
Abstract
Renal complications of influenza A virus infections are uncommon but can contribute to a deterioration in the patient’s condition, which include acute kidney injury (AKI) in critically ill patients, rhabdomyolysis, hemolytic uremic syndrome (HUS), acute glomerulonephritis (AGN), disseminated intravascular coagulation (DIC), Goodpasture’s syndrome, and acute tubulointerstitial nephritis (TIN). The clinical characteristics of AKI in critically ill patients with pandemic influenza A(H1N1) 2009 virus (A(H1N1)pdm09) infection are similar to uninfected patients. Underlying conditions associated with AKI include older age, diabetes mellitus, obesity, pregnancy, history of asthma, and chronic kidney disease. Histologic examination of the kidneys from patients with A(H1N1)pdm09 infection who died include acute tubular necrosis (ATN), myoglobin pigment, and DIC. A(H1N1)pdm09 is present in the kidneys of some patients. The clinical characteristics of patients with rhabdomyolysis associated with influenza A include younger age and the frequent occurrence of muscle symptoms. AKI occurs in approximately one third of patients with rhabdomyolysis due to influenza A. HUS is associated with A(H1N1)pdm09 as follows: Streptococcus pneumoniae-associated HUS following A(H1N1)pdm09 infection, HUS triggered by A(H1N1)pdm09 in patients with genetic complement dysregulation, and HUS associated with A(H1N1)pdm09 without known underlying disorder. AGN, Goodpasture’s syndrome, and acute TIN are extremely rare complications of influenza A virus infection. Although the pathogenesis underlying renal injuries due to influenza A virus has not been delineated, some hypotheses have been advanced, including ATN due to renal hypoperfusion or rhabdomyolysis, glomerular microthrombosis due to DIC, direct viral injury to the kidney, and an altered immune system with systemic mononuclear cell activation following influenza A virus infections.
Similar content being viewed by others

Introduction
Influenza is an acute respiratory disease of global importance that has caused epidemics and pandemics of human disease [24]. Although most influenza infections are self-limited, infants, young children, elderly adults, and people with underlying pulmonary and cardiac diseases are at increased risk for hospitalization for illnesses attributable to influenza, and some die from their complications [24, 41]. Various complications of seasonal influenza virus infection have been reported in the pulmonary, neurological, cardiac, and muscular systems [24, 73].
Renal complications of seasonal influenza A virus are uncommon and have been reported primarily as single cases or small series of patients, which include rhabdomyolysis [2, 4, 14, 25, 27, 32, 35, 40, 52, 70, 78, 79, 87, 96, 98, 99, 105], hemolytic uremic syndrome (HUS) [5, 26, 68, 97], disseminated intravascular coagulation (DIC) [26, 78, 102], acute renal failure (ARF) in critically ill patients [60, 78, 100, 101], and Goodpasture’s syndrome [37, 103] (Table 1).
Human infection of a novel influenza A (H1N1) virus of swine origin was first reported in Mexico during the spring of 2009 [31, 67]. Thereafter, the pandemic influenza A (H1N1) 2009 virus (A(H1N1)pdm09) spread rapidly and globally, resulting in the worldwide influenza pandemic [86, 104]. The clinical manifestations of A(H1N1)pdm09 and seasonal influenza are similar, and most illnesses associated with A(H1N1)pdm09 are mild with an overall case fatality rate of ≤0.5 % [86, 104]. However, in contrast to seasonal influenza, most of the serious illnesses caused by A(H1N1)pdm09 have occurred among children and non-elderly adults [104].
The complications of A(H1N1)pdm09 are similar to the complications of the seasonal influenza A virus, and include an exacerbation of underlying chronic illnesses, respiratory tract, neurologic, cardiac, and musculoskeletal complications, and secondary bacterial infections [74, 86]. Unlike seasonal influenza A infections, renal complications of A(H1N1)pdm09 have been increasingly reported, and include acute kidney injury (AKI) in critically ill patients, rhabdomyolysis, HUS, acute postinfectious glomerulonephritis (AGN), DIC, and acute tubulointerstitial nephritis (TIN) (Table 1).
Herein, we review the renal complications of influenza A virus infections, especially of A(H1N1)pdm09 infections, as have been described to date.
AKI in critically ill patients
AKI is the new consensus term for ARF [80] and has replaced ARF to emphasize that a continuum of kidney injury exists that begins long before sufficient loss of excretory kidney function can be measured with standard laboratory tests [10]. The Acute Dialysis Quality Initiative proposed the risk, injury, failure, loss, and end-stage (RIFLE) criteria as a definition for AKI [12]. The AKI network subsequently proposed the AKIN criteria with some minor modifications to the RIFLE criteria [57]. Both definitions have now been validated in thousands of patients and seemed to be similar [10].
AKI has emerged as a major public health problem and is common in critically ill patients [10, 80]. The occurrence of AKI is ≥36 % on the day after admission to an intensive care unit (ICU) [8], and the prevalence is ≥60 % during the ICU admission [39]. Moreover, critically ill patients with AKI incur an increased risk of hospital mortality [39].
AKI and ARF have rarely been reported in patients with seasonal influenza A virus infections as single case reports [60, 78, 101]. Watanabe et al. studied 45 hospitalized children with seasonal influenza A virus infection and reported that 24.4 % of the patients had renal involvement, of which 11 % exhibited ARF; all of the patients with renal involvement had sepsis and multiple organ dysfunction syndrome (MODS) [100].
AKI occurs in approximately one third of all hospitalized patients with A(H1N1)pdm09 [17, 28, 67]. Perez-Padilla et al. studied hospitalized patients with A(H1N1)pdm09 pneumonia in Mexico and reported that 6 of 18 patients developed AKI, and 5 patients died [67]. Demirjian et al. reported that 37 of 89 adult patients hospitalized with A (H1N1) pdm09 in the USA developed AKI; the majority of the patients were critically ill; 12 of 89 patients required renal replacement therapy (RRT); and 24 % of the patients with AKI died [28]. Brien et al. reported that 11 of 34 patients hospitalized with A(H1N1)pmd09 in Ireland developed AKI; 4 of 34 patients required RRT; and the most common cause of AKI was sepsis with acute tubular necrosis (ATN) [17].
AKI has been reported to occur in 7–66.7 % of critically ill patients with A(H1N1)pdm09 [1, 11, 21, 43, 47, 48, 54, 62, 69, 84, 90, 91, 95]. This diversity in the incidence of AKI results from different severities of infections and/or distinct definitions in AKI among studies. In patients with A(H1N1)pdm09 who are admitted to the ICU for mechanical ventilator support, the incidence of AKI, as defined by RIFLE or AKIN criteria, is 51–66.7 % [47, 62, 84]. Of the critically ill patients with A(H1N1)pdm09 and AKI, 15.6–51.6 % required RRT [1, 11, 21, 43, 47, 54, 62, 69, 90, 91]. The mortality rate of patients with A(H1N1)pdm09 and AKI is 25–92.3 % [1, 11, 21, 43, 47, 54, 62, 69, 90, 91], and AKI is associated with an increase in mortality [1, 28, 43, 54, 62, 69, 90, 91]. These clinical characteristics of AKI in critically ill patients with A(H1N1)pdm09 virus infections are similar to the characteristics of AKI in critically ill patients without A(H1N1)pdm09 infections [8, 39].
Underlying conditions, organ dysfunctions, and laboratory data have been reported as the risk factors for AKI (Table 2). Underlying conditions associated with AKI included older age [43, 84], diabetes mellitus [28, 69], obesity or elevated body mass index [28, 84], pregnancy [90], a history of asthma [84], and chronic kidney disease (CKD) [28, 43, 69]. Patients with organ dysfunction and AKI had the following characteristics: required ventilator support or had respiratory dysfunction [1, 54, 62, 69]; used vasopressor or had cardiovascular dysfunction [1, 69, 90]; had hematological dysfunction [28, 62, 90]; and had high Acute Physiology and Chronic Health Evaluation II [1, 54, 62, 84], Sequential Organ Failure Assessment [43, 54, 90], and Murray scores [90]. Patients with elevated creatine kinase (CK) levels [28, 69], severe acidosis, elevated C-reactive protein concentrations, or lactate dehydrogenase levels [1] were more likely to develop AKI.
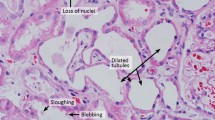
The precise pathogenic mechanisms underlying the development of AKI in patients with influenza A remain unclear; however, the following hypotheses have been postulated: ATN due to renal hypoperfusion or rhabdomyolysis, DIC, and direct viral injury to the kidney (although not proven) [84, 90, 99]. Based on histologic studies, Soto-Abraham et al. reported that one of five patients who died of A(H1N1)pdm09 had ATN [85]. Mauad et al. analyzed the autopsy findings of 21 patients who died of A(H1N1)pdm09 and showed that all patients exhibited mild-to-moderate forms of ATN; myoglobin pigment in the tubules existed in four patients and thrombotic angiopathy existed in another patient [55]. Bal et al. reported two patients with ATN and another patient with pathologic findings in the kidney consistent with DIC among nine patients who died of A(H1N1)pdm09 [9]. Nin et al. studied renal biopsies of four patients who died of A(H1N1)pdm09 and showed that two patients exhibited ATN with an increase in immunoreactivity for A(H1N1)pdm09 viral nucleoprotein in the distal tubules and in Bowman’s capsule epithelia [61]. Carmona et al. examined the autopsy specimens of five patients who died of A(H1N1)pdm09 and showed the following: ATN was present in all of the patients; there was no evidence of direct virus-induced kidney injury; and A(H1N1)pdm09 were in the cytoplasma of glomerular macrophages in the kidneys of four patients [20]. These studies indicate that ATN frequently occurs in patients who die of A(H1N1)pdm09, myoglobin pigments and glomerular microthrombosis due to DIC are present in some patients, and A(H1N1)pdm09 is present in the kidneys of some patients, although there is no evidence of direct viral injury to the kidney.
Rhabdomyolysis
Rhabdomyolysis is a potentially life-threatening syndrome characterized by the leak of muscle contents, including electrolytes, myoglobin, and other sarcoplasmic proteins, into the circulation [16, 46]. AKI is a potential complication of severe rhabdomyolysis, regardless of etiology, that develops in 33 % of patients [16, 46].
There are multiple potential causes of rhabdomyolysis, which are divided into the following eight categories: trauma, exertion, muscle hypoxia, genetic defects, body-temperature changes, metabolic and electrolyte disorders, drugs and toxins, and infections [16]. Viral infections as a cause of rhabdomyolysis have been described in a number of reports worldwide, of which influenza A and B viruses are the most common [46]. Although the pathogenesis underlying the development of rhabdomyolysis in influenza A virus infection has not been completely determined, it is presumed that direct viral invasion, viral toxins, or cytokines may induce myonecrosis causing rhabdomyolysis [46, 81].
Rhabdomyolysis in seasonal influenza A infection has been reported in 31 patients [2, 4, 14, 23, 25, 27, 29, 32, 33, 35, 40, 52, 58, 59, 70, 78, 79, 81, 87, 96, 98, 99, 105]. The clinical characteristics of the 31 patients included a relatively young age (median age, 28 years; range, 3–83 years), male predominance (18 of 31), and frequent occurrence of muscle symptoms (muscle pain, 24 of 31; muscle weakness, 18 of 31). Peak concentrations of CK ranged from 1,263 to 1,150,000 U/l (mean, 103,412 U/l). ARF occurred in 24 patients (77.4 %), and 21 patients with ARF underwent RRT. Although most patients recovered completely without any sequelae, three patients died secondary to MODS [25, 81, 105] and one patient with diabetes mellitus developed CKD [81]. Muscle biopsies showed a focal necrosis of muscle fibers without inflammation [4, 29, 58, 105] or degeneration of muscle fibers with an infiltration of inflammatory cells [4, 33, 40, 87]. These results suggest that rhabdomyolysis in patients with influenza A infections might result from muscle necrosis due to cytokines or viral myositis.
Although the true incidence of rhabdomyolysis in A(H1N1)pdm09 infections is unknown, a recent report showed that CK levels were elevated in 13.9 % of hospitalized patients with A(H1N1)pdm09 infections [19]. Another report demonstrated mild to moderate elevation of CK levels in 62 % of patients with A(H1N1)pdm09 pneumonia and respiratory failure [67]. Eleven patients with A(H1N1)pmd09 infections have been reported to develop rhabdomyolysis, and their clinical characteristics were similar to the patients with seasonal influenza A [7, 22, 30, 38, 50, 65, 75, 82, 88, 93]. The median age was 28 years (range, 8–59 years) and 6 of the 11 patients were males. The peak concentrations of CK were 1,371–1,127,000 U/l (mean, 206,355 U/l). AKI occurred in seven patients (63.6 %), and five patients with AKI underwent RRT. Nine patients recovered, but two patients died secondary to MODS [88, 93].
HUS
HUS is a disease characterized by nonimmune hemolytic anemia, thrombocytopenia, and renal impairment [64, 94]. Microvascular injury with endothelial cell damage is a pathologic characteristic of all forms of HUS [94]. The various etiologies of HUS allow classification into infection induced (Shiga- and verotoxin-producing bacteria, neuraminidase-producing Streptococcus pneumoniae, and human immunodeficiency virus), genetic (complement dysregulation and defective cobalamine metabolism), medication induced (calcineurin inhibitors, cytotoxic, chemotherapy agents, and quinine), and HUS associated with systemic diseases characterized by microvascular injury [15, 94]. The most common form of HUS is caused by verotoxin-producing Escherichia coli that cause prodromal acute enteritis and is termed diarrhea associated, typical HUS [64, 94].
Approximately 10 % of cases of HUS are classified as atypical when not caused by verotoxin-producing bacteria [63]. The primary causes of atypical HUS are defects in the regulation of the alternative complement pathway on vascular endothelial cells, including complement factor H (CFH), factor I, factor B, thrombomodulin, C3, membrane cofactor protein (MCP), and autoantibodies against factor H with or without CFH-related protein 1 and 3 deficiency [15].
HUS associated with seasonal influenza A virus is extremely rare; only four patients have been reported [5, 26, 68, 97], in whom two were postrenal transplant patients. Peterson and Olsen reported a 20-year-old female with posttransplant HUS related to influenza A virus infection [68]. The patient did not take calcineurin inhibitors, and refractory thrombocytopenia and hemolysis of the patient promptly subsided after removal of the graft kidney. Asaka et al. also described a 35-year-old male with posttransplant HUS following influenza A infection [5]. The patient recovered from HUS, despite continuing cyclosporine treatment without a dose reduction. Davison et al. reported a previously healthy, 14-year-old girl with HUS following influenza A infection [26]. The patient recovered from HUS with hemodialysis therapy alone. Watanabe reported a previously healthy, 3-year-old girl who presented HUS following influenza A infection. The girl had significant elevations in serum tumor necrosis factor-α and soluble interleukin-2 receptor levels [97].
HUS associated with A(H1N1)pdm09 has been reported in eight patients [3, 13, 18, 36, 51, 71, 72, 89] and can be classified into three categories: S. pneumoniae-associated HUS following A(H1N1)pdm09 infection; HUS triggered by A(H1N1)pdm09 in patients with genetic complement dysregulation; and HUS associated with A(H1N1)pdm09 without any known underlying disorders.
Lei et al. reported a 3-year-old girl with S. pneumoniae-associated HUS following A(H1N1)pdm09 infection in whom the erythrocyte cryptic Thomsen–Friedenreich (TF) antigen activation was positive [51]. S. pneumoniae is well-known as a causative pathogen of secondary bacterial pneumonia following influenza virus infection [51, 56]. Influenza virus alters the lungs of the host in a way that predisposes to adherence, invasion, and induction of disease by pneumococcus [56]. Access to receptors is a key factor and may be facilitated by the virus through epithelial damage, exposure or upregulation of existing receptors, or provoking damage. Alteration of the immune response by diminishing the ability of the host to clear pneumococcus or amplification of the inflammatory cascade likely contributes to the severity of the resulting infection [56]. Therefore, co-infection of S. pneumoniae should be sought in patients with HUS following influenza A virus infection.
Atypical HUS associated with complement dysregulation following A(H1N1)pdm09 has been reported in three patients. Bento et al. reported atypical HUS triggered by A(H1N1)pdm09 in a 17-year-old boy with a mutation in the gene (CD46) encoding MCP [13]. Rhee et al. described a 27-year-old male with atypical HUS, diffuse alveolar hemorrhage, and decreased complement factor C3 level following A(H1N1)pdm09 infection [72]. Al-Akash et al. reported recurrent posttransplant HUS triggered by A(H1N1)pdm09 in a 15-year-old male with a C3 gene mutation [3]. In addition, Çaltik et al. reported atypical HUS following A(H1N1) infection in a 15-year-old male with normal C3, CFH, and CHI levels who had experienced recurrent attacks of HUS four times with recovery of renal function [18]. The authors suggested the patient might have a CD46 gene mutation [18].
HUS associated with A(H1N1)pdm09 without any known underlying disorders has been reported in three patients. Printza et al. reported a case of A(H1N1)pdm09 that triggered atypical HUS in a 7-year-old boy with posterior reversible encephalopathy syndrome who had normal immune status findings, including C3, MCP, and ADAMTS-13, and did not have pneumonia [71]. Trachtman et al. described atypical HUS associated with A(H1N1)pdm09 in a 5-year-old girl [89]. Her blood and endotracheal tube cultures were negative for bacteria; the complement system was not studied. Golubovic et al. reported an 11-year-old boy with an A(H1N1)pdm09 virus infection complicated by atypical HUS [36]. The serum C3 and C4 levels were normal, and the blood, sputum, and throat swab cultures for bacteria did not reveal bacterial super-infection. Although the mechanisms of influenza A virus-associated HUS without complement dysregulation or S. pneumoniae co-infection have not been elucidated, an altered immune system such as systemic mononuclear cell activation following influenza A viral infection [5, 97], and unmasking of cryptic TF antigen caused by viral neuraminidase of influenza A [36] have been postulated.
AGN
AGN is an immunologic response of the kidney to infection that is commonly triggered by group A streptococci [45]. The typical clinical features of AGN include acute onset with gross hematuria, edema, hypertension and moderate proteinuria (acute nephritic syndrome) [45]. AGN can be caused by organisms other than group A streptococci including other strains of streptococci (group C and G), Gram-negative bacilli, mycobacteria, and viruses (influenza) [77]. Viruses have been incriminated in the evolution of acute immune complex-mediated glomerulonephritis [77].
Smith et al. prospectively studied 240 previously healthy personnel with non-streptococcal upper respiratory infections to define the incidence and clinicopathologic characteristics of virus-associated glomerulonephritis [83]. Among the 240 personnel, 9 had biopsy-proven, asymptomatic glomerulonephritis and 4 had serologic evidence of infection with seasonal influenza A [83].
AGN associated with A(H1N1)pdm09 has been reported in two publications. Jain et al. reported a 14-year-old boy who developed hypertension with macroscopic hematuria and marked proteinuria following A(H1N1)pdm09 [42]. The boy’s condition improved rapidly, with the exception of persistent microscopic hematuria. Kupferman et al. also described a 5-month-old girl who had generalized edema and hypertension with AKI, hematuria, proteinuria in the nephrotic range, and hypocomplementemia following A(H1N1)pdm09 infection [49]. His condition also improved rapidly. Although neither patient had a renal biopsy, the clinical and laboratory findings were strongly suggestive of AGN.
DIC
DIC is characterized by the widespread activation of tissue factor-dependent coagulation, insufficient control of coagulation by physiologic anticoagulation pathways, and plasminogen activator inhibitor-1-mediated attenuation of fibrinolysis and is most commonly precipitated by sepsis or trauma [34]. Histologic evidence of microvascular thrombosis and tissue injury in DIC has been reported in clinical, experimental, and autopsy findings [34]. DIC has also been associated with glomerular microthrombosis and acute renal failure [34]. Pro-inflammatory cytokines (tumor necrosis factor-α, interleukin-1 and interleukin-6) released early in the course of sepsis stimulate a procoagulant state that causes development of intravascular fibrin deposition, which results in DIC and organ dysfunction, including kidney dysfunction [34, 53, 100].
ARF due to DIC has been reported in patients with seasonal influenza A virus infection. Davison et al. reported the necropsy findings of 14-year-old boy with DIC and ARF who died from seasonal influenza A virus infection [26]. The histologic findings of the kidney showed fibrin deposition within glomerular capillaries and denuded endothelial cells nuclei of glomerular capillaries. Shenouda and Hatch reported a case of a 33-year-old female with DIC and ARF who died from influenza A viral pneumonia [78]. Whitaker et al. described six patients who developed ARF and DIC following seasonal influenza A virus infection [102]. Watanabe et al. studied 45 children hospitalized with seasonal influenza A virus infection and reported that five patients developed ARF, four of whom had DIC [100]. Bal et al. found DIC pathologic findings in the kidneys of a deceased 25-year-old pregnant woman who had A(H1N1)pdm09 [9].
Goodpasture’s syndrome
Goodpasture’s syndrome is an autoimmune disease that is frequently characterized by rapidly progressive glomerulonephritis, occasional pulmonary hemorrhage, and the presence of autoantibodies to the glomerular and alveolar basement membranes [44]. The target antigen is the α3NC1 domain of type IV collagen, which is expressed in target organs as an α345 network [66].
Influenza virus infection has been associated with Goodpasture’s syndrome. In 1919, Goodpasture described an 18-year-old male who died of pulmonary hemorrhage and a proliferative glomerulonephritis 6 weeks after influenza virus infection [37]; however, this patient with pulmonary-renal vasculitis syndrome likely had anti-neutrophil cytoplasmic antibody disease rather than true Goodpasture’s syndrome because there was a necrotizing small vessel vasculitis in the spleen and gut, which are unlikely in a case of Goodpasture’s syndrome [76]. Wilson et al. reported that a 43-year-old female developed pulmonary hemorrhage and a proliferative glomerulonephritis following influenza A virus infection [103]. The proliferative glomerular lesion of the patient contained immunofluorescent deposits of immunoglobulin G and complement in a linear pattern along the glomerular basement membrane [103].
Acute TIN
Acute TIN is a frequent cause of ARF and is characterized by the presence of an inflammatory call infiltrate in the interstitium of the kidney [92]. Although immune-allergic reactions to certain drugs are the most frequent etiology for acute TIN, viral infections are known to induce TIN [92]. Some viruses have been reported to cause TIN, including cytomegalovirus, hepatitis virus, human immunodeficiency virus, Epstein–Barr virus, and hantavirus [92]; however, acute TIN associated with influenza A virus infection has never been reported.
Ashtiani et al. recently described a case of acute TIN and incomplete renal Fanconi syndrome in a 4-year-old boy with A(H1N1)pdm09 infection who made a full recovery with supportive therapy alone [6]. The pathogenesis underlying the development of TIN in patients with virus infection is unknown; however, infection-related acute TIN usually leads to a sterile infiltrate, suggesting that immunologic disturbances might be responsible for TIN [6].
Conclusions
Renal complications of influenza A virus infection are uncommon but can lead to deterioration of the patient’s condition, including AKI in critically ill patients, rhabdomyolysis, HUS, AGN, DIC, Goodpasture’s syndrome, and acute TIN. Although the pathogenesis of renal injuries due to influenza A virus is incompletely described, some hypotheses have been postulated, including ATN due to renal hypoperfusion or rhabdomyolysis, glomerular microthrombosis due to DIC, direct viral injury to the kidney, and an altered immune system, such as systemic mononuclear cell activation following influenza A virus infection.
References
Abdulkader RC, Ho YL, de Sousa Santos S, Caires R, Arantes MF, Andrade L (2010) Characteristics of acute kidney injury in patients infected with the 2009 influenza A (H1N1) virus. Clin J Am Soc Nephrol 5:1916–1921
Abe M, Higuchi T, Okada K, Kaizu K, Matsumoto K (2006) Clinical study of influenza-associated rhabdomyolysis with acute renal failure. Clin Nephrol 66:166–170
Al-Akash SI, Almond PS, Savell VH Jr, Gharaybeh SI, Hogue C (2011) Eculizumab induces long-term remission in recurrent post-transplant HUS associated with C3 gene mutation. Pediatr Nephrol 26:613–619
Annerstedt M, Herlitz H, Mölne J, Oldfors A, Westberg G (1999) Rhabdomyolysis and acute renal failure associated with influenza virus type A. Scand J Urol Nephrol 33:260–264
Asaka M, Ishikawa I, Nakazawa T, Tomosugi N, Yuri T, Suzuki K (2000) Hemolytic uremic syndrome associated with influenza A virus infection in an adult renal allograft recipient: case report and review of the literature. Nephron 84:258–266
Ashtiani N, Mulder MF, van Wijk JA, Bokenkamp A (2012) A case of tubulointerstitial nephritis in a patient with an influenza H1N1 infection. Pediatr Nephrol 27:1985–1987
Ayala E, Kagawa FT, Wehner JH, Tam J, Upadhyay D (2009) Rhabdomyolysis associated with 2009 influenza A(H1N1). JAMA 302:1863–1864
Bagshaw SM, George C, Bellomo R, ANZICS Database Management Committee (2008) Early acute kidney injury and sepsis: a multicenter evaluation. Crit Care 12:R47
Bal A, Suri V, Mishra B, Bhalla A, Agarwal R, Abrol A, Ratho RK, Joshi K (2012) Pathology and virology findings in cases of fatal influenza A H1N1 virus infection in 2009-2010. Histopathology 60:326–335
Bellomo R, Kellum JA, Ranco C Acute kidney injury. Lancet. doi:10.1016/So14o-6736(11)61454-2
Bellomo R, Pettilä V, Webb SA, Bailey M, Howe B, Seppelt IM (2010) Acute kidney injury and 2009 H1N1 influenza-related critical illness. Contrib Nephrol 165:310–314
Bellomo R, Ronco C, Kellum JA, Mehta RL, Palevsky P, ADQI workgroup (2004) Acute renal failure—definition, outcome measures, animal models, fluid therapy and information technology needs: the Second International Consensus Conference of the Acute Dialysis Quality Initiative (ADQI) Group. Crit Care 8:R204–R212
Bento D, Mapril J, Rocha C, Marchbank KJ, Kavanagh D, Barge D, Strain L, Goodship TH, Meneses-Oliveira C (2010) Triggering of atypical hemolytic uremic syndrome by influenza A (H1N1). Ren Fail 32:753–756
Berry L, Braude S (1991) Influenza A infection with rhabdomyolysis and acute renal failure—a potentially fatal complication. Postgrad Med J 67:389–390
Besbas N, Karpman D, Landau D, Loirat C, Proesmans W, Remuzzi G, Rizzoni G, Taylor CM, Van de Kar N, Zimmerhackl LB, European Paediatric Research Group for HUS (2006) A classification of hemolytic uremic syndrome and thrombotic thrombocytopenic purpura and related disorders. Kidney Int 70:423–431
Bosh X, Poch E, Grau JM (2009) Rhabdomyolysis and acute kidney injury. N Engl J Med 361:62–72
Brien FJ, Jairam SD, Traynor CA, Kennedy CM, Power M, Denton MD, Magee C, Conlon PJ (2011) Pandemic H1N1 (2009) and renal failure: the experience of the Irish national territory referral centre. Ir J Med Sci 180:135–138
Caltik A, Akyüz SG, Erdogan O, Demircin G (2011) Hemolytic uremic syndrome triggered with a new pandemic virus: influenza A (H1N1). Pediatr Nephrol 26:147–148
Cao B, Li XW, Mao Y, Wang J, Lu HZ, Chen YS, Liang ZA, Liang L, Zhang SJ, Zhang B, Gu L, Lu LH, Wang DY, Wang C, National Influenza A Pandemic (H1N1) 2009 Clinical Investigation Group of China (2009) Clinical features of the initial cases of 2009 pandemic influenza A (H1N1) virus infection in China. N Engl J Med 361:2507–2517
Carmona F, Carlotti AP, Ramalho LN, Costa RS, Ramalho FS (2011) Evidence of renal infection in fatal cases of 2009 pandemic influenza A (H1N1). Am J Clin Pathol 136:416–423
Chacko J, Gangan B, Ashok E, Radha M, Hemanth HV (2010) Critically ill patients with 2009 H1N1 infection in an Indian ICU. Indian J Crit Care Med 14:77–82
Chen SC, Liu KS, Chang HR, Lee YT, Chen CC, Lee MC (2010) Rhabdomyolysis following pandemic influenza A (H1N1) infection. Neth J Med 68:317–319
Christenson JC, San Joaquin VH (1990) Influenza-associated rhabdomyolysis in a child. Pediatr Infect Dis J 9:60–61
Cox NJ, Subbarao K (1999) Influenza. Lancet 354:1277–1282
Cunningham E, Kohli R, Venuto RC (1979) Influenza-associated myoglobinuric renal failure. JAMA 242:2428–2429
Davison AM, Thomson D, Robson JS (1973) Intravascular coagulation complicating influenza A virus infection. Brit Med J 1:654–655
Dell KM, Schulman SL (1997) Rhabdomyolysis and acute renal failure in a child with influenza A infection. Pediatr Nephrol 11:363–365
Demirjian SG, Raina R, Bhimraj A, Navaneethan SD, Gordon SM, Schreiber MJ Jr, Guzman JA (2010) 2009 influenza A infection and acute kidney injury: incidence, risk factors and complication. Am J Nephrol 34:1–8
DiBona FJ, Morens DM (1977) Rhabdomyolysis associated with influenza A. Report of a case with unusual fluid and electrolyte abnormalities. J Pediatr 91:943–945
D’Silva D, Hewagama S, Doherty R, Korman TM, Buttery J (2009) Melting muscles: novel H1N1 influenza A associated rhabdomyolysis. Pediatr Infect Dis J 28:1138–1139
Echevarría-Zuno S, Mejiía-Aranguré JM, Mar-Obeso AJ, Grajales-Muñiz C, Robles-Pérez E, González-León M, Ortega-Alvarez MC, Gonzalez-Bonilla C, Rascón-Pacheco RA, Borja-Aburto VH (2009) Infection and death from influenza A H1N1 virus in Mexico: a retrospective analysis. Lancet 374:2072–2079
Fearnley RA, Lines SW, Lewington AJ, Bodenham AR (2011) Influenza A-induced rhabdomyolysis and acute kidney injury complicated by posterior reversible encephalopathy syndrome. Anaesthesia 66:738–742
Foulkes W, Rees J, Sewry C (1990) Influenza A and rhabdomyolysis. J Infect 21:303–304
Gando S (2010) Microvascular thrombosis and multiple organ dysfunction syndrome. Crit Care Med 38(2 Suppl):S35–S42
Goebel J, Harter HR, Boineau FG, El-Dahr SS (1997) Acute renal failure from rhabdomyolysis following influenza A in a child. Clin Pediatr 36:479–481
Golubovic E, Miljkovic P, Zivic S, Jovancic D, Kostic G (2011) Hemolytic uremic syndrome associated with novel influenza A H1N1 infection. Pediatr Nephrol 26:149–150
Goodpasture EW (1919) The significance of certain pulmonary lesions in relation to the etiology of influenza. Am J Med Sci 158:863–870
Gutierrez RL, Ellis MW, Decker CF (2010) Rhabdomyolysis and pandemic (H1N1) 2009 pneumonia in adult. Emerg Infect Dis 16:565
Hoste EA, Clermont G, Kerstein A, Venkataraman R, Angus DC, De Bacquer D, Kellum JA (2006) RIFLE criteria for acute kidney injury are associated with hospital mortality in critically ill patients: a cohort analysis. Crit Care 10:R73
Hotl P, Kibblewhite K (1995) Acute polymyositis and myoglobinuric renal failure associated with influenza A infection. N Z Med J 108:463
Izurieta HS, Thompson WW, Kramarz P, Shay DK, Davis RL, DeStefano F, Black S, Shinefield H, Fukuda K (2000) Influenza and the rates of hospitalization for respiratory disease among infants and young children. N Engl J Med 432:232–239
Jain T, Hemington L, Etuwewe B (2011) A case of post-infectious glomerulonephritis following infection with influenza A subtype H1N1. Pediatr Nephrol 26:151–152
Jung JY, Park BH, Hong SB, Koh Y, Suh GY, Jeon K, Koh SO, Kim JY, Cho JH, Choi HS, Park YB, Kim HC, Kim YS, Lim CY, Park MS (2011) Acute kidney injury in critically ill patients with pandemic influenza A pneumonia 2009 in Korea: a multicenter study. J Crit Care 26:577–585
Kalluri R (1999) Goodpasture syndrome. Kidney Int 55:1120–1122
Kanjanabuch T, Kittikowit W, Eiam-Ong S (2009) An update on acute postinfectious glomerulonephritis worldwide. Nat Rev Nephrol 5:259–269
Khan FY (2009) Rhabdomyolysis: a review of the literature. Neth J Med 67:272–283
Koegelenberg CF, Irusen EM, Cooper R, Diacon AH, Taljaard JJ, Mowlana A, von Groote-Bidlingmaier F, Bolliger CT (2010) High mortality from respiratory failure secondary to swine-origin influenza A (H1N1) in South Africa. Q J Med 103:319–325
Kumar A, Zarychanski R, Pinto R, Cook DJ, Marshall J, Lacroix J, Stelfox T, Bagshaw S, Choong K, Lamontagne F, Turgeon AF, Lapinsky S, Ahern SP, Smith O, Siddiqui F, Jouvet P, Khwaja K, McIntyre L, Menon K, Hutchison J, Hornstein D, Joffe A, Lauzier F, Singh J, Karachi T, Wiebe K, Olafson K, Ramsey C, Sharma S, Dodek P, Meade M, Hall R, Fowler RA, Canadian Critical Care Trials Group H1N1 Collaborative (2009) Critically ill patients with 2009 influenza A(H1N1) infection in Canada. JAMA 302:1872–1879
Kupferman JC, Trachtman H, Spitzer ED (2011) Acute glomerulonephritis and acute kidney injury associated with 2009 influenza A:H1N1 in an infant. Pediatr Nephrol 26:153–154
Lai CC, Wang CY, Lin HI (2010) Rhabdomyolysis and acute kidney injury associated with 2009 pandemic influenza A (H1N1). Am J Kidney Dis 55:615
Lei TH, Hsia SH, Wu CT, Lin JJ (2010) Streptococcus pneumoniae-associated haemolytic uremic syndrome following influenza A virus infection. Eur J Pediatr 169:237–239
Leebeek FW, Baggen MG, Mulder, Dingemans-Dumas AM (1995) Rhabdomyolysis associated with influenza A virus infection. Neth J Med 46:189–192
Levi M (2001) Pathogenesis and treatment of disseminated intravascular coagulation in the septic patient. J Crit Care 16:167–177
Martin-Loeches I, Papiol E, Rodríguez A, Diaz E, Zaragoza R, Granada RM, Socias L, Bonastre J, Valverdú M, Pozo JC, Luque P, Juliá-Narvaéz JA, Cordero L, Albaya A, Serón D, Rello J, H1N1 SEMICYUC Working Group (2011) Acute kidney injury in critical ill patients affected by influenza A (H1N) virus infection. Crit Care 15:R66
Mauad T, Hajjar LA, Callegari GD, da Silva LF, Schout D, Galas FR, Alves VA, Malheiros DM, Auler JO Jr, Ferreira AF, Borsato MR, Bezerra SM, Gutierrez PS, Caldini ET, Pasqualucci CA, Dolhnikoff M, Saldiva PH (2010) Lung pathology in fatal novel human influenza A (H1N1) infection. Am J Respir Crit Care Med 181:72–79
McCullers JA (2006) Insights into the interaction between influenza virus and pneumococcus. Clin Microbiol Rev 19:571–582
Mehta RL, Kellum JA, Shah SV, Molitoris BA, Ronco C, Warnock DG, Levin A, Acute Kidney Injury Network (2007) Acute Kidney Injury Network: report of an initiative to improve outcomes in acute kidney injury. Crit Care 11:R31
Minow RA, Gorbach S, Johnson BL Jr, Dornfeld L (1974) Myoglobinuria associated with influenza A infection. Ann Intern Med 80:359–361
Morgensen JL (1974) Myoglobinuria and renal failure associated with influenza. Ann Intern Med 80:362–363
Myking O, Schreiner A (1974) Influenza virus infection complicated by severe renal failure. Scand J Infect Dis 6:205–207
Nin N, Lorente JA, Sánchez-Rodríguez C, Granados R, Ver LS, Soto L, Hidalgo J, Fernández-Segoviano P, Ortín J, Esteban A (2011) Kidney histopathological findings in fatal pandemic 2009 influenza A (H1N1). Intensive Care Med 37:880–881
Nin N, Lorente JA, Soto L, Ríos F, Hurtado J, Arancibia F, Ugarte S, Echevarría E, Cardinal P, Saldarini F, Bagnulo H, Cortés I, Bujedo G, Ortega C, Frutos F, Esteban A (2011) Acute kidney injury in critically ill patients with 2009 influenza A (H1N1) viral pneumonia: an observational study. Intensive Care Med 37:768–774
Noris M, Remuzzi G (2009) Atypical hemolytic-uremic syndrome. N Engl J Med 361:1676–1687
Noris M, Remuzzi G (2005) Hemolytic uremic syndrome. J Am Soc Nephrol 16:1035–1050
Parikh M, Dolson G, Ramanathan V, Sangsiraprapha W (2010) Novel H1N1-associated rhabdomyolysis leading to acute renal failure. Clin Microbiol Infect 16:330–332
Pedchenko V, Vanacore R, Hudson B (2011) Goodpasture’s disease: molecular architecture of the autoantigen provides clues to etiology and pathogenesis. Curr Opin Nephrol Hypertens 20:290–296
Perez-Padilla R, de la Rosa-Zamboni D, Ponce de Leon S, Hernandez M, Quiñones-Falconi F, Bautista E, Ramirez-Venegas A, Rojas-Serrano J, Ormsby CE, Corrales A, Higuera A, Mondragon E, Cordova-Villalobos JA, INER Working Group on Influenza (2009) Pneumonia and respiratory failure from swine-origin influenza A (H1N1) in Mexico. N Engl J Med 361:680–689
Petersen VP, Olsen TS (1971) Late renal transplant failure due to the hemolytic-uremic syndrome. Acta Med Scand 189:377–380
Pettilä V, Webb SA, Bailey M, Howe B, Seppelt IM, Bellomo R (2011) Acute kidney injury in patients with influenza A (H1N1) 2009. Intensive Care Med 37:763–767
Pradère C, Planchard D, Plouzeau C, Merlet-Chicoine I, Valèro S, Paccalin M (2006) Acute tubular necrosis due to rhabdomyolysis resulting from influenza A infection. J Am Geriatr Soc 54:725
Printza N, Roilides E, Kotsiou M, Zafeiriou D, Hatzidimitriou V, Papachristou F (2011) Pandemic influenza A (H1N1) 2009-associated hemolytic uremic syndrome. Pediatr Nephrol 26:143–144
Rhee H, Song SH, Lee YJ, Choi HJ, Ahn JH, Seong EY, Lee SB, Kwak IS (2011) Pandemic H1N1 influenza A viral infection complicated by atypical hemolytic uremic syndrome and diffuse alveolar hemorrhage. Clin Exp Nephrol 15:948–952
Rothberg MB, Haessler SD, Brown RB (2008) Complications of viral influenza. Am J Med 121:258–264
Rothberg MB, Haessler SD (2010) Complications of seasonal and pandemic influenza. Crit Care Med 38(Suppl):e91–e97
Sato E, Nakamura T, Koide H (2011) Rhabdomyolysis induced by influenza A infection: case report and review of literature. Ther Apher Dial 15:208–209
Self S (2009) Goodpasture’s 1919 article on the etiology of influenza-the historical road to what we now call Goodpasture syndrome. Am J Med Sci 338:154
Silva FG (1998) Acute postinfectious glomerulonephritis and glomerulonephritis complicating persistent bacterial infection. In: Jennette JC, Olson JL, Schwartz MM, Silva FG (eds) Heptinstall’s pathology of the kidney, 5th edn. Lippincott-Raven, Philadelphia, pp 389–453
Shenouda A, Hatch FE (1976) Influenza A viral infection associated with acute renal failure. Am J Med 61:697–702
Simon NM, Rovner RN, Berlin BS (1970) Acute myoglobinuria associated with type A2 (Hong Kong) influenza. JAMA 212:1704–1705
Singbartl K, Kellum JA (2012) AKI in the ICU: definition, epidemiology, risk stratification, and outcomes. J Am Soc Nephrol 81:819–825
Singh U, Scheld M (1996) Infectious etiologies of rhabdomyolysis: three case reports and review. Clin Infect Dis 22:642–649
Slobogean BL, Reilly CW, Alvarez CM (2011) Recurrent viral-induced compartment syndrome. Pediatr Emerg Care 27:660–662
Smith MC, Cooke JH, Zimmerman DM, Bird JJ, Feaster BL, Morrison RE, Reimann BE (1979) Asymptomatic glomerulonephritis after nonstreptococcal upper respiratory infections. Ann Intern Med 91:697–702
Sood MM, Rigatto C, Zarychanski R, Komenda P, Sood AR, Bueti J, Reslerova M, Roberts D, Mojica J, Kumar A (2010) Acute kidney injury in critically ill patients infected with 2009 pandemic influenza A (H1N1): report from a Canadian province. Am J Kidney Dis 55:848–855
Soto-Abraham MV, Soriano-Rosas J, Díaz-Quiñónez A, Silva-Pereyra J, Vazquez-Hernandez P, Torres-López O, Roldán A, Cruz-Gordillo A, Alonso-Viveros P, Navarro-Reynoso F (2009) Pathological changes associated with the 2009 H1N1 virus. N Engl J Med 361:2001–2003
Sullivan SJ, Jacobson RM, Dowdle WR, Poland GA (2010) 2009 H1N1 influenza. Mayo Clin Proc 85:64–67
Swaringen JC, Seiler JG 3rd, Bruce RW Jr (2000) Influenza A induced rhabdomyolysis resulting in extensive compartment syndrome. Clin Orthop Relat Res 375:243–249
Tosun MS, Ertekin V, Orbak Z (2012) Rhabdomyolysis-induced acute renal failure associated with 2009 influenza A (H1N1) virus infection in a child with Crigler–Najjar syndrome. J Emerg Med 42:310–311
Trachtman H, Sethna C, Epstein R, D’Souza M, Rubin LG, Ginocchio CC (2011) Atypical hemolytic uremic syndrome associated with H1N1 influenza A virus infection. Pediatr Nephrol 26:145–146
Trimarchi H, Greloni G, Campolo-Girard V, Giannasi S, Pomeranz V, San-Roman E, Lombi F, Barcan L, Forrester M, Algranati S, Iriarte R, Rosa-Diez G (2010) H1N1 infection and the kidney in critically ill patients. J Nephrol 23:725–731
Ugarte S, Arancibia F, Soto R (2010) Influenza A pandemics: clinical and organizational aspects: the experience in Chile. Crit Care Med 38(Suppl):e133–e137
Ulinski T, Sellier-Leclerc AL, Tudorache E, Bensman A, Aoun B (2012) Acute tubulointerstitial nephritis. Pediatr Nephrol 27:1051–1057
Unverdi S, Akay H, Ceri M, Inal S, Altay M, Demiroz AP, Duranay M (2011) Acute kidney injury due to rhabdomyolysis in H1N1 influenza infection. Ren Fail 33:450–451
Van Why SK, Avner ED (2011) Hemolytic uremic syndrome. In: Kliegman RM, Stanton BF, St. Geme JW III, Schor NF, Behrman RE (eds) Nelson textbook of pediatrics, 19th edn. Elsevier, Philadelphia, pp 1791–1794
Venkata C, Sampathkumar P, Afessa B (2010) Hospitalized patients with 2009 H1N1 influenza infection: the Mayo Clinic experience. Mayo Clin Proc 85:798–805
Wakabayashi Y, Nakano T, Kikuno T, Ohwada T, Kikawada R (1994) Massive rhabdomyolysis associated with influenza A infection. Intern Med 33:450–453
Watanabe T (2001) Hemolytic uremic syndrome associated with influenza A virus infection. Nephron 89:359–360
Watanabe T (1998) Rhabdomyolysis and acute renal failure in acute necrotizing encephalopathy with influenza A. Pediatr Nephrol 12:85
Watanabe T (2001) Rhabdomyolysis and acute renal failure in children. Pediatr Nephrol 16:1072–1075
Watanabe T, Yoshikawa H, Abe Y, Yamazaki S, Uehara Y, Abe T (2003) Renal involvement in children with influenza A virus infection. Pediatr Nephrol 18:541–544
West SD, Brubskill NJ (2002) Complications associated with influenza infection. Postgrad Med J 78(100):107–108
Whitaker AN, Bunce I, Graeme ER (1974) Disseminated intravascular coagulation and acute renal failure in influenza A2 infection. Med J Aust 2:196–201
Wilson CB, Smith RC (1972) Goodpasture’s syndrome associated with influenza A2 virus infection. Ann Intern Med 76:91–94
Writing Committee of the WHO Consultation on Clinical Aspects of Pandemic (H1N1) 2009 Influenza, Bautista E, Chotpitayasunondh T, Gao Z, Harper SA, Shaw M, Uyeki TM, Zaki SR, Hayden FG, Hui DS, Kettner JD, Kumar A, Lim M, Shindo N, Penn C, Nicholson KG (2010) Clinical aspects of pandemic 2009 influenza A (H1N1) virus infection. N Engl J Med 362:1708–1719
Zamkoff K, Rosen N (1979) Influenza and myoglobinuria in brothers. Neurology 29:340–345
Conflict of interest
The author declares that there is no conflict of interest with regard to this manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Watanabe, T. Renal complications of seasonal and pandemic influenza A virus infections. Eur J Pediatr 172, 15–22 (2013). https://doi.org/10.1007/s00431-012-1854-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00431-012-1854-x