
Abstract
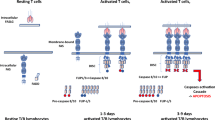
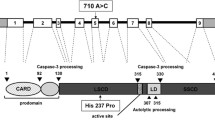
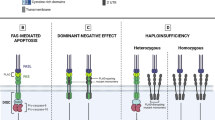
Autoimmune lymphoproliferative syndrome (ALPS) is characterized by lymphadenopathy, elevated numbers of T cells with αβ-T cell receptors but neither CD4 nor CD8 co-receptors, and impaired lymphocyte apoptosis in vitro. Defects in the Fas receptor are the most common cause of ALPS (ALPS Ia), but in rare cases other apoptosis proteins have been implicated, including caspase-10 (ALPS II). We investigated the role of variants of caspase-10 in ALPS. Of 32 unrelated probands with ALPS who did not have Fas defects, two were heterozygous for the caspase-10 missense mutation I406L. Like the previously reported ALPS II-associated mutation L285F, I406L impaired apoptosis when transfected alone and dominantly inhibited apoptosis mediated by wild type caspase-10 in a co-transfection assay. Other variants in caspase-10, V410I and Y446C, were found in 3.4 and 1.6% of chromosomes in Caucasians, and in 0.5 and <0.5% of African Americans, respectively. In contrast to L285F and I406L, these variants had no dominant negative effect in co-transfection assays into the H9 lymphocytic cell line. We found healthy individuals homozygous for V410I, challenging the earlier suggestion that homozygosity for V410I alone causes ALPS. Moreover, an association analysis suggested protection from severe disease by caspase-10 V410I in 63 families with ALPS Ia due to dominant Fas mutations (P<0.05). Thus, different genetic variations in caspase-10 can produce contrasting phenotypic effects.


Similar content being viewed by others
References
Barnhart BC, Alappat EC, Peter ME (2003) The CD95 type I/type II model. Semin Immunol 15:185–193
Bleesing JJH, Puck JM (2004) Autoimmune lymphoproliferative syndrome. In: ER Stiehm HO, Winkelstein J (eds) Immunologic disorders in infants and children. Elsevier, Saunders, Philadelphia pp 605–617
Bleesing JJH, Brown MR, Straus SE, Dale JK, Siegel RM, Johnson M, Lenardo MJ, Puck JM, Fleisher TA (2001) Immunophenotypic profiles in families with autoimmune lymphoproliferative syndrome. Blood 98:2466–2473
Clayton D (1999) A generalization of the transmission/disequilibrium test for uncertain-haplotype transmission. Am J Hum Genet 65:1170–1177
Drappa J, Vaishnaw AK, Sullivan KE, Chu J-L, Elkon KB (1996) Fas gene mutations in the Canale–Smith syndrome, an inherited lymphoproliferative disorder associated with autoimmunity. N Engl J Med 335:1643–1649
Fang BS, Sneller MC, Straus SE, Frenkel L, Dale JK, Rick ME (2000) Report of a factor VIII inhibitor in a patient with autoimmune lymphoproliferative syndrome. Am J Hematol 64:214–217
Fisher GH, Rosenberg FJ, Straus SE, Dale JK, Middleton LA, Lin AY, Strober W, Lenardo MJ, Puck JM (1995) Dominant interfering Fas gene mutations impair apoptosis in a human autoimmune lymphoproliferative syndrome. Cell 81:935–946
Gill JM, Quisel AM, Rocca PV, Walters DT (2003) Diagnosis of systemic lupus erythematosus. Am Fam Physician 68:2179–2186
Gronbæk K, Dalby T, Zeuthen J, Ralfkiaer E, Guldberg P (2000) The V410I (G1228A) variant of the caspase-10 gene is a common polymorphism of the Danish population. Blood 95:2184–2185
Holler N, Zaru R, Micheau O, Thome M, Attinger A, Valitutti S, Bodmer J-L, Schneider P, Seed B, Tschop J (2000) Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol 1:489–495
Horvath S, Xu X, Laird NM (2001) The family based association test method: strategies for studying general genotype–phenotype associations. Eur J Hum Genet 9:301–306
Hull KM, Drewe E, Aksentijevich I, Singh HK, Wong K, McDermott EM, Dean J, Powell RJ, Kastner DL (2002) The TNF receptor-associated periodic syndrome (TRAPS): emerging concepts of an autoinflammatory disorder. Medicine 81:349–368
Jackson CE, Fischer RE, Hsu AP, Anderson SM, Choi Y, Wang J, Dale JK, Fleisher TA, Middelton LA, Sneller MC, Lenardo MJ, Straus SE, Puck JM (1999) Autoimmune lymphoproliferative syndrome with defective Fas: genotype influences penetrance. Am J Hum Genet 64:1002–1014
Laird NM, Horvath S, Xu X (2000) Implementing a unified approach to family-based tests of association. Genet Epidemiol 19(Suppl 1):S36–S42
Lim MS, Straus SE, Dale JK, Fleisher TA, Stetler-Stevenson M, Strober W, Sneller MC, Puck JM, Lenardo MJ, Elenitoba-Johnson KSJ, Lin AY, Raffeld M, Jaffe ES (1998) Pathological findings in human autoimmune lymphoproliferative syndrome. Am J Pathol 153:1541–1550
Maruyama K, Tomita T, Shinozaki K, Kume H, Asada H, Saido TC, Ishiura S, Iwatsubo T, Obata K (1996) Familial Alzheimer’s disease-linked mutations at Val717 of amyloid precursor protein are specific for the increased secretion of Aβ42(43). Biochem Biophys Res Commun 227:730–735
Park WS, Lee JH, Shin MS, Park JY, Kim HS, Kim YS, Lee SN, Xiao W, Park CH, Lee SH, Yoo NJ, Lee JY (2002) Inactivating mutations of the caspase-10 gene in gastric cancer. Oncogene 21:2919–2925
Puck JM, Zhu S (2003) Immune disorders caused by defects in the caspase cascade. Curr Allergy Asthma Rep 3:378–384
Rieux-Laucat F, Le Deist F, Hivroz C, Roberts IAG, Debatin KM, Fischer A, de Villartay JP (1995) Mutations in Fas associated with human lymphoproliferative syndrome and autoimmunity. Science 268:1347–1349
Sneller MC, Wang J, Dale JK, Strober W, Middelton LA, Choi Y, Fleisher TA, Lim MS, Jaffe ES, Puck JM, Lenardo MJ, Straus SE (1997) Clincial, immunologic, and genetic features of an autoimmune lymphoproliferative syndrome associated with abnormal lymphocyte apoptosis. Blood 89:1341–1348
Sneller MC, Dale JK, Straus SE (2003) Autoimmune lymphoproliferative syndrome. Curr Opin Rheumatol 15:417–421
Straus SE, Jaffe ES, Puck JM, Dale JK, Elkon KB, Rösen-Wolff A, Peters AM, Sneller MC, Hallahan CW, Wang J, Fischer RE, Jackson CM, Lin AY, Baumler C, Siegert E, Marx A, Vaishnaw AK, Grodzicky T, Fleisher TA, Lenardo MJ (2001) The development of lymphomas in families with autoimmune lymphoproliferative syndrome with germline Fas mutations and defective lymphocyte apoptosis. Blood 98:194–200
Takahashi T, Tanaka M, Brannan CI, Jenkins NA, Copeland NG, Suda T, Nagata S (1994) Generalized lymphoproliferative disease in mice, caused by a point mutation in the Fas ligand. Cell 76:969–976
Vacek MM, Schäffer AA, Davis J, Fischer RE, Dale JK, Adams S, Straus SE, Puck JM (2006) HLA B44 is associated with decreased severity of autoimmune lymphoproliferative syndrome in patients with CD95 defects (ALPS type Ia). Clin Immunol 118:59–65
Vaishnaw AK, Toubi E, Ohsako S, Drappa J, Buys S, Estrada J, Sitarz A, Zemel L, Chu J-L, Elkon KB (1999) The spectrum of apoptotic defects and clinical manifestations, including systemic lupus erythematosus, in humans with CD95 (Fas/APO-1) mutations. Arthritis Rheum 42:1833–1842
Wang J, Zheng L, Lobito A, Chan FK-M, Dale J, Sneller M, Yao X, Puck JM, Straus SE, Lenardo MJ (1999) Inherited human Caspase 10 mutations underlie defective lymphocyte and dendritic cell apoptosis in autoimmune lymphoproliferative syndrome type II. Cell 98:47–58
Wang J, Chun HJ, Wong W, Spencer DM, Lenardo MJ (2001) Caspase-10 is an initiator caspase in death receptor signaling. Proc Natl Acad Sci USA 98:13884–13888
Watanabe-Fukunaga R, Brannan CI, Copeland NG, Jenkins NA, Nagata S (1992) Lymphoproliferation disorder in mice explained by defects in Fas antigen that mediates apoptosis. Nature 356:314–317
Wu J, Wilson J, He J, Xiang L, Schur PH, Mountz JD (1996) Fas ligand mutation in a patient with systemic lupus erythematosus and lymphoproliferative disease. J Clin Invest 98:1107–1113
Acknowledgements
The authors thank participating ALPS family members. They also thank Kusum Viswanathan for his diagnosis and referral of one of the probands, and Koneti Rao, Fred Gill and Faith Dugan for patient management, Dan Kastner for TNFRSF1A consultation, Maggie Brown and Tom Fleisher for cell surface phenotype analysis, Stacie Anderson and Martha Kirby for apoptosis assays, and Gretchen Gibney for assistance with data management. This research was supported by the Intramural Research Programs of the NHGRI, NIAID and NLM, National Institutes of Health.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Zhu, S., Hsu, A.P., Vacek, M.M. et al. Genetic alterations in caspase-10 may be causative or protective in autoimmune lymphoproliferative syndrome. Hum Genet 119, 284–294 (2006). https://doi.org/10.1007/s00439-006-0138-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-006-0138-9