-1753-8