
Abstract
Hereditary spastic paraplegias (HSP) are rare neurodegenerative diseases sharing the degeneration of the corticospinal tracts as the main pathological characteristic. They are considered one of the most heterogeneous neurological disorders. All modes of inheritance have been described for the 84 different loci and 67 known causative genes implicated up to now. Recent advances in molecular genetics have revealed clinico-genetic heterogeneity of these disorders including their clinical and genetic overlap with other diseases of the nervous system. The systematic analysis of a large set of genes, including exome sequencing, is unmasking unusual phenotypes or inheritance modes associated with mutations in HSP genes and related genes involved in various neurological diseases. A new nosology may emerge after integration and understanding of these new data to replace the current classification. Collectively, functions of the known genes implicate the disturbance of intracellular membrane dynamics and trafficking as the consequence of alterations of cytoskeletal dynamics, lipid metabolism and organelle structures, which represent in fact a relatively small number of cellular processes that could help to find common curative approaches, which are still lacking.
Similar content being viewed by others

Introduction
Hereditary spastic paraplegia (HSP) refers to a group of neurological diseases caused by corticospinal tract degeneration (Tallaksen et al. 2001; Fink 2003, 2013). Approximately, 1 to 10/100,000 people are affected by HSP, depending on the geographical area (Ruano et al. 2014). Patients suffer from the presence of pyramidal signs predominating in lower limbs (LL), which include spasticity (stiff legs) and exaggerated reflexes, associated to muscular weakness that can progress to spastic paralysis of the legs (paraplegia) (Harding 1983; Fink 2003). Pyramidal signs in the upper limbs (UL), as well as distal LL muscle wasting, may appear after long disease durations. Spasticity is usually more severe during gait than at rest. Patients present a swaying, scissor-like, shuffling gait. Age at onset is widely variable, from early childhood to late adulthood. An early sign of spastic paraplegia is the wearing down of the soles of the shoes at the toes and on the inner sides, because of the typical spasticity of adductor muscles and tiptoe gait.
Historically, cases are distinguished as pure or complicated on clinical grounds (Harding 1983), even if recent knowledge of these diseases has demonstrated that this is not always correlated with their genetic bases and can vary between patients in the same family. Pure forms are characterized by pyramidal signs, associated with muscle weakness and bladder dysfunction, but patients may also have decreased vibration sense at ankles or pes cavus. Patients rarely need a wheelchair but may use canes during the disease course, and they have usually, except in some clinico-genetic entities, a normal lifespan. In complicated forms, additional neurological signs are observed, such as cerebellar signs, neuropathy, mental/cognitive impairment, epilepsy, extrapyramidal and retinal signs, as well as extra-neurological signs such as gastroesophageal reflux, cataract and abnormal skin pigmentation. In complex forms, the functional handicap and lifespan will depend on the full clinical picture.
At present, therapeutic options are very limited. For all patients except those with inborn errors of metabolism, rehabilitation therapies with an interdisciplinary approach to maintain autonomy as much as possible, physiotherapy and training are the best treatment options. Regular physical therapy is important to maintain muscle strength and to preserve range of motion and, based on passive tendon stretching, gait and equilibrium rehabilitation. According to the functional repercussion of spasticity, medications such as oral baclofen, intramuscular botulinum toxin or intrathecal baclofen can be of some benefit to patients. Orthopedic options such as special shoes for pes cavus or achilles tendotomy for equinovarus are also proposed to allow a longer autonomous gait. Sphincter disturbances should be investigated by specialists and with a view to possible treatment with anticholinergics, antimuscarinic agents or botulinum toxin injections into the bladder (Fink 2013; Ginsberg et al. 2013). Additional symptoms of complex forms can also be treated, such as parkinsonism with levodopa (Anheim et al. 2009).
Exclusion diagnosis
There are various acquired and genetic causes that should be ruled out in patients with the symptom of spastic paraplegia without a family history (Table 1). Cerebral and spinal magnetic resonance imaging (MRI) investigations are important to rule out common neurological conditions and structural anomalies (e.g., spinal cord compression). For example, a frontal interhemispheric tumor may manifest as progressive spastic paraplegia with sphincter disturbances before other signs such as cognitive deterioration, headache or visual troubles appear. Disease progression, age at onset, additional symptoms and results from other supplementary investigations such as cerebrospinal fluid (CSF) analyses, blood biochemistry and serology, electroneuromyography and ophthalmological examination can give important clues to the diagnosis (Table 1). All these investigations will first exclude acquired causes of spastic paraplegia but will subsequently help with the diagnostic workflow to find the correct genetic diagnosis. Some apparently sporadic cases are in fact masked familial diseases. The absence of a family history in neurogenetic disorders is frequent in clinical practice and several explanations for apparent isolation are reduced penetrance, age-dependent penetrance, variable expressivity, de novo mutation, early death of the transmitting parent or underdiagnosis in pure dominant forms with mild symptoms, autosomal recessive inheritance in small kindreds or, more rarely, X-linked inheritance in affected men. Among other inherited neurogenetic conditions that must be ruled out are leukodystrophies, in the absence of inflammation but in the presence of MRI abnormalities. Biochemical analyses in serum and/or CSF can suggest neurometabolic diseases. Finally, dopa-responsive dystonias (DRD) are a group of autosomal dominant or recessive diseases, which may present with spasticity and can mimic HSP. The dystonic toe is well known and can be misdiagnosed as extensor plantar reflex (Furukawa et al. 2001). Diurnal fluctuations and high and sustained sensibility to levodopa are characteristic of DRD.
The exploration of rare genetic disorders is an important issue since some diseases associated with spasticity are treatable. In particular, spastic paraparesis can be one of the multiple presentations of inborn errors of metabolism in children and adults and in some cases the symptom spastic paraparesis remains the only symptom for years; therefore, these metabolic causes should be included in the general diagnostic approach to sporadic spastic paraparesis due to treatment options (e.g., diet for argininemia, biotin in biotinidase deficiency) (Tanyel and Mancano 1997; Sedel et al. 2007) (www.treatable-id.org).
Genetic aspects of HSP
Genetic analysis of HSP genes can be performed when, according to the clinical symptoms and signs, other important causes have been excluded. HSP genes are denoted Spastic Paraplegia Gene followed by a number according to their order of discovery (SPGn). Up to now, the clinical phenotype and age at onset were critical to prioritize molecular testing because of the heterogeneity of these diseases at the clinical and genetic levels (Supplementary Fig. S1). More than 25 novel causative genes have been reported in 2013–2014 due to next-generation sequencing methods, making this genetic workflow time-consuming (Martin et al. 2013; Oates et al. 2013; Boukhris et al. 2013; Landouré et al. 2013; Novarino et al. 2014; Dor et al. 2014; Esteves et al. 2014; Crow et al. 2014), even if there are some genes that are still more frequent than others and may be analyzed first, such as SPAST (SPG4) and KIAA1840 (SPG11) (see below). All classical modes of transmission can be found and there are at least 67 genes that, when mutated, can account for these diseases (Table 2) to which can be added additional genes for which spasticity can be present as part of the clinical presentation (Supplementary Table 1).
Autosomal dominant (AD) forms of HSP are mainly pure forms with ages at onset that can range from infancy to late adulthood. Mutations in SPAST (SPG4), ATL1 (SPG3), KIF5A (SPG10) and REEP1 (SPG31) are described as being responsible for around 50 % of all cases (Fig. 1) (Finsterer et al. 2012). SPAST point mutations and exonic rearrangements have been implicated in 10–40 % of the HSP patients (Hazan et al. 1999; Meijer et al. 2002; Beetz et al. 2006; Loureiro et al. 2013) and in up to 12 % of sporadic forms (Depienne et al. 2006).

The autosomal recessive (AR) forms appear to be particularly prevalent where consanguinity is common such as in the Middle East or Mediterranean countries (Coutinho et al. 1999; Boukhris et al. 2009; Ruano et al. 2014), and lesser frequent in central Europe, Japan (Takiyama 2014) and USA (with the exception of communities such as the Amish). They are also more complex in clinical terms, associated with greater genetic heterogeneity (Table 2) with an onset of symptoms that is generally early. Only two forms are associated with pure HSP, but this likely results from the assignment of few families each: SPG71 and SPG72. In complex forms, the associated signs may be subtle but important indicators of the mutated gene, such as cerebellar atrophy or cerebellar ataxia with optic atrophy in SPG7, developmental delay and short stature in SPG20 (Troyer syndrome), dysarthria, distal amyotrophy, premature aging and cognitive decline in SPG21 (Mast syndrome), peripheral neuropathy and abnormal skin and hair pigmentation in SPG23 (Lison syndrome) (Table 2). Mental retardation or intellectual deterioration, thin corpus callosum (TCC) and axonal neuropathy are highly suggestive of SPG11 (Stevanin et al. 2008a). Finally, spastic ataxia with dysarthria, nystagmus and retinal striations is suggestive of autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS). Mutations in the CYP7B1 (SPG5), SPG7, KIAA1840 (SPG11) and ZFYVE26 (SPG15) genes are among the most frequently found but their relative frequencies vary according to the geographical origin (Stevanin et al. 2008a; Paisan-Ruiz et al. 2008; Erichsen et al. 2009; Goizet et al. 2009a; Schüle et al. 2009b; Arnoldi et al. 2012; Klebe et al. 2012a; Pfeffer et al. 2014) (Fig. 1). Point mutations or rearrangements in KIAA1840 (SPG11) have been shown to account for approximately 20 % of AR-HSP (Stevanin et al. 2008a).
X-linked forms are rare and include two clinical entities well recognized by pediatricians (Table 2): SPG1, caused by mutations in the neural cell adhesion molecule L1CAM gene, and SPG2, which results from mutations in the gene encoding the proteolipid protein (PLP1), a myelin component. SPG2 can also account for late-onset cases in women (Sivakumar et al. 1999).
Phenotype–genotype correlations in HSP
Many studies have failed in the past to determine reliable phenotype–genotype correlations. However, the systematic analysis of a large set of genes, including exome sequencing, is regularly unmasking unusual phenotypes and inheritance modes associated with mutations in HSP genes and the nature of the mutation in some of them can now be correlated to a specific phenotype.
Instances where similar mutations are associated with a wide spectrum of HSP phenotypes; extension of the clinical picture previously observed
There are good examples of variable phenotypes among HSP subtypes, as SPG4 in which age at onset can vary from early childhood to asymptomatic status at old ages. As more families are reported with a mutation in a specific gene, the full spectrum of each genetic entity extends and there are now fewer than ten HSP loci/genes associated exclusively with pure forms of the disease, most of them accounting for single or only a few families so far (Table 2; Supplementary Table 1). This was the case, for example, with SPG10, which was initially thought to be a pure form but now also accounts for 10 % of the complex AD families (Goizet et al. 2009b). In SPG7, the occurrence of cerebellar ataxia and/or atrophy (Klebe et al. 2012a) or progressive external ophthalmoplegia (Wedding et al. 2014; Pfeffer et al. 2014) suggests that the analysis of this gene should be extended to other phenotypes. Patients with isolated optic neuropathy should also be tested for mutations on SPG7 (Klebe et al. 2012a).
Instances where the nature of the mutations of a specific HSP gene can determine the inheritance model and/or associated phenotype
One of the recent advances in HSP genetics is the identification of various modes of inheritance of the mutations in single HSP genes. This is what occurs for REEP2 mutations that have recently been implicated in three families with recessive or dominant transmission of a pure HSP, namely SPG72 (Novarino et al. 2014; Esteves et al. 2014). In one Portuguese family with AR inheritance, two mutations segregated in trans including a splice site mutation leading to a loss of function of the corresponding allele and a missense mutation responsible for reduced binding capacities to membranes of the protein formed from the second allele. In a French autosomal dominant family, the disease segregated with a heterozygous missense mutation that had a dominant negative effect on the capacity of the wild-type protein to bind membranes. In both cases, AD and AR mutations led to a complete loss of membrane binding capacities of the REEP2 protein with consequences for the tubular structure of the endoplasmic reticulum (ER) (Esteves et al. 2014). Recently, position p.Arg415 of Atlastin-1 (SPG3A) was shown to be a hotspot for missense mutations, first associated with incomplete penetrance with an AD inheritance pattern (D’Amico et al. 2004), and then with AR transmission (Varga et al. 2013) (Khan et al. 2014). Similarly, an unusual recessive or dominant inheritance has been suggested in SPAST (SPG4, Lindsey et al. 2000) and SPG7 (McDermott et al. 2001; Sánchez-Ferrero et al. 2013), respectively.
The nature of the mutation and/or its localization in the protein can sometimes impact both the inheritance model and the phenotype at the same time, so that the nature of the mutation can predict the phenotype. This was observed with KIF1A (SPG30), in which missense homozygous mutations located in the kinesin motor domain account for a relatively pure HSP (Erlich et al. 2011; Klebe et al. 2012b), whereas heterozygous mutations located in the ATP binding site of KIF1A were found in patients with severe mental retardation with axial hypotonia, peripheral spasticity and mild atrophy of cerebellum and or corpus callosum, a phenotype reminiscent of SPG11 (Hamdan et al. 2011; Chang et al. 2014). Homozygous KIF1A frameshift mutations lead to hereditary sensory neuropathy type IIC (Rivière et al. 2011). Similarly, heterozygous mutations in HSPD1 lead to SPG13 (Hansen et al. 2002), but homozygous missense mutations of the same gene are implicated in hypomyelinating leukodystrophy type 4 (Magen et al. 2008). Mutations in TFG are responsible for SPG57, an AR-HSP associated with optic atrophy and neuropathy (Beetz et al. 2013) but can also be responsible for AD motor and sensory neuropathy (Ishiura et al. 2012). Interestingly, the TFG mutations affect different domains of the protein: the coil–coil domain in the HSP family, the P/Q rich domain in the family with neuropathy, suggesting different pathological mechanisms. In addition, one patient with neuropathy had ubiquitin- and TDP43-positive cytoplasmic neuronal inclusions reminiscent of amyotrophic lateral sclerosis (ALS) (Supplementary Table 2), suggesting a toxic gain of function effect resulting in a dominant inheritance pattern. In contrast, biallelic mutations affect the capacity of TFG to self-assemble and then probably lead to a loss of function (Beetz et al. 2013). This is also the case with REEP1, in which frameshift mutations or missense mutations that abolish ER targeting and affect the capacity of the protein to bind ATL1 (Falk et al. 2002; Beetz et al. 2012) lead to HSP (Züchner et al. 2006; Beetz et al. 2008; Hewamadduma et al. 2009; Goizet et al. 2011), whereas in-frame deletions do not impact the capacity of the protein to bind ATL1 and lead to hereditary motor neuropathy type V (Beetz et al. 2012).
Instances where the mutations of a specific HSP gene lead to overlapping diseases
During the past few years, it has appeared that HSP and other neurological conditions are at opposite ends of a continuum of overlapping diseases. The clinical overlap of HSP with peripheral neuropathies, cerebellar ataxias or mental disabilities is not new since mutated HSP patients can have a clinical presentation associating symptoms specific to these groups of disorders. ARSACS is a good illustration of this clinical overlap between ataxias and spastic paraplegias (Bouhlal et al. 2011). It is sometimes difficult to decide which symptom is most prominent in the clinical presentation and this may also depend on the physician’s expertise. The overlap between HSP and ataxias was again recently highlighted by mutations in GBA2 (SPG46), which have been found in patients with spastic ataxia associated with cataract, having ataxia (Hammer et al. 2013; Votsi et al. 2014) or spasticity (Martin et al. 2013; Citterio et al. 2014) as the prominent clinical feature. Point mutations and exonic deletions in KIF1C have both been reported in HSP (Novarino et al. 2014) and spastic ataxia (Dor et al. 2014), illustrating the fact that both diseases are part of the same clinical spectrum. PNPLA6 mutations are found in patients with a wide range of phenotypes: Gordon Holmes spinocerebellar syndrome (ataxia with brisk reflexes and hypogonadism), Boucher-Neuhäuser syndrome (ataxia with chorioretinal dystrophy and hypogonadism), isolated cerebellar ataxia and isolated spastic paraplegia (SPG39) (Rainier et al. 2008; Synofzik et al. 2014).
Regarding motor neuron diseases and polyneuropathies (Fig. 2), HSP genes have been found mutated in patients with (i) peripheral nerve affections such as Charcot–Marie–Tooth (CMT) neuropathies, (ii) first and secondary motor neuron degeneration, such as ALS, and (iii) lower motor neuron disorders such as spinal muscular atrophy (SMA). For example, KIAA1840 (SPG11) mutations may mimic ALS5 when muscle wasting is marked in absence of other complicated signs, or complex HSP in presence of cerebellar and cognitive signs (Stevanin et al. 2007b, 2008a; Orlacchio et al. 2010; Daoud et al. 2012; Romagnolo et al. 2014). Similarly, mutations in TFG can also be associated with HSP (SPG57) and ALS-like presentations (Ishiura et al. 2012; Beetz et al. 2013). BIDC2 mutations are mainly responsible for SMA phenotypes but can cause HSP as well (Neveling et al. 2013; Oates et al. 2013). ERLIN2 mutations are responsible for SPG18 (Yıldırım et al. 2011; Alazami et al. 2011; Wakil et al. 2013) but also account for juvenile primary lateral sclerosis, another neurodegenerative disorder of the upper motor neuron overlapping HSP phenotype (Al-Saif et al. 2012). Finally, missense mutations in KIF5A affecting the kinesin motor domain, or in MARS (SPG70) encoding the methionyl-tRNA synthase essential for protein biosynthesis (Deniziak and Barciszewski 2001), are responsible for pure (Reid et al. 2000) or complex forms of HSP with neuropathy or amyotrophy (Tessa et al. 2008; Goizet et al. 2009b; Musumeci et al. 2011; Crimella et al. 2012; Collongues et al. 2013; Novarino et al. 2014; Liu et al. 2014) but also CMT (Crimella et al. 2012; Gonzalez et al. 2013; Liu et al. 2014). In MARS, the mutations identified in CMT-like disease and in HSP patients are located in different domains.

Clinico-genetic entities associated with hereditary spastic paraplegia (HSP) according to the motor neuron phenotypic presentation. When mutated, HSP genes can be associated with various phenotypes that overlap with upper and lower motor neuron diseases
Other examples are the FA2H and C9ORF12 genes, which can be found mutated in patients with neuronal brain iron accumulation (NBIA) or spastic paraplegia (Edvardson et al. 2008; Schneider and Bhatia 2010; Dick et al. 2010; Kruer et al. 2010; Hartig et al. 2011; Landouré et al. 2013). There are also two genes in which mutations are associated with complex HSP but also account for pontocerebellar hypoplasia: AMPD2 (SPG63) and EXOSC3 (Wan et al. 2012; Zanni et al. 2013; Novarino et al. 2014). GJC2 accounts for complex HSP associated with dysarthria, cerebellar ataxia and mental impairment (Orthmann-Murphy et al. 2009) and hypomyelinating leukodystrophy Pelizaeus–Merzbacher-like disease (Uhlenberg et al. 2004). Similarly, PGAP1 and C12ORF65 have been reported mutated in encephalomyopathies (Antonicka et al. 2010; Murakami et al. 2014), but they are also mutated in patients with complex HSP (Shimazaki et al. 2012; Tucci et al. 2013; Spiegel et al. 2014; Novarino et al. 2014).
Instances where mutations can either account for HSP or multisystemic disorders
Several genes mutated in HSP can also be responsible for multisystemic disorders in which spasticity can be part of the phenotype or even absent. Some of these genes cause developmental disorders such as those involving mutations in genes coding for adaptor protein complex 4 (AP4) (SPG47, 50, 51 and 52) (Moreno-De-Luca et al. 2011; Abou Jamra et al. 2011). KIAA0196 is mutated in pure AD-HSP (SPG8) patients (Valdmanis et al. 2007), but also in complex cases in which the spasticity decreased upon levodopa treatment (Bettencourt et al. 2013), and recently a homozygous splice site mutation leading to exon 27 skipping was involved in Ritscher–Schinzel syndrome, a developmental syndrome with craniofacial abnormalities, congenital heart defects and cerebellar brain malformations (Elliott et al. 2013).
Biallelic mutations in IBA57, encoding a Fe/S mitochondrial protein assembly factor, can be responsible for a slowly progressive childhood-onset HSP (Lossos et al. 2015), up to multiple mitochondrial dysfunction syndrome 3 (MMDS3; OMIM #615330), a severe lethal encephalopathy with multiple malformations, myopathy and hyperglycinemia (Ajit Bolar et al. 2013). Mutations in LYST were first described in Chediak–Higashi syndrome, a disease characterized by decreased pigmentation, photophobia, nystagmus and abnormal susceptibility to infection (Barbosa et al. 1997). Recently, one mutation was reported in a family with late-onset complex HSP with cerebellar ataxia, peripheral neuropathy and large peroxidase-positive granules in granulocytes (Shimazaki et al. 2014).
Finally, biallelic mutations in BSCL2 are responsible for congenital generalized lipodystrophy type 2, characterized by severe lipoatrophy, insulin resistance, hypertriglyceridemia and mental retardation (Magré et al. 2001). In contrast, heterozygous missense mutations in its N-glycosylation motif (p.Asn88Ser and p.Ser90Leu) result in a toxic gain of function by ER-stress-mediated cell death responsible for motor neuron diseases, including SPG17 and hereditary motor neuropathy type V (Windpassinger et al. 2004; Ito and Suzuki 2009).
Modifying factors
One striking observation is the large interfamilial but also intrafamilial phenotypic variability regarding age at onset and disease severity between patients, particularly in autosomal dominant HSP subtypes. Some SPG4 patients within the same family and carrying the same mutation may indeed remain asymptomatic throughout their life whereas others have an early onset and are severely affected and may present with additional features such as cognitive impairment and peripheral neuropathy. One major issue is therefore the identification of environmental or genetic modifiers in HSP. This has been especially studied in SPAST-positive patients; three modifier variants have been suggested so far: the rare p.Ser44Leu and p.Pro45Gln polymorphisms in SPAST and the p.Gly563Ala polymorphism located in HSPD1 (HSP60/SPG13) (Svenson et al. 2004; Bross et al. 2008). Large population studies are still lacking to validate these findings.
Phenotypic variability is well known in mitochondrial disorders. The mitochondrial MT-TI mutation segregating in a HSP family is associated with a disease severity that is correlated to the level of heteroplasmy: from pure HSP in the proband showing 55 % heteroplasmy levels in muscles, to a multisystemic disorder with cardiomyopathy in the brother with heteroplasmy levels of 90 % (Corona et al. 2002).
Sex-dependent penetrance or severity has been suggested for SPAST mutations on the basis of a significant excess of affected males (Starling et al. 2002; Proukakis et al. 2011) and/or an earlier age at onset (Mitne-Neto et al. 2007) This was also suggested for ATL1 mutations (Varga et al. 2013; Luo et al. 2014).
In other cases, a variant may be pathogenic when inherited as a recessive trait but may become a susceptibility factor for other neurological conditions at the heterozygous state. Recent observations in the gene encoding paraplegin are striking. SPG7 patients usually have pure or complex forms of HSP, mainly associated with cerebellar signs and optic atrophy. Interestingly, heterozygous carriers of SPG7 truncating mutations can develop a late-onset cerebellar syndrome/atrophy without spasticity, suggesting a predisposition for late-onset neurodegenerative disorders of heterozygous SPG7 carriers, mimicking an autosomal dominant inheritance when children are carrying two causative mutations. Moreover, patients with the heterozygous p.Asp411Ala mutation were reported with optic atrophy without ataxia or spasticity in a large autosomal dominant kindred. SPG7 has, therefore, to be considered in patients with late-onset cerebellar signs or optic atrophy, even in the absence of spasticity (Klebe et al. 2012a).
Genetic diagnosis of HSP
A genetic diagnosis workflow to be used in routine diagnosis is proposed in Supplementary Fig. 1 when new technologies are still not available. In sporadic and AD cases, ATL1 and SPAST genes should be tested first depending on age at onset. Of note, up to 12 % of sporadic cases are mutated in the SPAST gene (Depienne et al. 2006; Beetz et al. 2006) while up to 40 % are mutated among autosomal dominant forms. Since 50 % of the mutations in SPG4 are larger deletions, duplications or complex genomic rearrangements (Beetz et al. 2006; Depienne et al. 2007), a dedicated technique (array-CGH, MLPA) has to be used in parallel. In AR cases, the genes to be tested are based on the associated phenotype. Indeed, the relative frequency of SPG11 varies according to phenotype (Stevanin et al. 2008a). SPG11 accounts for <1 % of patients with a pure phenotype, 4.5 % of cases with spastic paraplegia and cognitive impairment without thinning of the corpus callosum (TCC), but up to 59 % of persons with early-onset progressive spasticity with mild intellectual disability and/or cognitive decline associated with TCC. The other HSP genes are analyzed depending on the inheritance mode, the clinical presentation and the results of additional examinations.
However, the number of causative genes is growing very rapidly thanks to the improvement of sequencing techniques, and the classical testing of one gene after the other is progressively being replaced by diagnostic kits allowing multiple genes to be tested in parallel. Access to all SPG variants with such techniques may open opportunities to analyze their modifier effects in addition to the causative mutation in the near future. This may also represent a challenge for the interpretation of their effects since multiple variants with potential effects will likely be identified in more than one candidate gene. The development of common databases and of bioinformatics but also biological pipelines for analysis of the pathogenic effects will be required and will sometimes complicate the diagnosis workflow.
When a mutation is identified, genetic counseling and prenatal and presymptomatic testing are possible options.
Physiopathology of HSP
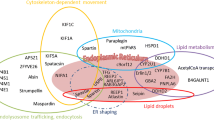
Known HSP genes encode proteins involved mainly in ER morphogenesis, microtubule dynamics and transport, mitochondrial quality control, lipid metabolism and endosomal/lysosomal functions (Table 3), and collectively suggest that HSP might be caused by impaired cellular trafficking (Stevanin et al. 2008b; Blackstone 2012).
Clear evidence of impaired trafficking comes from the involvement of KIF5A (SPG10), KIF1A (SPG30) and KIF1C (SAX2/SPAX2/SPG58), which are motor proteins (kinesins) involved in organelle/vesicle trafficking. In KIF5A, heterozygous missense mutations in the motor domain are associated with a reduced velocity along microtubules in gliding assays (Ebbing et al. 2008). There is also other evidence of trafficking disturbances in HSP. Neurons from knockout (KO) mice for Spast present with a marked impairment of microtubule dynamics along axons, accompanied by axonal swelling and cargo stalling (Tarrade et al. 2006; Kasher et al. 2009; Fassier et al. 2013). Spastin (SPAST/SPG4) is a microtubule-severing protein, which links cytoskeletal dynamics to membrane remodeling in several cellular processes. Abnormal axonal swellings have been also reported in Plp1, Fa2h and Kif5a KO mice and/or fly models (Edgar et al. 2004; Potter et al. 2011; Füger et al. 2012; Karle et al. 2012). Axon swellings with accumulation of membranous material in axons have also been observed in Spg7 KO mice (Ferreirinha et al. 2004) and in nerve biopsies of SPG11 patients (Hehr et al. 2007). Finally, axonal trafficking of vesicles was shown to be impaired in neurons derived from induced pluripotent stem cells (IPSC) of SPG11 patients (Pérez-Brangulí et al. 2014).
The best known example of mitochondrial dysfunction in HSP is related to the SPG7 subtype. The corresponding gene is the first to have been identified in HSPs, in 1998 (Casari et al. 1998) and encodes paraplegin. Paraplegin is a conserved subunit of the ATP-dependent m-AAA protease of the inner membrane of the mitochondria involved in the quality control of multiple proteins of the respiratory pathway. SPG7 is associated with multiple mitochondrial DNA deletions, suggesting that functions of other mitochondrial proteins involved in either mitochondrial DNA replication itself or pathways of mitochondrial quality control are altered (Wedding et al. 2014). As a consequence, SPG7 patients present with reduced mitochondrial respiration rates and increased sensitivity to oxidative stress (Atorino et al. 2003). Spg7 KO mice show axonal swellings with accumulation of membranous material and mitochondria in distal axons (Ferreirinha et al. 2004) reminiscent of what is observed in Spg4/Spast KO mice and therefore making the link between mitochondrial alterations and intracellular trafficking defects. Recently, reduced levels and activities of mitochondrial 4Fe-4S mitochondrial proteins have been observed in a family with a combination of spastic paraplegia, optic atrophy, and peripheral neuropathy (SPOAN) due to IBA57 mutations (Lossos et al. 2015). Finally, impaired mitochondrial motility was shown in neurons derived from iPSC of SPG3A patients (Zhu et al. 2014), again linking trafficking and mitochondrial functions; mitochondrial distribution is dependent on microtubule cytoskeleton and tubular ER functions.
The number of HSP proteins involved in the functions of the ER is growing. Six genes related to these functions are mutated in HSP (Goyal and Blackstone 2013). Atlastin-1 is a GTPase able to promote ER tubule homotypic fusion by forming trans-oligomeric complexes between two adjacent ER tubules (Orso et al. 2009). Rismanchi et al. (2008) showed ER morphology effects of the mutant atlastin-1 while ER-Golgi trafficking was largely unaffected. Another subgroup of proteins acts in ER shaping: ARL6IP1 (SPG61), reticulon 2 (SPG12), REEP1 (SPG31), REEP2 (SPG72) and RAB3GAP2 (Montenegro et al. 2012; Novarino et al. 2014; Esteves et al. 2014). Of note, spastin, atlastin-1 and REEP1 have been found to interact with each other and to act on microtubule interactions with the tubules of the ER (Park et al. 2010).
The secretory pathway is also altered. Spatacsin (SPG11) and spastizin (SPG15) account for proteins involved in the formation of lysosomes (Chang et al. 2014) and interact with components of the AP5 complex involved in membrane sorting of late endosomes (Hirst et al. 2013). Another adaptor protein complex, AP4, is also involved in neurodevelopmental diseases overlapping with HSP: SPG50, SPG51, SPG47 and SPG52 (Moreno-De-Luca et al. 2011; Abou Jamra et al. 2011). Accumulation of giant lysosomes and autophagosomes was observed in patient’s cells and in a Drosophila KO for Lyst, suggesting that LYST plays a role in homotypic fusion of these organelles (Rahman et al. 2012). BICD2 is an adaptor protein necessary for retrograde transport of vesicles from ER to Golgi (Heffernan and Simpson 2014). Finally, the NIPA1 protein (SPG6) is a neuron-specific transmembrane protein principally localized in the early endosomal compartment and on the plasma membrane, and its ortholog in Drosophila (Spict) was shown to interact with bone morphogenetic protein (BMP) receptors and promote their internalization from the membrane (Wang et al. 2007). BMP signaling is necessary for normal microtubule cytoskeleton assembly, and NIPA1 mutants are less efficient in the lysosomal degradation of BMP receptors, therefore, interfering with distal axonal functions (Tsang et al. 2009).
There are some subtypes of HSP that affect multiple brain regions and are associated with an early onset of the symptoms, which include psychomotor delay. These disorders include the SPGs affecting the AP4 complex as well as SPG1 and SPG2. In SPG3A and SPG11, the early onset of the disease may also suggest an abnormal development but information is lacking to confirm this point. A good example of abnormal development is SPG1. L1CAM function is necessary for correct formation of the corticospinal tract. Indeed, mice lacking L1cam mimic the human phenotype and present with defects in axonal guidance in the corticospinal tracts and reduced decussation (Cohen et al. 1998). Of note, abnormal development leading to psychomotor delay has also been suspected in HSP due to mutations in PGAP1 (SPG67) (Novarino et al. 2014).
Some genes expressed almost exclusively in glial cells have been identified in HSP, such as SLC16A2 coding for MCT8, a thyroid hormone transporter expressed by astrocytes during embryonic development in mammals. Several other proteins involved in HSP are expressed predominantly in non-neuronal cells, such as MAG and PLP1, two components of myelin, and FA2H, an enzyme involved in the hydroxylation of sphingolipids, galactolipids and other fatty acids (Hiroko 2010). Mice lacking Fa2h show a degeneration of myelin sheaths at 18 months (Zöller et al. 2008). PLP1/DM20 is expressed in oligodendrocytes and oligodendrocyte progenitor cells (OPC). Despite normal myelination, mice lacking Plp1 have physically fragile myelin and a decrease of its cholesterol content (Werner et al. 2013). It can be suggested that PLP1/DM20 may stabilize and maintain the myelin sheath. Moreover, these mice have an alteration of fast retrograde and anterograde transport (Edgar et al. 2004).
Lipid metabolism is an emerging pathway in HSP, but its importance is growing daily and has opened an entirely novel perspective on the pathogenesis of this group of diseases. There is increasing evidence that lipids have critical roles as signaling mediators and effectors, and that lipid composition of neuronal membranes affects crucial processes such as exocytosis and ion channel functions, and contributes to the formation of membrane domains. As an example, the loss of the B4GALNT1, an enzyme of the catabolism of complex gangliosides, changes the cholesterol and phospholipid content of membranes (Ohmi et al. 2011). B4galnt1 KO mice show an age-dependent neurodegenerative phenotype, central and peripheral axonal degeneration, reduced myelin volume and loss of axo-glial junctions. This phenotype was rescued by B4GALNT1 expression in neurons but not by B4GALNT1 glial expression, indicating that neuronal rather that glial gangliosides are important for integrity of the CNS (Yao et al. 2014). On the other hand, SPG5 encodes an enzyme of the bilic acid and neurosteroid formation (CYP7B1). SPG5 physiopathology may result from an accumulation of toxic substrates and the absence of neurosteroids (Leoni and Caccia 2011; Theofilopoulos et al. 2014). In Gba2 KO mice, the accumulation of glucocerebrosides has been shown in brain, liver and testis (Gonzalez-Carmona et al. 2012), but only infertility has been explored in these mice and whether this accumulation is toxic in the brain is not proved. Lipid storage may also be affected in some of these diseases since Atlastin-1, REEP1, Spartin and Seipin modulate lipid droplet structures (Renvoisé et al. 2012; Klemm et al. 2013; Falk et al. 2014).
Altogether, the functional knowledge gained from the known functions of the HSP genes and from the analysis of various animal models of these diseases suggest that the pathology results from disturbance of intracellular membrane trafficking and may account for the ‘dying-back’ mechanism observed in neuropathological human cases (Supplementary Table 2) (Deluca et al. 2004). The relationship between most of the genes involved in HSP has recently been pinpointed by the publication of an HSP interactome that may be useful to incriminate further causative genes in the future (Novarino et al. 2014). Very recently, nucleotide metabolism and autophagy were also novel functions reported as possibly altered in HSP (Oz-Levi et al. 2012; Novarino et al. 2014).
Conclusions
The increasing number of genes and the extension of the clinical picture associated with each genetic entity are building a new nosology that may modify the way molecular diagnosis and treatment of these diseases is done. Although it is often impossible to identify the mutated HSP gene in an individual patient on the basis of clinical criteria, the relative frequencies and clinical characteristics still help to elaborate an effective diagnostic strategy after careful exclusion of other causes. The strategy is simple in certain cases because of the high relative frequencies of two major genes: SPG4, which represents up to 40 % of pure AD forms, and SPG11, which accounts for up to 59 % of cases with a TCC transmitted in an AR manner. However, numerous studies have shown that the classic subdivision of HSP into pure and complex forms, still in use in clinical practice, is imperfect. In addition, the clinical and genetic overlap of various neurodegenerative diseases suggests that HSP is (or indeed the HSPs are) in a continuum with other neurological diseases and that the phenotype of a given patient will depend on multiple factors, including the mutated gene, the nature of the mutation and its location in the protein, the zygosity of the mutation, modifier variants and the environment. When considering all factors, it becomes clear that the dogma linking one gene to one phenotype has to be replaced by one patient—one disease, which will fit with personalized medicine in the future. On the other hand, there is indication for a possible unification of genetic forms from the cell biological and, thus, potentially therapeutic point of view. In particular, the functions of several recently identified HSP proteins suggest that they may participate in the same molecular pathway of lipid metabolism, which may lead to common therapies. Interestingly, disturbances in lipid metabolism also offered the unprecedented opportunity to identify biomarkers for HSP, as in SPG5 (25 and 27 hydroxy-cholesterol) (Schüle et al. 2010) and SPG26 (testosterone or GM2/GM3 levels) (Boukhris et al. 2013; Harlalka et al. 2013) (Table 2; Supplementary Table 1), prerequisite for therapeutic trials. Finally, elucidation of the underlying pathogenic mechanisms will also help to develop more effective therapeutic agents. Preclinical trials in Spast KO flies and mice (ex vivo) and SPG4 human embryonic stem cells indicated that microtubule-binding agents might have therapeutic value (Fassier et al. 2013; Denton et al. 2014). Similarly, these drugs proved to be efficient in rescuing axon growth defects in SPG3A iPSC (Zhu et al. 2014) which open therapeutic avenues for HSP subtypes related to abnormal trafficking.
References
Abou Jamra R, Philippe O, Raas-Rothschild A et al (2011) Adaptor protein complex 4 deficiency causes severe autosomal-recessive intellectual disability, progressive spastic paraplegia, shy character, and short stature. Am J Hum Genet 88:788–795. doi:10.1016/j.ajhg.2011.04.019
Ajit Bolar N, Vanlander AV, Wilbrecht C et al (2013) Mutation of the iron-sulfur cluster assembly gene IBA57 cause severe myopathy and encephalopathy. Hum Mol Genet 22:2590–2602
Alazami AM, Adly N, Al Dhalaan H, Alkuraya FS (2011) A nullimorphic ERLIN2 mutation defines a complicated hereditary spastic paraplegia locus (SPG18). Neurogenetics 12:333–336. doi:10.1007/s10048-011-0291-8
Al-Saif A, Bohlega S, Al-Mohanna F (2012) Loss of ERLIN2 function leads to juvenile primary lateral sclerosis. Ann Neurol 72:510–516. doi:10.1002/ana.23641
Anheim M, Lagier-Tourenne C, Stevanin G et al (2009) SPG11 spastic paraplegia. A new cause of juvenile parkinsonism. J Neurol 256:104–108. doi:10.1007/s00415-009-0083-3
Antonicka H, Ostergaard E, Sasarman F et al (2010) Mutations in C12orf65 in patients with encephalomyopathy and a mitochondrial translation defect. Am J Hum Genet 87:115–122. doi:10.1016/j.ajhg.2010.06.004
Arnoldi A, Crimella C, Tenderini E et al (2012) Clinical phenotype variability in patients with hereditary spastic paraplegia type 5 associated with CYP7B1 mutations. Clin Genet 81(2):150–157. doi:10.1111/j.1399-0004.2011.01624.x
Atorino L, Silvestri L, Koppen M et al (2003) Loss of m-AAA protease in mitochondria causes complex I deficiency and increased sensitivity to oxidative stress in hereditary spastic paraplegia. J Cell Biol 163:777–787. doi:10.1083/jcb.200304112
Barbosa MD, Barrat FJ, Tchernev VT et al (1997) Identification of mutations in two major mRNA isoforms of the Chediak–Higashi syndrome gene in human and mouse. Hum Mol Genet 6:1091–1098
Bauer P, Leshinsky-Silver E, Blumkin L et al (2012) Mutation in the AP4B1 gene cause hereditary spastic paraplegia type 47 (SPG47). Neurogenetics 13:73–76. doi:10.1007/s10048-012-0314-0
Beetz C, Nygren AOH, Schickel J et al (2006) High frequency of partial SPAST deletions in autosomal dominant hereditary spastic paraplegia. Neurology 67:1926–1930. doi:10.1212/01.wnl.0000244413.49258.f5
Beetz C, Schüle R, Deconinck T et al (2008) REEP1 mutation spectrum and genotype/phenotype correlation in hereditary spastic paraplegia type 31. Brain 131:1078–1086. doi:10.1093/brain/awn026
Beetz C, Pieber TR, Hertel N et al (2012) Exome sequencing identifies a REEP1 mutation involved in distal hereditary motor neuropathy type V. Am J Hum Genet 91:139–145. doi:10.1016/j.ajhg.2012.05.007
Beetz C, Johnson A, Schuh AL et al (2013) Inhibition of TFG function causes hereditary axon degeneration by impairing endoplasmic reticulum structure. Proc Natl Acad Sci 110:5091–5096. doi:10.1073/pnas.1217197110
Bettencourt C, Morris HR, Singleton AB et al (2013) Exome sequencing expands the mutational spectrum of SPG8 in a family with spasticity responsive to l-DOPA treatment. J Neurol 260:2414–2416. doi:10.1007/s00415-013-7044-6
Blackstone C (2012) Cellular pathways of hereditary spastic paraplegia. Annu Rev Neurosci 35:25–47. doi:10.1146/annurev-neuro-062111-150400
Blumen SC, Bevan S, Abu-Mouch S et al (2003) A locus for complicated hereditary spastic paraplegia maps to chromosome 1q24-q32. Ann Neurol 54:796–803. doi:10.1002/ana.10768
Bouhlal Y, Amouri R, El Euch-Fayeche G, Hentati F (2011) Autosomal recessive spastic ataxia of Charlevoix-Saguenay: an overview. Parkinsonism Relat Disord 17:418–422. doi:10.1016/j.parkreldis.2011.03.005
Bouhouche A, Benomar A, Bouslam N et al (2006) Mutation in the epsilon subunit of the cytosolic chaperonin-containing t-complex peptide-1 (Cct5) gene causes autosomal recessive mutilating sensory neuropathy with spastic paraplegia. J Med Genet 43:441–443. doi:10.1136/jmg.2005.039230
Boukhris A, Stevanin G, Feki I et al (2009) Tunisian hereditary spastic paraplegias: clinical variability supported by genetic heterogeneity. Clin Genet 75(6):527–536. doi:10.1111/j.1399-0004.2009.01176.x
Boukhris A, Schule R, Loureiro JL et al (2013) Alteration of ganglioside biosynthesis responsible for complex hereditary spastic paraplegia. Am J Hum Genet 93:118–123. doi:10.1016/j.ajhg.2013.05.006
Bross P, Naundrup S, Hansen J et al (2008) The Hsp60-(p. V98I) mutation associated with hereditary spastic paraplegia SPG13 compromises chaperonin function both in vitro and in vivo. J Biol Chem 283:15694–15700. doi:10.1074/jbc.M800548200
Caballero Oteyza A, Battaloğlu E, Ocek L et al (2014) Motor protein mutations cause a new form of hereditary spastic paraplegia. Neurology 82(22):2007–2016. doi:10.1212/WNL.0000000000000479
Casari G, De Fusco M, Ciarmatori S et al (1998) Spastic paraplegia and OXPHOS impairment caused by mutations in paraplegin, a nuclear-encoded mitochondrial metalloprotease. Cell 93:973–983
Chang J, Lee S, Blackstone C (2014) Spastic paraplegia proteins spastizin and spatacsin mediate autophagic lysosome reformation. J Clin Invest. 124(12):5249–5262. doi:10.1172/JCI77598
Citterio A, Arnoldi A, Panzeri E et al (2014) Mutations in CYP2U1, DDHD2 and GBA2 genes are rare causes of complicated forms of hereditary spastic paraparesis. J Neurol 261(2):373–381. doi:10.1007/s00415-013-7206-6
Cohen NR, Taylor JS, Scott LB et al (1998) Errors in corticospinal axon guidance in mice lacking the neural cell adhesion molecule L1. Curr Biol 8:26–33
Collongues N, Depienne C, Boehm N et al (2013) Novel SPG10 mutation associated with dysautonomia, spinal cord atrophy, and skin biopsy abnormality. Eur J Neurol 20:398–401. doi:10.1111/j.1468-1331.2012.03803.x
Corona P, Lamantea E, Greco M et al (2002) Novel heteroplasmic mtDNA mutation in a family with heterogeneous clinical presentations. Ann Neurol 51:118–122. doi:10.1002/ana.10059
Coutinho P, Barros J, Zemmouri R et al (1999) Clinical heterogeneity of autosomal recessive spastic paraplegias: analysis of 106 patients in 46 families. Arch Neurol 56:943–949
Cremers FP, Pfeiffer RA, van de Pol TJ et al (1987) An interstitial duplication of the X chromosome in a male allows physical fine mapping of probes from the Xq13-q22 region. Hum Genet 77:23–27
Crimella C, Baschirotto C, Arnoldi A et al (2012) Mutations in the motor and stalk domains of KIF5A in spastic paraplegia type 10 and in axonal Charcot-Marie-Tooth type 2. Clin Genet 82:157–164. doi:10.1111/j.1399-0004.2011.01717.x
Crow YJ, Zaki MS, Abdel-Hamid MS et al (2014) Mutations in ADAR1, IFIH1, and RNASEH2B presenting as spastic paraplegia. Neuropediatrics 45(6):386–393. doi:10.1055/s-0034-1389161
D’Amico A, Tessa A, Sabino A et al (2004) Incomplete penetrance in an SPG3A-linked family with a new mutation in the atlastin gene. Neurology 62:2138–2139
Dan P, Edvardson S, Bielawski J, Hama H, Saada A (2011) 2-Hydroxylated sphingomyelin profiles in cells from patients with mutated fatty acid 2-hydroxylase. Lipids Health Dis 10:84. doi:10.1186/1476-511X-10-84
Daoud H, Zhou S, Noreau A et al (2012) Exome sequencing reveals SPG11 mutations causing juvenile ALS. Neurobiol Aging 33(839):e5–e9. doi:10.1016/j.neurobiolaging.2011.11.012
De Bot ST, Schelhaas HJ, Kamsteeg E-J, van de Warrenburg BPC (2012) Hereditary spastic paraplegia caused by a mutation in the VCP gene. Brain J Neurol 135:e223; author reply e224. doi:10.1093/brain/aws201
Deluca GC, Ebers GC, Esiri MM (2004) The extent of axonal loss in the long tracts in hereditary spastic paraplegia. Neuropathol Appl Neurobiol 30:576–584. doi:10.1111/j.1365-2990.2004.00587.x
Deniziak MA, Barciszewski J (2001) Methionyl-tRNA synthetase. Acta Biochim Pol 48:337–350
Denton KR, Lei L, Grenier J, Rodionov V, Blackstone C, Li XJ (2014) Loss of spastin function results in disease-specific axonal defects in human pluripotent stem cell-based models of hereditary spastic paraplegia. Stem Cells 32(2):414–423. doi:10.1002/stem.1569
Depienne C, Tallaksen C, Lephay JY et al (2006) Spastin mutations are frequent in sporadic spastic paraparesis and their spectrum is different from that observed in familial cases. J Med Genet 43:259–265. doi:10.1136/jmg.2005.035311
Depienne C, Fedirko E, Forlani S et al (2007) Exon deletions of SPG4 are a frequent cause of hereditary spastic paraplegia. J Med Genet 44:281–284. doi:10.1136/jmg.2006.046425
Dick KJ, Eckhardt M, Paisán-Ruiz C et al (2010) Mutation of FA2H underlies a complicated form of hereditary spastic paraplegia (SPG35). Hum Mutat 31:E1251–E1260. doi:10.1002/humu.21205
Dor T, Cinnamon Y, Raymond L et al (2014) KIF1C mutations in two families with hereditary spastic paraparesis and cerebellar dysfunction. J Med Genet 51:137–142. doi:10.1136/jmedgenet-2013-102012
Dumitrescu AM, Liao X-H, Best TB et al (2004) A novel syndrome combining thyroid and neurological abnormalities is associated with mutations in a monocarboxylate transporter gene. Am J Hum Genet 74:168–175. doi:10.1086/380999
Dursun U, Koroglu C, Kocasoy Orhan E et al (2009) Autosomal recessive spastic paraplegia (SPG45) with mental retardation maps to 10q24.3-q25.1. Neurogenetics 10:325–331. doi:10.1007/s10048-009-0191-3
Ebbing B, Mann K, Starosta A et al (2008) Effect of spastic paraplegia mutations in KIF5A kinesin on transport activity. Hum Mol Genet 17:1245–1252. doi:10.1093/hmg/ddn014
Edgar JM, McLaughlin M, Yool D et al (2004) Oligodendroglial modulation of fast axonal transport in a mouse model of hereditary spastic paraplegia. J Cell Biol 166:121–131. doi:10.1083/jcb.200312012
Edvardson S, Hama H, Shaag A et al (2008) Mutations in the fatty acid 2-hydroxylase gene are associated with leukodystrophy with spastic paraparesis and dystonia. Am J Hum Genet 83:643–648. doi:10.1016/j.ajhg.2008.10.010
Elliott AM, Simard LR, Coghlan G et al (2013) A novel mutation in KIAA0196: identification of a gene involved in Ritscher-Schinzel/3C syndrome in a First Nations cohort. J Med Genet 50:819–822. doi:10.1136/jmedgenet-2013-101715
Engert JC, Bérubé P, Mercier J et al (2000) ARSACS, a spastic ataxia common in northeastern Québec, is caused by mutations in a new gene encoding an 11.5-kb ORF. Nat Genet 24:120–125. doi:10.1038/72769
Erichsen AK, Koht J, Stray-Pedersen A et al (2009) Prevalence of hereditary ataxia and spastic paraplegia in southeast Norway: a population-based study. Brain 132:1577–1588. doi:10.1093/brain/awp056
Erlich Y, Edvardson S, Hodges E et al (2011) Exome sequencing and disease-network analysis of a single family implicate a mutation in KIF1A in hereditary spastic paraparesis. Genome Res 21:658–664. doi:10.1101/gr.117143.110
Esteves T, Durr A, Mundwiller E et al (2014) Loss of association of REEP2 with membranes leads to hereditary spastic paraplegia. Am J Hum Genet 94:268–277. doi:10.1016/j.ajhg.2013.12.005
Falk J, Bonnon C, Girault J-A, Faivre-Sarrailh C (2002) F3/contactin, a neuronal cell adhesion molecule implicated in axogenesis and myelination. Biol Cell 94:327–334
Falk J, Rohde M, Bekhite MM et al (2014) Functional mutation analysis provides evidence for a role of REEP1 in lipid droplet biology. Hum Mutat 35:497–504. doi:10.1002/humu.22521
Fassier C, Tarrade A, Peris L et al (2013) Microtubule-targeting drugs rescue axonal swellings in cortical neurons from spastin knockout mice. Dis Model Mech 6:72–83. doi:10.1242/dmm.008946
Ferreirinha F, Quattrini A, Pirozzi M et al (2004) Axonal degeneration in paraplegin-deficient mice is associated with abnormal mitochondria and impairment of axonal transport. J Clin Invest 113:231–242. doi:10.1172/JCI20138
Fink JK (2003) The hereditary spastic paraplegias: nine genes and counting. Arch Neurol 60(8):1045–1049
Fink JK (2013) Hereditary spastic paraplegia: clinico-pathologic features and emerging molecular mechanisms. Acta Neuropathol 126(3):307–328. doi:10.1007/s00401-013-1115-8
Finsterer J, Löscher W, Quasthoff S et al (2012) Hereditary spastic paraplegias with autosomal dominant, recessive, X-linked, or maternal trait of inheritance. J Neurol Sci 318:1–18. doi:10.1016/j.jns.2012.03.025
Füger P, Sreekumar V, Schüle R et al (2012) Spastic paraplegia mutation N256S in the neuronal microtubule motor KIF5A disrupts axonal transport in a Drosophila HSP model. PLoS Genet 8:e1003066. doi:10.1371/journal.pgen.1003066
Furukawa Y, Graf WD, Wong H et al (2001) Dopa-responsive dystonia simulating spastic paraplegia due to tyrosine hydroxylase (TH) gene mutations. Neurology 56:260–263
Ginsberg D, Cruz F, Herschorn S et al (2013) OnabotulinumtoxinA is effective in patients with urinary incontinence due to neurogenic detrusor overactivity [corrected] regardless of concomitant anticholinergic use or neurologic etiology. Adv Ther 30:819–833. doi:10.1007/s12325-013-0054-z
Goizet C, Boukhris A, Maltete D et al (2009a) SPG15 is the second most common cause of hereditary spastic paraplegia with thin corpus callosum. Neurology 73:1111–1119. doi:10.1212/WNL.0b013e3181bacf59
Goizet C, Boukhris A, Mundwiller E et al (2009b) Complicated forms of autosomal dominant hereditary spastic paraplegia are frequent in SPG10. Hum Mutat 30:E376–E385. doi:10.1002/humu.20920
Goizet C, Depienne C, Benard G et al (2011) REEP1 mutations in SPG31: frequency, mutational spectrum, and potential association with mitochondrial morpho-functional dysfunction. Hum Mutat 32:1118–1127. doi:10.1002/humu.21542
Gonzalez M, McLaughlin H, Houlden H et al (2013) Exome sequencing identifies a significant variant in methionyl-tRNA synthetase (MARS) in a family with late-onset CMT2. J Neurol Neurosurg Psychiatry 84:1247–1249. doi:10.1136/jnnp-2013-305049
Gonzalez-Carmona MA, Sandhoff R, Tacke F et al (2012) Beta-glucosidase 2 knockout mice with increased glucosylceramide show impaired liver regeneration. Liver Int 32:1354–1362. doi:10.1111/j.1478-3231.2012.02841.x
Goyal U, Blackstone C (2013) Untangling the web: mechanisms underlying ER network formation. Biochim Biophys Acta 1833:2492–2498. doi:10.1016/j.bbamcr.2013.04.009
Hamdan FF, Gauthier J, Araki Y et al (2011) Excess of de novo deleterious mutations in genes associated with glutamatergic systems in nonsyndromic intellectual disability. Am J Hum Genet 88:306–316. doi:10.1016/j.ajhg.2011.02.001
Hammer MB, Eleuch-Fayache G, Schottlaender LV et al (2013) Mutations in GBA2 cause autosomal-recessive cerebellar ataxia with spasticity. Am J Hum Genet 92:245–251. doi:10.1016/j.ajhg.2012.12.012
Hansen JJ, Dürr A, Cournu-Rebeix I et al (2002) Hereditary spastic paraplegia SPG13 is associated with a mutation in the gene encoding the mitochondrial chaperonin Hsp60. Am J Hum Genet 70:1328–1332. doi:10.1086/339935
Harding AE (1983) Classification of the hereditary ataxias and paraplegias. Lancet 1:1151–1155
Harlalka GV, Lehman A, Chioza B et al (2013) Mutations in B4GALNT1 (GM2 synthase) underlie a new disorder of ganglioside biosynthesis. Brain 136:3618–3624. doi:10.1093/brain/awt270
Hartig MB, Iuso A, Haack T et al (2011) Absence of an orphan mitochondrial protein, C19orf12, causes a distinct clinical subtype of neurodegeneration with brain iron accumulation. Am J Hum Genet 89:543–550. doi:10.1016/j.ajhg.2011.09.007
Hanein S, Durr A, Ribai P et al (2007) A novel locus for autosomal dominant “uncomplicated” hereditary spastic paraplegia maps to chromosome 8p21.1-q13.3. Hum Genet 122:261–273
Hanein S, Martin E, Boukhris A et al (2008) Identification of the SPG15 gene, encoding spastizin, as a frequent cause of complicated autosomal recessive spastic paraplegia including Kjellin syndrome. Am J Hum Genet 82:992–1002
Hazan J, Fonknechten N, Mavel D et al (1999) Spastin, a new AAA protein, is altered in the most frequent form of autosomal dominant spastic paraplegia. Nat Genet 23:296–303. doi:10.1038/15472
Heffernan LF, Simpson JC (2014) The trials and tubule-ations of Rab6 involvement in Golgi-to-ER retrograde transport. Biochem Soc Trans 42:1453–1459. doi:10.1042/BST20140178
Hehr U, Bauer P, Winner B et al (2007) Long-term course and mutational spectrum of spatacsin-linked spastic paraplegia. Ann Neurol 62:656–665. doi:10.1002/ana.21310
Hewamadduma C, McDermott C, Kirby J et al (2009) New pedigrees and novel mutation expand the phenotype of REEP1-associated hereditary spastic paraplegia (HSP). Neurogenetics 10:105–110. doi:10.1007/s10048-008-0163-z
Hiroko H (2010) Fatty acid 2-Hydroxylation in mammalian sphingolipid biology. Biochim Biophys Acta 1801:405–414. doi:10.1016/j.bbalip.2009.12.004
Hirst J, Borner GHH, Edgar J et al (2013) Interaction between AP-5 and the hereditary spastic paraplegia proteins SPG11 and SPG15. Mol Biol Cell 24(16):2558–2569. doi:10.1091/mbc.E13-03-0170
Hodgkinson CA, Bohlega S, Abu-Amero SN et al (2002) A novel form of autosomal recessive pure hereditary spastic paraplegia maps to chromosome 13q14. Neurology 59:1905–1909
Ilgaz-Aydinlar E, Rolfs A, Serteser M, Parman Y (2014) Mutation in FAM134B causing hereditary sensory neuropathy with spasticity in a Turkish family. Muscle Nerve 49:774–775. doi:10.1002/mus.24145
Ishiura H, Sako W, Yoshida M et al (2012) The TRK-fused gene is mutated in hereditary motor and sensory neuropathy with proximal dominant involvement. Am J Hum Genet 91:320–329. doi:10.1016/j.ajhg.2012.07.014
Ito D, Suzuki N (2009) Seipinopathy: a novel endoplasmic reticulum stress-associated disease. Brain 132:8–15. doi:10.1093/brain/awn216
Jouet M, Rosenthal A, Armstrong G et al (1994) X-linked spastic paraplegia (SPG1), MASA syndrome and X-linked hydrocephalus result from mutations in the L1 gene. Nat Genet 7:402–407. doi:10.1038/ng0794-402
Karle KN, Möckel D, Reid E, Schöls L (2012) Axonal transport deficit in a KIF5A(−/−) mouse model. Neurogenetics 13:169–179. doi:10.1007/s10048-012-0324-y
Kasher PR, De Vos KJ, Wharton SB et al (2009) Direct evidence for axonal transport defects in a novel mouse model of mutant spastin-induced hereditary spastic paraplegia (HSP) and human HSP patients. J Neurochem 110:34–44. doi:10.1111/j.1471-4159.2009.06104.x
Khan TN, Klar J, Tariq M et al (2014) Evidence for autosomal recessive inheritance in SPG3A caused by homozygosity for a novel ATL1 missense mutation. Eur J Hum Genet 22(10):1180–1184. doi:10.1038/ejhg.2014.5
Klebe S, Depienne C, Gerber S et al (2012a) Spastic paraplegia gene 7 in patients with spasticity and/or optic neuropathy. Brain 135:2980–2993. doi:10.1093/brain/aws240
Klebe S, Lossos A, Azzedine H et al (2012b) KIF1A missense mutations in SPG30, an autosomal recessive spastic paraplegia: distinct phenotypes according to the nature of the mutations. Eur J Hum Genet 20:645–649. doi:10.1038/ejhg.2011.261
Klemm RW, Norton JP, Cole RA et al (2013) A conserved role for atlastin GTPases in regulating lipid droplet size. Cell Rep 3:1465–1475. doi:10.1016/j.celrep.2013.04.015
Kruer MC, Paisán-Ruiz C, Boddaert N et al (2010) Defective FA2H leads to a novel form of neurodegeneration with brain iron accumulation (NBIA). Ann Neurol 68:611–618. doi:10.1002/ana.22122
Kurth I, Pamminger T, Hennings JC et al (2009) Mutations in FAM134B, encoding a newly identified Golgi protein, cause severe sensory and autonomic neuropathy. Nat Genet 41:1179–1181. doi:10.1038/ng.464
Landouré G, Zhu P-P, Lourenço CM et al (2013) Hereditary spastic paraplegia type 43 (SPG43) is caused by mutation in C19orf12. Hum Mutat 34:1357–1360. doi:10.1002/humu.22378
Leoni V, Caccia C (2011) Oxysterols as biomarkers in neurodegenerative diseases. Chem Phys Lipids 164:515–524. doi:10.1016/j.chemphyslip.2011.04.002
Liguori R, Giannoccaro MP, Arnoldi A et al (2014) Impairment of brain and muscle energy metabolism detected by magnetic resonance spectroscopy in hereditary spastic paraparesis type 28 patients with DDHD1 mutations. J Neurol 261(9):1789–1793. doi:10.1007/s00415-014-7418-4
Lin P, Li J, Liu Q et al (2008) A missense mutation in SLC33A1, which encodes the acetyl-CoA transporter, causes autosomal-dominant spastic paraplegia (SPG42). Am J Hum Genet 83:752–759. doi:10.1016/j.ajhg.2008.11.003
Lindsey JC, Lusher ME, McDermott CJ et al (2000) Mutation analysis of the spastin gene (SPG4) in patients with hereditary spastic paraparesis. J Med Genet 37(10):759–765
Liu Y-T, Laurá M, Hersheson J et al (2014) Extended phenotypic spectrum of KIF5A mutations: from spastic paraplegia to axonal neuropathy. Neurology 83:612–619. doi:10.1212/WNL.0000000000000691
Lossos A, Stümpfig C, Stevanin G et al (2015) Fe/S protein assembly gene IBA57 mutation causes hereditary spastic paraplegia. Neurology 84(7):659–667. doi:10.1212/WNL.0000000000001270
Loureiro JL, Brandão E, Ruano L et al (2013) Autosomal dominant spastic paraplegias: a review of 89 families resulting from a Portuguese survey. JAMA Neurol 70:481–487. doi:10.1001/jamaneurol.2013.1956
Luo Y, Chen C, Zhan Z et al (2014) Mutation and clinical characteristics of autosomal-dominant hereditary spastic paraplegias in china. Neurodegener Dis 14(4):176–183. doi:10.1159/000365513
Macedo-Souza LI, Kok F, Santos S et al (2008) Reevaluation of a large family defines a new locus for X-linked recessive pure spastic paraplegia (SPG34) on chromosome Xq25. Neurogenetics 9:225–226. doi:10.1007/s10048-008-0130-8
Magen D, Georgopoulos C, Bross P et al (2008) Mitochondrial hsp60 chaperonopathy causes an autosomal-recessive neurodegenerative disorder linked to brain hypomyelination and leukodystrophy. Am J Hum Genet 83:30–42. doi:10.1016/j.ajhg.2008.05.016
Magré J, Delépine M, Khallouf E et al (2001) Identification of the gene altered in Berardinelli-Seip congenital lipodystrophy on chromosome 11q13. Nat Genet 28:365–370. doi:10.1038/ng585
Mannan AU, Krawen P, Sauter SM et al (2006) ZFYVE27 (SPG33), a novel spastin-binding protein, is mutated in hereditary spastic paraplegia. Am J Hum Genet 79:351–357. doi:10.1086/504927
Martin E, Schüle R, Smets K et al (2013) Loss of function of glucocerebrosidase GBA2 is responsible for motor neuron defects in hereditary spastic paraplegia. Am J Hum Genet 92:238–244. doi:10.1016/j.ajhg.2012.11.021
McDermott CJ, Dayaratne RK, Tomkins J et al (2001) Paraplegin gene analysis in hereditary spastic paraparesis (HSP) pedigrees in northeast England. Neurology 56(4):467–471
Meijer IA, Hand CK, Cossette P et al (2002) Spectrum of SPG4 mutations in a large collection of North American families with hereditary spastic paraplegia. Arch Neurol 59:281–286
Meijer IA, Cossette P, Roussel J et al (2004) A novel locus for pure recessive hereditary spastic paraplegia maps to 10q22.1-10q24.1. Ann Neurol 56:579–582. doi:10.1002/ana.20239
Mitne-Neto M, Kok F, Beetz C et al (2007) A multi-exonic SPG4 duplication underlies sex-dependent penetrance of hereditary spastic paraplegia in a large Brazilian pedigree. Eur J Hum Genet 15(12):1276–1279
Montenegro G, Rebelo AP, Connell J et al (2012) Mutations in the ER-shaping protein reticulon 2 cause the axon-degenerative disorder hereditary spastic paraplegia type 12. J Clin Invest 122:538–544. doi:10.1172/JCI60560
Moreno-De-Luca A, Helmers SL, Mao H et al (2011) Adaptor protein complex-4 (AP-4) deficiency causes a novel autosomal recessive cerebral palsy syndrome with microcephaly and intellectual disability. J Med Genet 48:141–144. doi:10.1136/jmg.2010.082263
Murakami Y, Tawamie H, Maeda Y et al (2014) Null mutation in PGAP1 impairing Gpi-anchor maturation in patients with intellectual disability and encephalopathy. PLoS Genet 10:e1004320. doi:10.1371/journal.pgen.1004320
Musumeci O, Bassi MT, Mazzeo A et al (2011) A novel mutation in KIF5A gene causing hereditary spastic paraplegia with axonal neuropathy. Neurol Sci 32:665–668. doi:10.1007/s10072-010-0445-8
Neveling K, Martinez-Carrera LA, Hölker I et al (2013) Mutations in BICD2, which encodes a golgin and important motor adaptor, cause congenital autosomal-dominant spinal muscular atrophy. Am J Hum Genet 92:946–954. doi:10.1016/j.ajhg.2013.04.011
Novarino G, Fenstermaker AG, Zaki MS et al (2014) Exome Sequencing links corticospinal motor neuron disease to common neurodegenerative disorders. Science 343:506–511. doi:10.1126/science.1247363
Oates EC, Rossor AM, Hafezparast M et al (2013) Mutations in BICD2 cause dominant congenital spinal muscular atrophy and hereditary spastic paraplegia. Am J Hum Genet 92:965–973. doi:10.1016/j.ajhg.2013.04.018
Ohmi Y, Tajima O, Ohkawa Y et al (2011) Gangliosides are essential in the protection of inflammation and neurodegeneration via maintenance of lipid rafts: elucidation by a series of ganglioside-deficient mutant mice. J Neurochem 116:926–935. doi:10.1111/j.1471-4159.2010.07067.x
Orlacchio A, Kawarai T, Gaudiello F et al (2005) New locus for hereditary spastic paraplegia maps to chromosome 1p31.1-1p21.1. Ann Neurol 58:423–429. doi:10.1002/ana.20590
Orlacchio A, Patrono C, Gaudiello F et al (2008) Silver syndrome variant of hereditary spastic paraplegia: a locus to 4p and allelism with SPG4. Neurology 70:1959–1966. doi:10.1212/01.wnl.0000294330.27058.61
Orlacchio A, Babalini C, Borreca A et al (2010) SPATACSIN mutations cause autosomal recessive juvenile amyotrophic lateral sclerosis. Brain 133(Pt 2):591–598. doi:10.1093/brain/awp325
Orso G, Pendin D, Liu S et al (2009) Homotypic fusion of ER membranes requires the dynamin-like GTPase atlastin. Nature 460:978–983. doi:10.1038/nature08280
Orthmann-Murphy JL, Salsano E, Abrams CK et al (2009) Hereditary spastic paraplegia is a novel phenotype for GJA12/GJC2 mutations. Brain 132:426–438. doi:10.1093/brain/awn328
Oz-Levi D, Ben-Zeev B, Ruzzo EK et al (2012) Mutation in TECPR2 reveals a role for autophagy in hereditary spastic paraparesis. Am J Hum Genet 91:1065–1072. doi:10.1016/j.ajhg.2012.09.015
Paisan-Ruiz C, Dogu O, Yilmaz A et al (2008) SPG11 mutations are common in familial cases of complicated hereditary spastic paraplegia. Neurology 70:1384–1389. doi:10.1212/01.wnl.0000294327.66106.3d
Park SH, Zhu P-P, Parker RL, Blackstone C (2010) Hereditary spastic paraplegia proteins REEP1, spastin, and atlastin-1 coordinate microtubule interactions with the tubular ER network. J Clin Invest 120:1097–1110. doi:10.1172/JCI40979
Patel H, Cross H, Proukakis C et al (2002) SPG20 is mutated in Troyer syndrome, an hereditary spastic paraplegia. Nat Genet 31:347–348. doi:10.1038/ng937
Pérez-Brangulí F, Mishra HK, Prots I et al (2014) Dysfunction of spatacsin leads to axonal pathology in SPG11-linked hereditary spastic paraplegia. Hum Mol Genet 23:4859–4874. doi:10.1093/hmg/ddu200
Pfeffer G, Gorman GS, Griffin H et al (2014) Mutations in the SPG7 gene cause chronic progressive external ophthalmoplegia through disordered mitochondrial DNA maintenance. Brain 137:1323–1336. doi:10.1093/brain/awu060
Potter KA, Kern MJ, Fullbright G et al (2011) Central nervous system dysfunction in a mouse model of Fa2h deficiency. Glia 59:1009–1021. doi:10.1002/glia.21172
Proukakis C, Moore D, Labrum R, Wood NW, Houlden H (2011) Detection of novel mutations and review of published data suggests that hereditary spastic paraplegia caused by spastin (SPAST) mutations is found more often in males. J Neurol Sci 306(1–2):62–65. doi:10.1016/j.jns.2011.03.043
Rahman M, Haberman A, Tracy C et al (2012) Drosophila mauve mutants reveal a role of LYST homologs late in the maturation of phagosomes and autophagosomes. Traffic 13:1680–1692. doi:10.1111/tra.12005
Rainier S, Chai J-H, Tokarz D et al (2003) NIPA1 gene mutations cause autosomal dominant hereditary spastic paraplegia (SPG6). Am J Hum Genet 73:967–971. doi:10.1086/378817
Rainier S, Bui M, Mark E et al (2008) Neuropathy target esterase gene mutations cause motor neuron disease. Am J Hum Genet 82:780–785. doi:10.1016/j.ajhg.2007.12.018
Reid E, Dearlove AM, Osborn O et al (2000) A locus for autosomal dominant “pure” hereditary spastic paraplegia maps to chromosome 19q13. Am J Hum Genet 66:728–732
Reid E, Kloos M, Ashley-Koch A et al (2002) A kinesin heavy chain (KIF5A) mutation in hereditary spastic paraplegia (SPG10). Am J Hum Genet 71:1189–1194. doi:10.1086/344210
Renvoisé B, Stadler J, Singh R et al (2012) Spg20−/− mice reveal multimodal functions for Troyer syndrome protein spartin in lipid droplet maintenance, cytokinesis and BMP signaling. Hum Mol Genet 21:3604–3618. doi:10.1093/hmg/dds191
Ribai P, Stevanin G, Bouslam N et al (2006) A new phenotype linked to SPG27 and refinement of the critical region on chromosome. J Neurol 253:714–719. doi:10.1007/s00415-006-0094-2
Rismanchi N, Soderblom C, Stadler J et al (2008) Atlastin GTPases are required for Golgi apparatus and ER morphogenesis. Hum Mol Genet 17:1591–1604. doi:10.1093/hmg/ddn046
Rivière J-B, Ramalingam S, Lavastre V et al (2011) KIF1A, an axonal transporter of synaptic vesicles, is mutated in hereditary sensory and autonomic neuropathy type 2. Am J Hum Genet 89:219–230. doi:10.1016/j.ajhg.2011.06.013
Romagnolo A, Masera S, Mattioda A et al (2014) Atypical hereditary spastic paraplegia mimicking multiple sclerosis associated with a novel SPG11 mutation. Eur J Neurol 21:e14–e15. doi:10.1111/ene.12297
Rosenthal A, Jouet M, Kenwrick S (1992) Aberrant splicing of neural cell adhesion molecule L1 mRNA in a family with X-linked hydrocephalus. Nat Genet 2:107–112. doi:10.1038/ng1092-107
Ruano L, Melo C, Silva MC, Coutinho P (2014) The global epidemiology of hereditary ataxia and spastic paraplegia: a systematic review of prevalence studies. Neuroepidemiology 42:174–183. doi:10.1159/000358801
Sánchez-Ferrero E, Coto E, Beetz C et al (2013) SPG7 mutational screening in spastic paraplegia patients supports a dominant effect for some mutations and a pathogenic role for p. A510V. Clin Genet 83(3):257–262. doi:10.1111/j.1399-0004.2012.01896.x
Saugier-Veber P, Munnich A, Bonneau D et al (1994) X-linked spastic paraplegia and Pelizaeus-Merzbacher disease are allelic disorders at the proteolipid protein locus. Nat Genet 6:257–262. doi:10.1038/ng0394-257
Schneider SA, Bhatia KP (2010) Three faces of the same gene: FA2H links neurodegeneration with brain iron accumulation, leukodystrophies, and hereditary spastic paraplegias. Ann Neurol 68:575–577. doi:10.1002/ana.22211
Schüle R, Bonin M, Dürr A et al (2009a) Autosomal dominant spastic paraplegia with peripheral neuropathy maps to chr12q23-24. Neurology 72:1893–1898. doi:10.1212/WNL.0b013e3181a6086c
Schüle R, Schlipf N, Synofzik M et al (2009b) Frequency and phenotype of SPG11 and SPG15 in complicated hereditary spastic paraplegia. J Neurol Neurosurg Psychiatry 80:1402–1404. doi:10.1136/jnnp.2008.167528
Schüle R, Siddique T, Deng H-X et al (2010) Marked accumulation of 27-hydroxycholesterol in SPG5 patients with hereditary spastic paresis. J Lipid Res 51:819–823. doi:10.1194/jlr.M002543
Schuurs-Hoeijmakers JHM, Geraghty MT, Kamsteeg E-J et al (2012) Mutations in DDHD2, encoding an intracellular phospholipase A(1), cause a recessive form of complex hereditary spastic paraplegia. Am J Hum Genet 91:1073–1081. doi:10.1016/j.ajhg.2012.10.017
Schwartz CE, May MM, Carpenter NJ et al (2005) Allan-Herndon-Dudley syndrome and the monocarboxylate transporter 8 (MCT8) gene. Am J Hum Genet 77:41–53. doi:10.1086/431313
Sedel F, Fontaine B, Saudubray JM, Lyon-Caen O (2007) Hereditary spastic paraparesis in adults associated with inborn errors of metabolism: a diagnostic approach. J Inherit Metab Dis 30:855–864. doi:10.1007/s10545-007-0745-1
Seri M, Cusano R, Forabosco P et al (1999) Genetic mapping to 10q23.3-q24.2, in a large Italian pedigree, of a new syndrome showing bilateral cataracts, gastroesophageal reflux, and spastic paraparesis with amyotrophy. Am J Hum Genet 64:586–593. doi:10.1086/302241
Shimazaki H, Takiyama Y, Ishiura H et al (2012) A homozygous mutation of C12orf65 causes spastic paraplegia with optic atrophy and neuropathy (SPG55). J Med Genet 49:777–784. doi:10.1136/jmedgenet-2012-101212
Shimazaki H, Honda J, Naoi T et al (2014) Autosomal-recessive complicated spastic paraplegia with a novel lysosomal trafficking regulator gene mutation. J Neurol Neurosurg Psychiatry 85:1024–1028. doi:10.1136/jnnp-2013-306981
Simpson MA, Cross H, Proukakis C et al (2003) Maspardin is mutated in mast syndrome, a complicated form of hereditary spastic paraplegia associated with dementia. Am J Hum Genet 73:1147–1156. doi:10.1086/379522
Sivakumar K, Sambuughin N, Selenge B et al (1999) Novel exon 3B proteolipid protein gene mutation causing late-onset spastic paraplegia type 2 with variable penetrance in female family members. Ann Neurol 45:680–683
Słabicki M, Theis M, Krastev DB et al (2010) A genome-scale DNA repair RNAi screen identifies SPG48 as a novel gene associated with hereditary spastic paraplegia. PLoS Biol 8:e1000408. doi:10.1371/journal.pbio.1000408
Spiegel R, Mandel H, Saada A et al (2014) Delineation of C12orf65-related phenotypes: a genotype–phenotype relationship. Eur J Hum Genet 22:1019–1025. doi:10.1038/ejhg.2013.284
Starling A, Rocco P, Passos-Bueno MR, Hazan J, Marie SK, Zatz M (2002) Autosomal dominant (AD) pure spastic paraplegia (HSP) linked to locus SPG4 affects almost exclusively males in a large pedigree. J Med Genet 39(12):e77
Steinmüller R, Lantigua-Cruz A, Garcia-Garcia R et al (1997) Evidence of a third locus in X-linked recessive spastic paraplegia. Hum Genet 100:287–289. doi:10.1007/s004390050507
Stevanin G, Paternotte C, Coutinho P et al (2007a) A new locus for autosomal recessive spastic paraplegia (SPG32) on chromosome 14q12-q21. Neurology 68:1837–1840. doi:10.1212/01.wnl.0000262043.53386.22
Stevanin G, Santorelli FM, Azzedine H et al (2007b) Mutations in SPG11, encoding spatacsin, are a major cause of spastic paraplegia with thin corpus callosum. Nat Genet 39:366–372. doi:10.1038/ng1980
Stevanin G, Azzedine H, Denora P et al (2008a) Mutations in SPG11 are frequent in autosomal recessive spastic paraplegia with thin corpus callosum, cognitive decline and lower motor neuron degeneration. Brain 131:772–784. doi:10.1093/brain/awm293
Stevanin G, Ruberg M, Brice A (2008b) Recent advances in the genetics of spastic paraplegias. Curr Neurol Neurosci Rep 8:198–210
Svenson IK, Kloos MT, Gaskell PC et al (2004) Intragenic modifiers of hereditary spastic paraplegia due to spastin gene mutations. Neurogenetics 5:157–164. doi:10.1007/s10048-004-0186-z
Synofzik M, Gonzalez MA, Lourenco CM et al (2014) PNPLA6 mutations cause Boucher-Neuhauser and Gordon Holmes syndromes as part of a broad neurodegenerative spectrum. Brain 137:69–77. doi:10.1093/brain/awt326
Takiyama Y (2014) Japan Spastic Paraplegia Research Consortium (JASPAC). Brain Nerve 66(10):1210–1217. doi:10.11477/mf.1416200012
Tallaksen CM, Dürr A, Brice A (2001) Recent advances in hereditary spastic paraplegia. Curr Opin Neurol 14:457–463
Tamagaki A, Shima M, Tomita R et al (2000) Segregation of a pure form of spastic paraplegia and NOR insertion into Xq11.2. Am J Med Genet 94:5–8. doi:10.1002/1096-8628(20000904)94:1<5::AID-AJMG2>3.0.CO;2-O
Tanyel MC, Mancano LD (1997) Neurologic findings in vitamin E deficiency. Am Fam Physician 55:197–201
Tarrade A, Fassier C, Courageot S et al (2006) A mutation of spastin is responsible for swellings and impairment of transport in a region of axon characterized by changes in microtubule composition. Hum Mol Genet 15:3544–3558. doi:10.1093/hmg/ddl431
Tessa A, Silvestri G, de Leva MF et al (2008) A novel KIF5A/SPG10 mutation in spastic paraplegia associated with axonal neuropathy. J Neurol 255:1090–1092. doi:10.1007/s00415-008-0840-8
Tesson C, Nawara M, Salih MAM et al (2012) Alteration of fatty-acid-metabolizing enzymes affects mitochondrial form and function in hereditary spastic paraplegia. Am J Hum Genet 91:1051–1064. doi:10.1016/j.ajhg.2012.11.001
Theofilopoulos S, Griffiths WJ, Crick PJ et al (2014) Cholestenoic acids regulate motor neuron survival via liver X receptors. J Clin Invest 124:4829–4842. doi:10.1172/JCI68506
Tiranti V, Corona P, Greco M et al (2000) A novel frameshift mutation of the mtDNA COIII gene leads to impaired assembly of cytochrome c oxidase in a patient affected by Leigh-like syndrome. Hum Mol Genet 9:2733–2742. doi:10.1093/hmg/9.18.2733
Tsang HTH, Edwards TL, Wang X et al (2009) The hereditary spastic paraplegia proteins NIPA1, spastin and spartin are inhibitors of mammalian BMP signalling. Hum Mol Genet 18:3805–3821. doi:10.1093/hmg/ddp324
Tsaousidou MK, Ouahchi K, Warner TT et al (2008) Sequence alterations within CYP7B1 implicate defective cholesterol homeostasis in motor-neuron degeneration. Am J Hum Genet 82:510–515. doi:10.1016/j.ajhg.2007.10.001
Tucci A, Liu Y-T, Preza E et al (2013) Novel C12orf65 mutations in patients with axonal neuropathy and optic atrophy. J Neurol Neurosurg Psychiatry 85:486–492. doi:10.1136/jnnp-2013-306387
Uhlenberg B, Schuelke M, Rüschendorf F et al (2004) Mutations in the gene encoding gap junction protein alpha 12 (connexin 46.6) cause Pelizaeus–Merzbacher-like disease. Am J Hum Genet 75:251–260. doi:10.1086/422763
Valdmanis PN, Meijer IA, Reynolds A et al (2007) Mutations in the KIAA0196 gene at the SPG8 locus cause hereditary spastic paraplegia. Am J Hum Genet 80:152–161. doi:10.1086/510782
Valente EM, Brancati F, Caputo V et al (2002) Novel locus for autosomal dominant pure hereditary spastic paraplegia (SPG19) maps to chromosome 9q33-q34. Ann Neurol 51:681–685. doi:10.1002/ana.10204
Varga R-E, Schüle R, Fadel H et al (2013) Do not trust the pedigree: reduced and sex-dependent penetrance at a novel mutation hotspot in ATL1 blurs autosomal dominant inheritance of spastic paraplegia. Hum Mutat 34:860–863. doi:10.1002/humu.22309
Vazza G, Zortea M, Boaretto F et al (2000) A new locus for autosomal recessive spastic paraplegia associated with mental retardation and distal motor neuropathy, SPG14, maps to chromosome 3q27-q28. Am J Hum Genet 67:504–509. doi:10.1086/303017
Verkerk AJMH, Schot R, Dumee B et al (2009) Mutation in the AP4M1 gene provides a model for neuroaxonal injury in cerebral palsy. Am J Hum Genet 85:40–52. doi:10.1016/j.ajhg.2009.06.004
Verny C, Guegen N, Desquiret V et al (2011) Hereditary spastic paraplegia-like disorder due to a mitochondrial ATP6 gene point mutation. Mitochondrion 11:70–75. doi:10.1016/j.mito.2010.07.006
Votsi C, Zamba-Papanicolaou E, Middleton LT et al (2014) A novel GBA2 gene missense mutation in spastic ataxia. Ann Hum Genet 78:13–22. doi:10.1111/ahg.12045
Wakil SM, Bohlega S, Hagos S et al (2013) A novel splice site mutation in ERLIN2 causes hereditary spastic paraplegia in a Saudi family. Eur J Med Genet 56:43–45. doi:10.1016/j.ejmg.2012.10.003
Wan J, Yourshaw M, Mamsa H et al (2012) Mutations in the RNA exosome component gene EXOSC3 cause pontocerebellar hypoplasia and spinal motor neuron degeneration. Nat Genet 44:704–708. doi:10.1038/ng.2254
Wang X, Shaw WR, Tsang HTH et al (2007) Drosophila spichthyin inhibits BMP signaling and regulates synaptic growth and axonal microtubules. Nat Neurosci 10:177–185. doi:10.1038/nn1841
Wang X, Yang Y, Wang X, Li C, Jia J (2014) A novel KIAA0196 (SPG8) mutation in a Chinese family with spastic paraplegia. Chin Med J (Engl) 127(10):1987–1989
Watts GDJ, Wymer J, Kovach MJ et al (2004) Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat Genet 36:377–381. doi:10.1038/ng1332
Wedding IM, Koht J, Tran GT et al (2014) Spastic paraplegia type 7 is associated with multiple mitochondrial DNA deletions. PLoS One 9:e86340. doi:10.1371/journal.pone.0086340
Werner HB, Krämer-Albers E-M, Strenzke N et al (2013) A critical role for the cholesterol-associated proteolipids PLP and M6B in myelination of the central nervous system. Glia 61:567–586. doi:10.1002/glia.22456
Windpassinger C, Auer-Grumbach M, Irobi J et al (2004) Heterozygous missense mutations in BSCL2 are associated with distal hereditary motor neuropathy and Silver syndrome. Nat Genet 36:271–276. doi:10.1038/ng1313
Yao D, McGonigal R, Barrie JA et al (2014) Neuronal expression of GalNAc transferase is sufficient to prevent the age-related neurodegenerative phenotype of complex ganglioside-deficient mice. J Neurosci 34:880–891. doi:10.1523/JNEUROSCI.3996-13.2014
Yıldırım Y, Orhan EK, Iseri SAU et al (2011) A frameshift mutation of ERLIN2 in recessive intellectual disability, motor dysfunction and multiple joint contractures. Hum Mol Genet 20:1886–1892. doi:10.1093/hmg/ddr070
Zanni G, Scotton C, Passarelli C et al (2013) Exome sequencing in a family with intellectual disability, early onset spasticity, and cerebellar atrophy detects a novel mutation in EXOSC3. Neurogenetics 14:247–250. doi:10.1007/s10048-013-0371-z
Zhao X, Alvarado D, Rainier S et al (2001) Mutations in a newly identified GTPase gene cause autosomal dominant hereditary spastic paraplegia. Nat Genet 29:326–331. doi:10.1038/ng758
Zhao G, Hu Z, Shen L et al (2008) A novel candidate locus on chromosome 11p14.1-p11.2 for autosomal dominant hereditary spastic paraplegia. Chin Med J (Engl) 121:430–434
Zhu P-P, Denton KR, Pierson TM et al (2014) Pharmacologic rescue of axon growth defects in a human iPSC model of hereditary spastic paraplegia SPG3A. Hum Mol Genet 23:5638–5648. doi:10.1093/hmg/ddu280
Zivony-Elboum Y, Westbroek W, Kfir N et al (2012) A founder mutation in Vps37A causes autosomal recessive complex hereditary spastic paraparesis. J Med Genet 49:462–472. doi:10.1136/jmedgenet-2012-100742
Zöller I, Meixner M, Hartmann D et al (2008) Absence of 2-hydroxylated sphingolipids is compatible with normal neural development but causes late-onset axon and myelin sheath degeneration. J Neurosci 28:9741–9754. doi:10.1523/JNEUROSCI.0458-08.2008
Zortea M, Vettori A, Trevisan CP et al (2002) Genetic mapping of a susceptibility locus for disc herniation and spastic paraplegia on 6q23.3-q24.1. J Med Genet 39:387–390
Züchner S, Wang G, Tran-Viet K-N et al (2006) Mutations in the novel mitochondrial protein REEP1 cause hereditary spastic paraplegia type 31. Am J Hum Genet 79:365–369. doi:10.1086/505361
Acknowledgments
The authors are grateful to Drs. Odile Boesflug-Tanguy, Christel Depienne, Frédéric Darios, Cyril Goizet, Annick Toutain and Khalid H. El-Hachimi for helpful discussions. The authors’ work was partially funded by the French National Agency for Research (to GS), the European Union (Omics call of the 7th framework program, NEUROMICS, to GS; E-rare program, NEUROLIPID, to GS), the Tom Wahlig Foundation, the VERUM Foundation (to GS), the Vestre Viken Health trust (to JK) and the program “Investissements d’Avenir” ANR-10-IAIHU-06 (to GS). CT was the recipient of a PhD fellowship from the French Ministry for Research from the Doctoral School of the Ecole Pratique des Hautes Etudes.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Tesson, C., Koht, J. & Stevanin, G. Delving into the complexity of hereditary spastic paraplegias: how unexpected phenotypes and inheritance modes are revolutionizing their nosology. Hum Genet 134, 511–538 (2015). https://doi.org/10.1007/s00439-015-1536-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-015-1536-7