
Abstract
This is the second part of a two-part review on soft tissue tumours which may be encountered in the mediastinum. This review is based on the 2013 WHO classification of soft tissue tumours and the 2015 WHO classification of tumours of the lung, pleura, thymus and heart and provides an updated overview of mesenchymal tumours that have been reported in the mediastinum.
Similar content being viewed by others


Introduction
In this second part of a two-part review, the 2013 WHO classification of soft tissue tumours and the 2015 WHO classification of tumours of the thymus form the basis to summarize current knowledge about soft tissue tumours in the mediastinum. This comprehensive review covers published reports from the English and Japanese literature. In the first of these reviews, general aspects, adipocytic, fibroblastic/myofibroblastic, fibrohistiocytic tumours and soft tissue tumours arising as somatic components in germ cell tumours, were discussed. We now turn to mediastinal smooth muscle, skeletal muscle, vascular, chondro-osseous, nerve sheath and miscellaneous tumours of uncertain differentiation, including undifferentiated sarcomas. Since most of the tumours described here closely resemble soft tissue tumours elsewhere in the body, we will focus our review on defining criteria, epidemiology, clinical findings and prognosis, rather than histological descriptions. For in-depth coverage of the histological aspects of these tumours, the reader is referred to specific literature.
Smooth muscle tumours
Smooth muscle cell tumours resemble normal smooth muscle and retain many of their immunohistochemical markers. Fewer benign smooth muscle cell tumours (leiomyomas) than malignant smooth muscle tumours (leiomyosarcomas) have been reported in the mediastinum.
To qualify as a primary mediastinal leiomyoma, tumours should arise from the somatic soft tissue of the mediastinum, whilst esophageal and large vessel primaries should be excluded. If these criteria are applied, primary mediastinal leiomyoma is very rare with approximately 20 reported cases in the literature [14, 19, 136, 139, 149, 162, 180, 205, 227]. Mediastinal leiomyoma is twice as common in females as in males and is mainly seen in the posterior mediastinum of older adults (age range 23–75 years; median age 50 years). These tumours may remain asymptomatic, or patients may complain of dyspnea or chest pain. The tumours may grow to a large size (up to 1,600 g). The morphology of mediastinal leiomyoma is similar to its counterparts elsewhere in the body and is defined as a neoplasm resembling normal smooth muscle cells and devoid of pleomorphism and necrosis and with a very low mitotic rate (<1 mitoses/50 HPF). Immunohistochemical stains will show positivity for smooth muscle actin and desmin in the majority of cases. Surgical removal is curative, and in the reports with follow-up, no recurrences or deaths due to tumour were described.
The main differential diagnosis of leiomyoma is leiomyosarcoma, which is distinguished by increased mitotic activity, pleomorphism and necrosis.
Mediastinal leiomyosarcoma is a very rare tumour with less than 40 cases described as case reports and in two small series [2, 13, 34, 37, 52, 72, 79, 82, 90, 106, 107, 111, 133, 163, 187, 188, 207, 228]. Mediastinal leiomyosarcoma occurs in adult patients (26–88 years) of either sex. A disproportionate number of cases arise in the posterior mediastinum, where tumours are often clinically asymptomatic. Resected tumours measured up to 18 cm, often without clear relationship to neighbouring anatomic structures. In some cases, an origin from major vessels seemed likely [188, 228], whilst in others, major vessels appeared rather entrapped within the tumour. Mediastinal leiomyosarcoma is typically a non-encapsulated, circumscribed mass, which may infiltrate the heart, lungs, thoracic vertebrae or spinal canal [34, 37, 107, 133]. Patients were treated by surgical resection, sometimes combined with chemo- and/or radiotherapy. Local recurrence and distant metastasis occur in a significant proportion of patients. The differential diagnosis of leiomyosarcoma comprises other spindle cell sarcomas, in particular synovial sarcoma and malignant peripheral nerve sheath tumours (MPNST). Whilst well-differentiated leiomyosarcoma will generally stain for both smooth muscle actin and desmin, less differentiated cases may show diminished staining or even absence of muscular markers, requiring additional investigations (e.g. calponin, caldesmon, TLE, S-100 protein and/or nestin immunohistochemistry or fluorescence in situ hybridisation (FISH) analyses) to rule out MPNST or synovial sarcoma. Rare cases of liposarcoma may contain a smooth muscle component (‘lipoleiomyosarcoma’) [62, 68]. In these cases, the lipomatous and smooth muscle components are admixed, and the dual differentiation is obvious. Leiomyosarcoma as a secondary non-germ cell sarcoma is extremely rare, and leiomyosarcoma has not been described as the sole mesenchymal component [35].
Pericytic (perivascular) tumours
Less than ten cases of glomus tumour, including a single malignant variant, have been described in the mediastinum [20, 22, 30, 55, 64, 83, 108]. All cases were located in the posterior or superior compartments of the mediastinum in young or middle-aged females. The single malignant case, which resulted in this patient’s death, occurred in a 74-year-old female. Pain and dyspnea or cough were presenting symptoms. All benign cases were successfully treated by surgery without evidence of disease at follow-up.
A single case of (infantile) myofibromatosis, which in the WHO classification is included in the spectrum of pericytic tumours, located in the central mediastinum and involving the hilar region of the lung was described in a 4-year-old male infant and was treated by pneumonectomy [181].
Angioleiomyoma, which is included in the pericytic group of tumours in the current WHO classification, is considered a benign proliferation of vascular smooth muscle cells and has on rare occasion been reported in the mediastinum [85, 123, 161, 209, 225]. The reported tumours were (with one exception) small and were either discovered incidentally or caused symptoms by compression of nerves in the posterior mediastinum. In reports with follow-up, angioleiomyoma behaved in a benign fashion.
Skeletal muscle tumours
Mediastinal mesenchymal tumours with skeletal muscle differentiation include rhabdomyoma, rhabdomyosarcoma, rhabdomyosarcoma as a component of MPNST (so-called Triton tumour) or as a somatic-type malignancy in germ cell tumours.
Rhabdomyoma is an uncommon benign skeletal muscle tumour. In the paediatric population, it is considered a hamartoma arising in the heart of children with tuberous sclerosis. Five cases of extra-cardiac non-syndromic rhabdomyoma have been described in the mediastinum. All patients were elderly adults (range 52–80 years; four males, one female) [21, 101, 126, 182, 233]. Symptomatic cases presented with non-specific signs. All cases had at some connection with the cervical area, one case was multifocal with a second cervical tumour [233]. It has been suggested that rhabdomyoma may originate from the third and fourth branchial pouches or from myoid cells in the thymus [182].
Primary mediastinal rhabdomyosarcoma is exceedingly rare. The nine reported cases with adequate documentation were all located in the anterior mediastinum [17, 31, 152, 160, 175, 192]. Seven cases occurred in males. Five cases were observed in young adults, two cases in children (4 months and 9 years of age) [31, 175] and two in older individuals. Six cases were diagnosed as alveolar rhabdomyosarcoma, which was confirmed by genetic analysis in two cases [31, 175]. Two cases were of the embryonal subtype, and one case was diagnosed as pleomorphic rhabdomyosarcoma. In the older literature, additional sparse reports of mediastinal rhabdomyosarcoma are on record. However, these cases predate immunohistochemical and molecular typing and cannot be reliably accepted as rhabdomyosarcomas [192]. The prognosis of the reported cases of primary mediastinal rhabdomyosarcoma was poor.
The differential diagnosis of the rare primary mediastinal rhabdomyosarcoma is the much more common rhabdomyosarcoma as a component of a germ cell tumour [23, 35, 69, 120]. In germ cell tumours with somatic-type malignancy, the prognosis is determined by the somatic (i.e. sarcomatous) component and in the setting of rhabdomyosarcoma is dismal. Rhabdomyoblastic differentiation has been reported in both low-grade (thymoma) [29, 129, 174] and high-grade (thymic carcinoma, carcinosarcoma) [41, 48, 145] thymic epithelial tumours and thus enters the differential diagnosis of rhabdomyosarcoma [29, 41, 48, 129, 145, 174]. Identification of an epithelial component by appropriate immunohistochemical staining will aid the accurate diagnosis. Rhabdomyoblastic differentiation may also occur in MPNST, which are then called malignant Triton tumours. Nine cases have been reported in the mediastinum (reviewed by Ren et al. [166]). The typical spindle cell MPSNT component usually predominates, whilst cells with rhabdomyoblastic differentiation commonly form a minor component of the tumour. These tumours carry a very poor prognosis.
Vascular tumours
Amongst mesenchymal neoplasms in the mediastinum, vascular (endothelial) tumours are not uncommon with well over 100 cases reported in the literature.
Benign and intermediate-grade vascular tumours
Benign blood vessel tumours (hemangioma variants) have been reported as individual cases and have been summarized in several reviews [32, 39, 127, 130]. Endothelial tumours of intermediate grade (hemangioendotheliomas) are characterized by local infiltrative growth and rare metastases.
Mediastinal hemangioma may originate in the soft tissue of the mediastinum or may arise in the thymus [5, 32, 154]. Mediastinal hemangiomas span a wide age range, from the newborn to the elderly, with most cases occurring in young adults. Of the reported cases, 50–75 % were located in the anterior mediastinum [32, 130]. Up to 50 % of cases were asymptomatic. Patients with symptomatic tumours presented with pain, cough and rarely with signs of compression of vital structures by the tumour, such as Horner’s syndrome, neurological signs and superior vena cava obstruction [32]. Tumour size ranged from a few centimetres to 20 cm [32, 130] and included both capillary and cavernous hemangioma subtypes. In the series by Moran and Suster, associated histological features included fatty metaplasia, fibrosis, smooth muscle overgrowth and inflammation [130]. Surgical resection is curative in most cases, although recurrence may rarely occur [32].
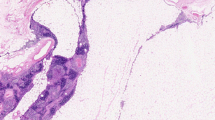
Lymphangioma has been reported as a cystic mass in the mediastinum of children and is very uncommon in adults (Fig. 1)

Lymphangioma. Anterior mediastinal mass considered to be a benign cystic lesion was discovered during routine check-up in an 81-year-old male. The mass increased in size from 16 to 22 mm in a period of 8 years. No recurrence 1 year after the surgery. a Non-enhanced CT axial image shows a small homogeneous mass with attenuation. b Axial fat-saturated T2-weighted MR image shows a hyperintense signal with small internal septations. c HE stain of resected tumour. Irregular thin-walled vascular structures in adipose tissue. d D2-40 stain, supporting the lymphatic differentiation of the lining endothelium
[61, 147, 156]. Lymphangiomas are not considered true neoplasms but rather malformations of the lymphatic vasculature with progressive cystic change resulting from obstruction of lymphatic drainage [80]. The progressive growth of these lesions causes dyspnea. Surgical resection is curative in most cases, although recurrences may require repeated surgery [156].
Vascular endothelial tumours of intermediate malignancy (with local aggressive behaviour and/or rare metastases) have been reported in the mediastinum as case reports and case series. These include kaposiform hemangioendothelioma (KHE) and a single case of composite hemangioendothelioma [24].
Less than 15 cases of exclusively mediastinal KHE have been reported, in addition to a number of cases where large KHEs extended contiguously from the neck into the mediastinum [51, 77, 81, 89, 114, 140, 210, 211, 220, 226, 234]. The reported mediastinal KHE cases were exclusively seen in infants (0–16 months of age) with almost equal sex distribution. Thrombocytopenic consumptive coagulopathy (Kasabach-Merritt syndrome) was present in almost all of the reported cases and is a major cause of fatality in this tumour. Mediastinal KHE is histologically similar to its soft tissue counterparts and is characterized by poorly circumscribed nodules of tightly packed small capillary-sized vessels, Kaposi sarcoma-like areas with spindled cells and absence of HHV-8 immunoreactivity. A component of larger lymphatic vessels is often present. The prognosis of KHE is variable. Complete surgical removal may provide a cure but may not be possible in cases with extensive infiltration. Variable success has been achieved by medical treatment with interferon and chemotherapy. The prognosis is often determined by the haematological complications rather than the local tumour effects. In the mediastinal cases with follow-up information, six patients were alive without evidence of disease, two patients were alive with disease and two patients died of disease.
A single case of composite hemangioendothelioma, a rare vascular neoplasm of intermediate grade with variable histological components, was described in the central mediastinal area of a 50-year-old female. There was evidence of disease 13 months after surgical resection [24].
Malignant vascular tumours
Malignant vascular tumours comprising epithelioid hemangioendothelioma (EHE) and angiosarcoma (AS) have been reported in the mediastinum.
Mediastinal EHE is a malignant endothelial neoplasm with approximately 30 cases published mostly as case reports [7, 16, 25, 27, 56, 60, 88, 103, 109, 115, 121, 143, 151, 191, 202, 215, 216, 229] and a single series of 12 primary mediastinal cases described by Suster et al. [191] All cases occurred in adults (median age 46 years, age range 19–69 years) with twice as many cases reported in males as in females. A considerable proportion of EHE was discovered incidentally on imaging studies for other indications. Symptomatic patients complained of chest pain, cough and dyspnea. All reported cases were located in the anterior or anterior–superior mediastinum and a number of reports document involvement of or an origin from large veins (superior vena cava and innominate vein in particular). The tumours were often partially encapsulated but often at least focally contained infiltrative areas. Mediastinal EHE showed the typical histology with epithelioid endothelial cells, often with intra-cytoplasmic lumina (‘blister cells’), set in a variable hyalinized myxo-collagenous or chondroid matrix. Osteoclastic giant cells and metaplastic bone were reported in several cases [191]. Intra-nuclear cytoplasmic inclusions are another feature typically observed in EHE. A recurrent genetic aberration in EHE, including mediastinal cases, is translocation of the CAMTA1 gene on chromosome 1p and fusion with the WWTR1 gene on chromosome 3q [7, 53]. A subset of EHE harbour a different genetic abnormality in which a YAP1-TFE3 fusion gene is generated from YAP1 sequences on chromosome 11 and TFE1 sequences from the X-chromosome [9]. To date, the YAP1-TFE3 fusion gene has not been reported in mediastinal EHE.
The differential diagnosis of EHE includes epithelioid angiosarcoma (EAS) and metastatic adenocarcinoma. The intra-cytoplasmic lumina in EHE may easily be mistaken for glandular differentiation, which in conjunction with the cytokeratin expression seen in up to 40 % of EHE may lead to an erroneous diagnosis of metastatic carcinoma. The diagnosis of EHE can be established by recognition of the vascular differentiation by immunohistochemistry (with CD31, CD34, ERG or FLI staining) (Fig. 2).

Epithelioid hemangioendothelioma. Anterior mediastinal mass in 56-year-old female. Angiosarcoma was diagnosed on needle biopsy. The tumour was subsequently excised and diagnosed as epithelioid hemangioendothelioma. The presence of a CAMTA1-WWTR1 translocation was confirmed by RT-PCR. The patient remains free of recurrence 23 years post-surgery. a The HE stain shows myxo-collagenous matrix with epithelioid cells with intra-cytoplasmic vascular spaces consistent with so-called blister cells. b Membranous CD31 staining confirming the vascular nature of the tumour cells
Epithelioid angiosarcoma shows high-grade cytology and usually lacks the typical myxoid, hyaline or chondroid stroma of EHE. Features more commonly associated with EAS include capillary vessels, vascular lakes and papillary growth [7]. The genetic changes identified in the EHE have only been identified in a single case of EAS and thus may serve to distinguish the two entities. The prognosis of mediastinal EHE is not unfavourable; of the 15 cases with follow-up, 13 patients showed no evidence of disease following surgical removal, one patient died of complications of the surgical procedure, and one patient died of the tumour 9 years after initial presentation (no surgery was performed). A subdivision into low- and high-grade EHE on the basis of cytonuclear features has been suggested [7].

Ganglioneuroma. Paraspinal tumour in the mediastinum of a 45-year-old female, extending from the cervical region to the diaphragm. The mass was excised, the patient remained disease free. a CT image showing the cervical aspect of the paraspinal mass. b HE stain of the tumour, revealing a ‘schwannian’ background with scattered ganglion cells. c Neurofilament stain revealing elongated slender cytoplasmic extensions
Mediastinal AS has almost exclusively been reported in the anterior mediastinum. Less than 40 cases are on record [7, 42, 67, 69, 93, 151, 195, 217, 230]. Although older reports frequently described a favourable outcome even after marginal resection, it should be noted that many of these did not include immunohistochemical confirmation, raising the possibility that the reports may have incorporated entities that would currently not be diagnosed as AS. Furthermore, the prognosis and the demographics of the patients in older series are in marked contrast with more recently described cases. In a recent series of mediastinal EAS, 12 of 13 patients succumbed to the disease [7], whilst in older reports with follow-up data, no evidence of disease was documented in seven of ten patients after surgical removal (follow-up 6–36 months) [42, 67, 69, 93, 195, 217]. Similarly, in a more recent series, there was a distinct male predominance with a median age of 67.5 years, whilst in the older reports, an equal gender distribution with a median age of 42 years was recorded. The demographic and prognostic data of the recent series are more concordant with the high tumour-related mortality reported for AS in other organs [57]. In contrast to EHE, all cases of AS were symptomatic with pain being the most common presenting symptom. Most AS measured less than 10 cm in largest dimension and showed an infiltrative growth pattern. There are too few well-documented cases to draw firm conclusions on treatment and prognosis. The main differential diagnoses are other vascular neoplasms, in particular EHE. However, AS is an infiltrative neoplasm with high-grade cytology and usually lacks the peculiar fibro-myxoid stroma of EHE. It is not uncommon to observe AS as a component of somatic type malignancy in mediastinal germ cell tumours [35, 47, 69, 120, 131, 171, 230].
Chondro-osseous tumours
To qualify as a primary mediastinal osteo- or chondrosarcoma, an origin from bony structures, chest wall soft tissue, tracheo-bronchial structures and a putative relationship with a germ cell tumour must be excluded. Bearing these restrictions in mind, about ten cases of primary extraskeletal mediastinal osteosarcoma cases occurring in all compartments of the mediastinum have been reported [28, 70, 84, 86, 110, 186, 196, 208, 221]. However, similar to neoplasms described in previous paragraphs, several reports predate current concepts and ideally would require substantiation. Although reported cases spanned a wide age range (19–77 years), six cases occurred in young adults (19–30 years). Four patients were females, and six were males. A single case was most likely a radiation induced osteosarcoma following radiotherapy for Hodgkin’s disease [28]. Most patients presented with chest pain or dyspnea. Synchronous secondary deposits were reported in several cases [110, 186, 196, 221]. Tumours ranged in size from 4.5 to 16 cm and were often partially encapsulated. Of eight patients with follow-up data, five died of disease, one patient was alive with disease and two patients showed no evidence of disease after therapy, including one patient who developed a recurrence, which was treated with radiotherapy. Osteosarcoma has only rarely been reported as somatic type malignancy in germ cell tumours [131, 204].
Less than ten cases of extraskeletal mediastinal chondrosarcoma have been reported, six cases were described in a single series [189] and the remaining cases as case reports. Tumour location in the anterior as well as in the posterior mediastinum was described for both conventional and mesenchymal chondrosarcomas. Patients ranged from 11 to 36 years with no obvious sex predilection [91, 189]. Tumours ranged in size from 5 to 15 cm and were mostly encapsulated. The mesenchymal chondrosarcomas contained areas of primitive small cells and more mature cartilaginous areas, whilst the conventional chondrosarcomas were composed of hyaline cartilage with increased cellularity and atypical chondrocytes. In the cases with follow-up, recurrences occurred in five patients, two patients died of disease (one after a recurrence) and two patients were alive without disease (one after a recurrence). Chondrosarcoma arising in a germ cell tumour appears to be extremely uncommon with only a single reported case in the mediastinum [125].
The histological differential diagnosis of mediastinal osteosarcoma and chondrosarcoma is very limited. Osseous metaplasia may occur in other tumours including thymoma [183] but is unlikely to be confused with the atypical cells of osteosarcoma. Bone or cartilage tissue of variable maturity may be a component of a mediastinal teratoma but does also not show the irregular texture and cellular atypia of osteo- and chondrosarcomas. Osteosarcomatous differentiation has also been reported in malignant peripheral nerve sheath tumours [44].
Ewing sarcoma/primitive neuroectodermal tumour (PNET)
In the WHO classification, Ewing sarcoma (ES)/PNET is included in the classification of bone tumours despite the fact that up to 20 % of cases are extraskeletal (extra-osseous Ewing sarcoma, eES) and are located in various soft tissue sites and even in parenchymal organs. Furthermore, the cell of origin and differentiation of ES/PNET remain currently unknown. As stated above for chondro-osseous tumours, an origin from bony structures such as vertebrae and chest wall must be excluded in order to qualify as primary mediastinal eES/PNET, leading to the exclusion of several reported cases where tumours extended to these structures or where insufficient details were given [112, 231]. Less than 20 convincing cases of eES/PNET have been reported in the mediastinum [3, 50, 74, 75, 100, 102, 119, 153, 164, 169, 178, 231]. The cases mostly occurred in young individuals (median age 28 years, range 5–66 years) with equal gender distribution and in all compartments of the mediastinum. The presenting symptoms were non-specific; pain and dyspnea were frequent. Some tumours extended through the foramina and caused nerve compression symptoms [100, 119, 169]. The characteristic genetic translocation involving the EWSR1 gene on chromosome 22 was confirmed in several cases [75, 119]. Multimodal therapy was given in a number of cases with variable results. Long-term cure was achieved in isolated cases.
The differential diagnosis is broad and includes eES/PNET with variant translocations and eES-like tumours unrelated to EWS (small blue round cell tumours), as well as synovial sarcomas and undifferentiated pleomorphic sarcomas with a small cell morphology. In the anterior mediastinum, T lymphoblastic lymphoma (TLBL) is a major differential diagnostic consideration, especially since both tumours express CD99. Immunohistochemical staining for lymphocyte markers and TdT will exclude lymphoma. Small cell neuroendocrine carcinoma (both primary mediastinal and metastatic) can be excluded by its high-grade cytologic features and expression of neuroendocrine and (sometimes weak) epithelial markers and TTF1. In the posterior mediastinum, neuroblastoma must be excluded, which is usually CD99 negative and does not show EWSR1 gene translocations.
Nerve sheath tumours and tumours derived from the autonomic nervous system
Neural tumours are the most common mesenchymal tumours in the mediastinum with an estimated incidence of 12–19 % of all mediastinal tumours. Aspects of these tumours have been the subject of several comprehensive reviews [65, 116, 122, 165, 167]; the various entities will therefore only be covered in general terms in this review, for specific details the reader is referred to these reviews. Neural mediastinal tumours may be categorized into tumours of nerve sheath—schwannian derivation, which are predominantly seen in adults and those of the autonomic (neuronal–ganglionic/sympathetic–parasympathetic) nervous system, which are much more common in the paediatric age group. Tumours which are thought to arise form embryonic remnants of the neural tube, such as rare examples of ependymoma (Table 1), constitute a third category [43, 54, 222].
Schwannomas are the most common of the mediastinal neural tumours and are considered to arise from spinal nerves. They are therefore mainly seen in the posterior mediastinum, although cases located in the anterior mediastinum are also on record [193, 206]. A significant proportion of cases are asymptomatic and discovered incidentally by routine imaging investigations. Symptomatic cases present with chest pain, cough or compression symptoms, in particular in tumours extending through spinal foramina. There is no gender predilection for schwannoma; they are most common in young- to middle-aged adults. Similar to other locations, these are circumscribed encapsulated tumours, which may undergo cystic degeneration and degenerative (‘ancient’) changes. Surgical removal is curative with extremely low recurrence rates.
The cellular variant of schwannoma, which may be mistaken for a malignant tumour owing to its increased cellularity and lack of Verocay bodies, is particularly common in the mediastinum. Its benign behaviour has been confirmed in several series [58, 113, 218, 223]. A more aggressive variant of schwannoma, previously designated melanotic schwannoma, has been shown to have a high recurrence and metastatic rate with associated 27 % mortality in a single series, prompting the prefix ‘malignant’ to its name [201]. Malignant melanotic schwannoma may be associated with the Carney complex and typically originates from spinal nerves in the posterior mediastinum or retroperitoneum.
A single hybrid nerve sheath tumour sharing features of perineurioma and schwannoma has been reported in the posterior mediastinum. These hybrid tumours are commonly found in superficial soft tissues and require immunohistochemistry to confirm their hybrid nature [157]. Because of their coexpression of vimentin and EMA, they need to be distinguished from meningioma, type A thymoma and follicular dendritic cell sarcoma. A single case of myxoid neurothekeoma/nerve sheath myxoma in the superior mediastinum was recently reported [176].
Neurofibroma, similar to schwannoma, occurs mainly in the posterior mediastinum. Up to 45 % of neurofibromas occur in patients with neurofibromatosis type I (von Recklinghausen’s disease; NF1), in this setting, the tumours occur at a younger age and are often multiple [116]. Plexiform neurofibroma is pathognomonic for NF1 and carries a risk of transformation to MPNST. The anatomical location and ill-defined borders (in contrast to schwannoma) of plexiform neurofibroma hamper removal resulting in a higher recurrence rate.
Malignant peripheral nerve sheath tumours are high-grade sarcomas which typically arise in association with larger nerve trunks and carry a poor prognosis. MPNST, in particular those arising in NF1-associated neurofibromas, can occur in young patients [45, 46]. Similar to other nerve sheath tumours, mediastinal MPNST is typically located in the posterior compartment, although cases may also arise in the anterior mediastinum [92, 99, 148, 166, 232]. Complete surgical resection may provide a cure [105, 124], but overall, these are aggressive neoplasms with high rates of local recurrence and metastases. Variants of MPNST, which have also been reported in the mediastinum, may show divergent mesenchymal differentiation with components of skeletal muscle (rhabdomyosarcoma, the so-called Triton tumour) [105], osteosarcoma and chondrosarcoma [44]. MPNST, including the Triton tumour variant, has been reported as a somatic component of mediastinal germ cell tumours [33, 118, 131].
Granular cell tumour (GrCT) shows differentiation along neuroectodermal lines and shares similarities with Schwann cells. GrCT occurs in the posterior mediastinum, in keeping with its proposed neuroectodermal/neural derivation. Less than 20 cases of GrCT have been reported in the mediastinum [1, 4, 8, 15, 40, 76, 87, 95, 98, 117, 137, 159, 168, 170, 179, 184, 185, 224], including cases with malignant behaviour or atypical features suggestive of malignant GrCT [40, 76, 137, 185]. Benign mediastinal GrCT occurred twice as frequently in females as in males; malignant GrCT (four cases) were equally distributed in males and females. Benign GrCT were seen in young- to middle-aged adults (range 11–53 years, median age 27 years) with an average size of 4 cm and often discovered incidentally. Malignant GrCTs were symptomatic and occurred in older individuals (range 59–66 years, median age 63 years) and were larger (range 5.4–15 cm, average size 10.3 cm).
A very rare tumour with proposed neuroectodermal differentiation based on electron microscopic comparative studies, melanotic neuroectodermal tumour of infancy (retinal anlage tumour, melanotic progronoma), which most commonly occurs in the jaws of infants has twice been reported in the mediastinum [36, 128]. These tumours show a biphasic morphology with small cells of supposed neural origin which are immunoreactive for neuronal/neuroendocrine markers such as synaptophysin and chromogranin, surrounded by larger cells with (neuro)ectodermal differentiation evidenced by staining reactions with EMA, cytokeratin and HMB45. Two cases have been reported in the mediastinum; one case presented in a 7-month-old infant and second case was reported in a 23-year-old female who died of metastases [36, 128].
Neuroblastic tumours with variable ganglionic differentiation originating from sympathetic ganglia range from benign ganglioneuromas through to malignant neuroblastoma and the intermediate form, ganglioneuroblastoma [116]. In line with their postulated cell of origin, most of these tumours develop in the posterior mediastinum. Malignant neuroblastic–ganglionic tumours (neuroblastoma and ganglioneuroblastoma) are very rarely encountered in the adult population [11], whilst ganglioneuroma is seen in children and young adults and is a benign and often asymptomatic tumour (Fig. 3) [116]. Whilst a significant proportion of neuroblastomas present in the mediastinum, it has been suggested that these tumours differ from the more common adrenal neuroblastomas by their pathogenesis and a more favourable prognosis [116]. A single case of neuroblastoma as a somatic component of a mediastinal germ cell tumour was reported, in which the neuroblastemous component metastasized and resulted in the demise of the patient [144].
Mediastinal paraganglionic tumours of the autonomous nervous system have not infrequently been reported in the mediastinum as case series and single case reports, many of which have been included in recent reviews [66, 104, 132, 141, 142, 146, 150]. Mediastinal paragangliomas may occur in the anterior mediastinum, where they are associated with aorticopulmonary, vagal, subclavian, carotid and coronary vessel (branchiomeric) paraganglia. Branchiomeric (aorticopulmonary) paraganglia (chemodectomas) are rarely functional [219].
Paragangliomas located in the posterior compartment derive from the sympathetic chain and are located in the costovertebral sulcus and secrete catecholamines in up to 50 % of cases, resulting in systemic symptoms (‘extra-adrenal pheochromocytoma’). Mediastinal paraganglioma occurs over a wide age range; those located in the anterior mediastinum are slightly more common in females, whilst paravertebral paraganglioma has slightly more frequently been reported in young adult males [66, 132]. The biological behaviour of these tumours cannot reliably be predicted based on morphological features. Up to a quarter of cases may metastasize [104]. Owing to the close vicinity of branchomeric paraganglia to mediastinal vessels and organs and associated surgical considerations, the prognosis of this subgroup is worse than for paravertebral paraganglioma.
Remnants of the embryonic neural tube may on very rare occasions give rise to tumours located in the mediastinum. Six cases of mediastinal ependymoma, including one case of myxopapillary ependymoma, have been published, which were summarized in a case report by Estrozi et al. [54]. All cases occurred in the posterior mediastinum of adult females (age range 35–71 years; median age 40.5 years). The tumours ranged in size from 5.0 to 9.0 cm. Of four cases with follow-up, three patients were alive without disease after surgical resection, and one patient suffered metastases to the pleura and mediastinal lymph nodes.
Tumours of uncertain differentiation
The WHO classification of soft tissue tumours includes a chapter on neoplasms with unclear histogenesis, some of which have been reported to occur in the mediastinum (Table 2) and are described in the following section. It is conceivable that a number of these tumours may not actually have a normal tissue counterpart but instead reflect the consequence of genetic aberrations in which translocations and other genetic errors culminate in histological patterns, which are not encountered in normal tissue. A number of the entities included in this section of the WHO classification are site-specific, such as intra-muscular myxoma, and are not encountered in the mediastinum; those that have been reported in the mediastinum are listed in Table 2 and are described in the following section. The term ‘malignant mesenchymoma’ which was used in the past for sarcomas with two or more types of differentiation is no longer recommended since the respective tumours are now classified as sarcomas with areas of divergent differentiation (e.g. dedifferentiated liposarcoma or Triton tumour).
Angiomatoid fibrous histiocytoma
Despite its name, angiomatoid fibrous histiocytoma is generally considered a tumour of uncertain histogenesis rather than a true fibrohistiocytic tumour (see part I of this review). Two cases of angiomatoid fibrous histiocytoma, which were both resected without further disease activity, have been reported in the mediastinum [12, 38].
Ossifying fibromyxoid tumour
Two cases of ossifying fibromyxoid tumour (OFT) have been reported in the mediastinum, both in the anterior compartment [49, 197]. One case presented as a large mass (1,052 g) in a 50-year-old female who complained of chest pain [197]. No evidence of recurrence or metastasis was noted 1-year after surgery on follow-up. The second case presented as an 11-cm tumour in a 59-year-old female; no follow-up data was provided [49].
Myoepithelial tumours including parachordoma
There have been no convincing reports of myoepithelial tumours in the mediastinum. A single case of a myoepthelial tumour located in the inferior mediastinum in close association with the carina was suggested to originate from heart muscle and thus cannot be considered a bona fide tumour of mediastinal soft tissue [10].
Synovial sarcoma
Synovial sarcoma (SySa) is a high-grade spindle cell sarcoma of uncertain histogenesis, often with epithelial differentiation, most commonly occurring in deep soft tissue of the extremities in young adults. SySa is one of the more frequently encountered sarcomas in the mediastinum.
Salah et al. performed a literature survey of mediastinal SySa and identified 40 cases in the English language with appropriate descriptive details [173], including 15 patients described in a series by Suster et al. [190]. The majority of cases occurred in young adults (age range 3–83 years; median age 30.5 years) with a male predominance (2.9:1). Seventy percent of cases were reported in the anterior mediastinum with tumour size ranging from 5 to 20 cm (median size 11 cm). Pain and dyspnea were the most common presenting symptoms.
Most cases were monophasic spindle cell SySa on histology. A specific chromosomal translocation, t(X;18)(p11;q11), involving the SS18 (SYT, SSXT) gene on chromosome 18 and the SSX1, SSX2 or SSX4 gene on the X-chromosome is a characteristic feature of SySa and has been demonstrated in mediastinal SySa [94, 172, 203].
Complete surgical resection was achieved in 57.5 % of patients, which was combined with either radiation or chemotherapy in eight patients. Progression occurred in 67 % of patients. Eleven of 30 (37 %) patients with follow-up data died of disease, nine (30 %) were alive with disease and nine (27 %) patients were alive with no evidence of disease; disease status was unknown for two patients. Complete resection was identified as the sole factor influencing survival.
The differential diagnosis of mediastinal SySa comprises other spindle cell sarcomas, in particular MPNST, malignant solitary fibrous tumour (SFT) and pleomorphic sarcoma NOS. Type A thymoma and sarcomatoid carcinoma may also enter the differential diagnosis.
Type A thymoma is usually composed of uniform non-hyperchromatic epithelial spindle cells, whilst the cells in synovial sarcoma are hyperchromatic and mitotically active. The glandular and epithelial components in biphasic synovial sarcoma may be mistaken for elements of type A thymoma.
Immunohistochemistry is very useful. Although synovial sarcoma is usually positive for cytokeratin and EMA, staining is typically weak and/or focal, whilst type A thymoma is typically diffusely cytokeratin positive. TLE1 and CD56 are commonly expressed in SySa and not found in type A thymoma, whilst bcl2 expression is found in both. Demonstration of the t(X;18) translocation by FISH is diagnostic of SySa, and this genetic abnormality has not been identified in other tumours.
MPNST may be very difficult to distinguish from synovial sarcoma without molecular analysis, as these tumours share some morphological and immunohistochemical features. S-100 staining may be seen synovial sarcoma, and conversely MPNST is frequently completely negative for S-100. Similarly, staining for TLE1 may also be positive in MPNST and SFT. SFT may be distinguished from SySa by CD34 and STAT-6 immunohistochemistry, as both markers are not expressed in SySa.
Epithelioid sarcoma
A single case of mediastinal epithelioid sarcoma was identified in the literature. The tumour was located in the posterior mediastinum of a 1-year-old male infant. After radiotherapy and chemotherapy, no evidence of disease was recorded after a follow-up period of 44 months [71].
Alveolar soft part sarcoma (ASPS)
Two well-documented cases of primary mediastinal ASPS have been reported to date. ASPS is a rare tumour with a distinctive histology and characteristic genetic translocation which arises in the deep soft tissue of the lower limb girdle, most commonly in young individuals. ASPS ultimately carries a poor prognosis, and metastases may occur after long time intervals. The two mediastinal cases described in a report by Flieder et al. were located in the anterior mediastinum of a 23-year-old male and in the posterior mediastinum of a 22-year-old male [59]. One of these patients presented with synchronous metastatic disease in the lungs and brain, and the second patient was lost to follow-up after 14 years without evidence of recurrence or metastasis. A third putative case of primary mediastinal ASPS was reported in the anterior mediastinum of a 13-year-old female. However, during work-up, a tumour was later identified in the thigh, which most likely represents the primary location [177].
Clear cell sarcoma of soft tissue
Two cases of clear cell sarcoma (CCS; melanoma of soft parts) have been reported as primary tumours in the mediastinum [194, 199]. A 15-cm tumour was excised from the superior mediastinum of a 59-year-old female in the report by Tirabosco et al. [199]. Although the histology was representative for CCS, immunohistochemical staining for HMB45 and Melan-A was negative. The diagnosis was supported by demonstration of a translocation involving the EWSR1 gene by FISH analysis; the partner gene was not identified. Metastatic disease developed 16 months post-surgery; no further follow-up was provided. A second case of CCS was resected by VATS from the superior mediastinum of a 63-year-old male [194]. A tumour of 8.5 cm was removed and showed the typical histology for CCS with positive staining for HMB45 and S-100 protein. Melan-A was negative and no genetic analysis was performed. At 15 months of follow-up, there was no evidence of disease.
Extraskeletal myxoid chondrosarcoma
One case of mediastinal extraskeletal myxoid chondrosarcoma was included in the series of chondrosarcoma of Suster et al. [189].
Extrarenal rhabdoid tumour
Extrarenal rhaboid (ERRT) tumour is an exceedingly rare tumour, which is histologically similar to its renal and central nervous system counterparts. ERRT occurs typically in young infants and carries an exceptionally poor prognosis. The SMARCB1 (INI1) gene, located on chromosome 22q11, is mutated or deleted in ERRT. Despite its rarity in somatic soft tissue, several cases, mainly included in series, have been reported in the mediastinum [73, 155, 158, 198]. All five patients with sufficient clinical details died of disease. One female patient was diagnosed at the age of 26 years, and two paediatric patients were infants (8 months and a congenital case).
PEComa
Tumours with assumed perivascular differentiation and an epithelioid morphology (PEComa) include angiomyolipoma, which contains a variable component of mature fat and most commonly arises in the kidney. PEComa cells have a distinct morphology and immunophenotype with evidence of smooth muscle and melanocytic differentiation with variable staining for smooth muscle actin, desmin and melanocytic markers such as Melan-A and HMB45. Since their first description, PEComas have been described in many anatomical locations. PEComa may arise in the setting of tuberous sclerosis and lymphangioleiomyomatosis.
To date, less than 15 cases of PEComa have been reported in the mediastinum, which have all behaved in a benign fashion [6, 18, 26, 63, 78, 96, 97, 134, 200, 212–214]. Three quarters of the reported cases were discovered incidentally by imaging studies for other reasons, symptomatic cases mainly presented with shortness of breath. The marked female predominance seen in non-mediastinal cases was less pronounced in the mediastinal tumours (2:1). The cases were reported over a wide age range (22─63 years; median age 54.4 years). Anatomically, the cases were evenly distributed in the anterior (n = 5) and posterior (n = 4) mediastinum with single cases in other compartments, with sizes ranging from 4 to 15 cm (average size 7.8 cm). Three of reported cases are likely to have arisen in association with tuberous sclerosis/lymphangioleiomyomatosis [134, 200, 212].
Desmoplastic small round cell tumour
A single case of desmoplastic small round cell tumour (DSRCT) was reported in the anterior mediastinum of a 22-year-old male [138].
Unclassified/undifferentiated sarcoma
High-grade undifferentiated sarcomas were previously often classified as malignant fibrous histiocytoma (MFH). Following the current understanding that there is insufficient support for histiocytic (or any other specific) differentiation, these tumours are now categorized as undifferentiated sarcomas in the WHO classification. So-called ‘MFH’ cases have been described in the mediastinum but do not show any further characteristic features. A series of 34 so-called MFH cases in the mediastinum were reviewed by Murakawa et al. [135], and further individual case reports haven been published. As in other soft tissue sites, these tumours are seen over a wide age range but mainly in older adults, with an equal gender distribution. As would be expected, these tumours have a very poor prognosis despite therapeutic intervention.
References
Abenoza P, Sibley RK (1987) Granular cell myoma and schwannoma: fine structural and immunohistochemical study. Ultrastruct Pathol 11:19–28
Abiko T, Sato S, Futamata T, Kato R (2005) A case of pleomorphic leiomyosarcoma of the posterior mediastinum. Nippon Kokyukigeka Gakkai Zasshi 19:819–822
Ahmad R, Mayol B, Davis M, Rougraff B (1999) Extraskeletal Ewing’s sarcoma. Cancer 85:725–731
Aisner SC, Chakravarthy AK, Joslyn JN, Coughlin TR (1988) Bilateral granular cell tumors of the posterior mediastinum. Ann Thorac Surg 46:688–689
Akiba T, Morikawa T, Hirayama S, Ohki T (2012) Thymic haemangioma presenting with a left innominate vein aneurysm: insight into the aetiology. Interact Cardiovasc Thorac Surg 15:925–927. doi:10.1093/icvts/ivs340
Amir AM, Zeebregts CJ, Mulder HJ (2004) Anterior mediastinal presentation of a giant angiomyolipoma. Ann Thorac Surg 78:2161–2163. doi:10.1016/S0003-4975(03)01512-1
Anderson T, Zhang L, Hameed M, Rusch V, Travis WD, Antonescu CR (2015) Thoracic epithelioid malignant vascular tumors: a clinicopathologic study of 52 cases with emphasis on pathologic grading and molecular studies of WWTR1-CAMTA1 fusions. Am J Surg Pathol 39:132–139. doi:10.1097/PAS.0000000000000346
Angeles RM, Papari M, Malecki Z (2005) Pathologic quiz case: a 43-year-old woman with an incidentally detected posterior mediastinal mass. Granular cell tumor of the posterior mediastinum. Arch Pathol Lab Med 129:e27–e28. doi:10.1043/1543-2165(2005)129<e27:PQCAYW>2.0.CO;2
Antonescu CR, Le Loarer F, Mosquera JM, Sboner A, Zhang L, Chen CL, Chen HW, Pathan N, Krausz T, Dickson BC, Weinreb I, Rubin MA, Hameed M, Fletcher CD (2013) Novel YAP1-TFE3 fusion defines a distinct subset of epithelioid hemangioendothelioma. Genes Chromosomes Cancer 52:775–784. doi:10.1002/gcc.22073
Arab M, Danel C, d’Attellis N, Juvin K, Hernigou A, Fabiani JN, Riquet M (2005) A rare inferior middle mediastinal tumor resection under extra-corporeal circulation. Ann Thorac Surg 79:1413–1415. doi:10.1016/j.athoracsur.2003.10.019
Argani P, Erlandson RA, Rosai J (1997) Thymic neuroblastoma in adults: report of three cases with special emphasis on its association with the syndrome of inappropriate secretion of antidiuretic hormone. Am J Clin Pathol 108:537–543
Asakura S, Tezuka N, Inoue S, Kihara N, Fujino S (2001) Angiomatoid fibrous histiocytoma in mediastinum. Ann Thorac Surg 72:283–285
Auliac JB, Cuvelier A, Peillon C, Louvel JP, Metayer J, Muir JF (1999) Mediastinal leiomyosarcoma. Rev Mal Respir 16:210–213
Baldo X, Sureda C, Gimferrer JM, Belda J (1997) Primary mediastinal leiomyoma: an angiographic study and embolisation of the feeding vessels to improve the surgical approach. Eur J Cardiothorac Surg 11:574–576
Bean SM, Eloubeidi MA, Eltoum IA, Cerfolio RJ, Jhala DN (2005) Preoperative diagnosis of a mediastinal granular cell tumor by EUS-FNA: a case report and review of the literature. Cytojournal 2:8. doi:10.1186/1742-6413-2-8
Begbie SD, Bell DR, Nevell DF (1997) Mediastinal epithelioid hemangioendothelioma in a patient with type IV Ehlers-Danlos syndrome: a case report and review of the literature. Am J Clin Oncol 20:412–415
Begin LR, Schurch W, Lacoste J, Hiscott J, Melnychuk DA (1994) Glycogen-rich clear cell rhabdomyosarcoma of the mediastinum. Potential diagnostic pitfall. Am J Surg Pathol 18:302–308
Bertrand G, Bidabe MC, George P, Dubin P, Touzard C (1984) Angiomyolipoma of the central mediastinum. An apparently undescribed entity. Ann Chir 38:679–681
Bhagotra R, Kapoor R, Singhal VS (1990) Leiomyoma of the mediastinum. Indian J Chest Dis Allied Sc 32:133–136
Boor A, Jurkovic I, Janik M, Vajo J, Kocan P, Ponist J, Dudrikova K, Krajcar R, Stubna J (2000) Benign glomus tumor of the superior posterior mediastinum. Cesk Patol 36:156–159
Box JC, Newman CL, Anastasiades KD, Lucas GW, Latouff OM (1995) Adult rhabdomyoma: presentation as a cervicomediastinal mass (case report and review of the literature). Am Surg 61:271–276
Brindley GV Jr (1949) Glomus tumor of the mediastinum. J Thorac Surg 18:417–420
Caballero C, Gomez S, Matias-Guiu X, Prat J (1992) Rhabdomyosarcomas developing in association with mediastinal germ cell tumours. Virchows Arch A Pathol Anat Histopathol 420:539–543
Cakir E, Demirag F, Gulhan E, Oz G, Tastepez I (2009) Mediastinal composite hemangioendothelioma. A rare tumor at an unusual location. Tumori 95:98–100
Campos J, Otero E, Dominguez MJ, Gonzalez-Quintela A (2007) Epithelioid hemangioendothelioma in the posterior mediastinum. Eur J Intern Med 18:331–332. doi:10.1016/j.ejim.2006.11.010
Candas F, Berber U, Yildizhan A, Yiyit N, Gorur R, Isitmangil T (2013) Anterior mediastinal angiomyolipoma. Ann Thorac Surg 95:1431–1432. doi:10.1016/j.athoracsur.2012.07.066
Carassai P, Caput M (2010) Report of a case of epithelioid hemangioendothelioma of the anterior mediastinum metastatic to pleura. Pathologica 102:112–114
Catanese J, Dutcher JP, Dorfman HD, Andres DF, Wiernik PH (1988) Mediastinal osteosarcoma with extension to lungs in a patient treated for Hodgkin’s disease. Cancer 62:2252–2257
Chetty R, Reddi A (2003) Rhabdomyomatous multilocular thymic cyst. Am J Clin Pathol 119:816–821. doi:10.1309/QDJC-A1HX-QLHL-CFTM
Choi YJ, Yang KH, Gang SJ, Kim BK, Kim SM (1991) Malignant glomus tumor originating in the superior mediastinum—an immunohistochemical and ultrastructural study. J Korean Med Sci 6:157–163
Chu WP (2013) Anterior mediastinal alveolar rhabdomyosarcoma in an infant: rare site for a common paediatric tumour. Hong Kong Med J 19:458–459. doi:10.12809/hkmj133714
Cohen AJ, Sbaschnig RJ, Hochholzer L, Lough FC, Albus RA (1987) Mediastinal hemangiomas. Ann Thorac Surg 43:656–659
Comiter CV, Kibel AS, Richie JP, Nucci MR, Renshaw AA (1998) Prognostic features of teratomas with malignant transformation: a clinicopathological study of 21 cases. J Urol 159:859–863
Conner WC, Fink GW, McGinnis KM, Alfieris GM (2004) Surgical management of leiomyosarcoma of the mediastinum. Ann Thorac Surg 77:334–336
Contreras AL, Punar M, Tamboli P, Tu SM, Pisters L, Moran C, Czerniak BA, Guo CC (2010) Mediastinal germ cell tumors with an angiosarcomatous component: a report of 12 cases. Hum Pathol 41:832–837. doi:10.1016/j.humpath.2009.11.008
D’Abrera VS, Burfitt-Williams W (1973) A melanotic neuroectodermal neoplasm of the posterior mediastinum. J Pathol 111:165–172. doi:10.1002/path.1711110304
D’Aiuto M, Veronesi G, Pompilio G, Gasparri R, Presicci F, Galetta D, Biglioli P, Spaggiari L (2005) Extended right pneumonectomy with partial left atrial resection for primary leiomyosarcoma of the mediastinum. J Thorac Cardiovasc Surg 129:694–695. doi:10.1016/j.jtcvs.2004.07.057
Davies KA, Cope AP, Schofield JB, Chu CQ, Mason JC, Krausz T, Smith P, Denman AM, Walport MJ (1995) A rare mediastinal tumour presenting with systemic effects due to IL-6 and tumour necrosis factor (TNF) production. Clin Exp Immunol 99:117–123
Davis JM, Mark GJ, Greene R (1978) Benign blood vascular tumors of the mediastinum. Report of four cases and review of the literature. Radiology 126:581–587. doi:10.1148/126.3.581
De Luca G, Luciano A, Benincasa G, Sessa R, Petteruti F (2013) Giant malignant granular cell tumor (GCT) of the posterior mediastinum. J Thorac Oncol 8:1107–1108. doi:10.1097/JTO.0b013e318299ad62
de Queiroga EM, Chikota H, Bacchi CE, Moran CA, Suster S (2004) Rhabdomyomatous carcinoma of the thymus. Am J Surg Pathol 28:1245–1250
Deroux A, Destors M, Coudurier M, Lantuejoul S, Aubert M, Girard N, Moro-Sibilot D (2012) A case of mediastinal angiosarcoma. Rev Mal Respir 29:1120–1123. doi:10.1016/j.rmr.2012.04.009
Doglioni C, Bontempini L, Iuzzolino P, Furlan G, Rosai J (1988) Ependymoma of the mediastinum. Arch Pathol Lab Med 112:194–196
Ducatman BS, Scheithauer BW (1984) Malignant peripheral nerve sheath tumors with divergent differentiation. Cancer 54:1049–1057
Ducatman BS, Scheithauer BW, Piepgras DG, Reiman HM, Ilstrup DM (1986) Malignant peripheral nerve sheath tumors. A clinicopathologic study of 120 cases. Cancer 57:2006–2021
Ducatman BS, Scheithauer BW, Piepgras DG, Reiman HM (1984) Malignant peripheral nerve sheath tumors in childhood. J Neuro-Oncol 2:241–248
Ehrenreich T, Freund AJ, Shapiro HN (1953) Hemangio-endothelioma arising in a mediastinal teratoma. Dis Chest 23:294–303
Eimoto T, Kitaoka M, Ogawa H, Niwa H, Murase T, Tateyama H, Inagaki H, Soji T, Wang HJ (2002) Thymic sarcomatoid carcinoma with skeletal muscle differentiation: report of two cases, one with cytogenetic analysis. Histopathology 40:46–57
Ekfors TO, Kulju T, Aaltonen M, Kallajoki M (1998) Ossifying fibromyxoid tumour of soft parts: report of four cases including one mediastinal and one infantile. APMIS 106:1124–1130
El Weshi A, Allam A, Ajarim D, Al Dayel F, Pant R, Bazarbashi S, Memon M (2010) Extraskeletal Ewing’s sarcoma family of tumours in adults: analysis of 57 patients from a single institution. Clin Oncol (R Coll Radiol) 22:374–381. doi:10.1016/j.clon.2010.02.010
Enjolras O, Wassef M, Mazoyer E, Frieden IJ, Rieu PN, Drouet L, Taieb A, Stalder JF, Escande JP (1997) Infants with Kasabach-Merritt syndrome do not have “true” hemangiomas. J Pediatr 130:631–640
Eroglu A, Kurkcuoglu C, Karaoglanoglu N, Gursan N (2002) Primary leiomyosarcoma of the anterior mediastinum. Eur J Cardiothorac Surg 21:943–945
Errani C, Zhang L, Sung YS, Hajdu M, Singer S, Maki RG, Healey JH, Antonescu CR (2011) A novel WWTR1-CAMTA1 gene fusion is a consistent abnormality in epithelioid hemangioendothelioma of different anatomic sites. Genes Chromosomes Cancer 50:644–653. doi:10.1002/gcc.20886
Estrozi B, Queiroga E, Bacchi CE, Faria Soares de Almeida V, Lucas de Carvalho J, Lageman GM, Rosado-de-Christenson M, Suster S (2006) Myxopapillary ependymoma of the posterior mediastinum. Ann Diagn Pathol 10:283–287. doi:10.1016/j.anndiagpath.2006.03.015
Fergeson JO, Clagett OT, Mc DJ (1954) Glomus tumor of the mediastinum; review of the literature and report of case. Surgery 36:320–326
Ferretti GR, Chiles C, Woodruff RD, Choplin RH (1998) Epithelioid hemangioendothelioma of the superior vena cava: computed tomography demonstration and review of the literature. J Thorac Imaging 13:45–48
Fletcher C, Bridge J, Lee J-C (2013) Extra-pleural solitary fibrous tumour, 4th edn, WHO classification of tumours of soft tissue and bone. IARC Press, Lyon, pp 80–82
Fletcher CD, Davies SE, McKee PH (1987) Cellular schwannoma: a distinct pseudosarcomatous entity. Histopathology 11:21–35
Flieder DB, Moran CA, Suster S (1997) Primary alveolar soft-part sarcoma of the mediastinum: a clinicopathological and immunohistochemical study of two cases. Histopathology 31:469–473
Flucke U, Vogels RJ, de Saint Aubain Somerhausen N, Creytens DH, Riedl RG, van Gorp JM, Milne AN, Huysentruyt CJ, Verdijk MA, van Asseldonk MM, Suurmeijer AJ, Bras J, Palmedo G, Groenen PJ, Mentzel T (2014) Epithelioid hemangioendothelioma: clinicopathologic, immunohistochemical, and molecular genetic analysis of 39 cases. Diagn Pathol 9:131. doi:10.1186/1746-1596-9-131
Fokkema JP, Paul MA, Vrouenraets BC (2014) Mediastinal lymphangioma in an adult. Ann R Coll Surg Engl 96:e24–e25. doi:10.1308/003588414X13946184901209
Folpe AL, Weiss SW (2002) Lipoleiomyosarcoma (well-differentiated liposarcoma with leiomyosarcomatous differentiation): a clinicopathologic study of nine cases including one with dedifferentiation. Am J Surg Pathol 26:742–749
Fukuzawa J, Shimizu T, Sakai E, Ido A, Fujita Y, Tsuji T, Ohki Y, Kimura T, Fujita M, Onodera S (1992) Case report of angiomyolipoma of the posterior upper mediastinum. Nihon Kyobu Shikkan Gakkai Zasshi 30:464–467
Gaertner EM, Steinberg DM, Huber M, Hayashi T, Tsuda N, Askin FB, Bell SW, Nguyen B, Colby TV, Nishimura SL, Miettinen M, Travis WD (2000) Pulmonary and mediastinal glomus tumors—report of five cases including a pulmonary glomangiosarcoma: a clinicopathologic study with literature review. Am J Surg Pathol 24:1105–1114
Gale AW, Jelihovsky T, Grant AF, Leckie BD, Nicks R (1974) Neurogenic tumors of the mediastinum. Ann Thorac Surg 17:434–443
Gallivan M, Chun B, Rowden G, Lack E (1980) Intrathoracic paravertebral malignant paraganglioma. Arch Pathol Lab Med 104:46–51
Gibbs AR, Johnson NF, Giddings JC, Powell DE, Jasani B (1984) Primary angiosarcoma of the mediastinum: light and electron microscopic demonstration of factor VIII-related antigen in neoplastic cells. Hum Pathol 15:687–691
Gomez-Roman JJ, Val-Bernal JF (1997) Lipoleiomyosarcoma of the mediastinum. Pathology 29:428–430
Gonzalez-Vela JL, Savage PD, Manivel JC, Torkelson JL, Kennedy BJ (1990) Poor prognosis of mediastinal germ cell cancers containing sarcomatous components. Cancer 66:1114–1116
Greenwood SM, Meschter SC (1989) Extraskeletal osteogenic sarcoma of the mediastinum. Arch Pathol Lab Med 113:430–433
Gross E, Rao BN, Pappo A, Bowman L, Shearer P, Kaste S, Greenwald C, Michalkiewicz E, Pratt C (1996) Epithelioid sarcoma in children. J Pediatr Surg 31:1663–1665
Gupta S, Jindal SK, Vashisht R, Singh H, Malik SK (1983) Leiomyosarcoma of the mediastinum. Eur J Respir Dis 64:69–71
Gururangan S, Bowman LC, Parham DM, Wilimas JA, Rao B, Pratt CB, Douglass EC (1993) Primary extracranial rhabdoid tumors. Clinicopathologic features and response to ifosfamide. Cancer 71:2653–2659
Halefoglu AM (2013) Extraskeletal Ewing’s sarcoma presenting as a posterior mediastinal mass. Arch Bronconeumol 49:82–84. doi:10.1016/j.arbres.2012.02.020
Halliday J, Soon SY, Monaghan H, Walker WS, Zamvar V (2010) Extraskeletal Ewing’s sarcoma presenting as a mediastinal mass. Ann Thorac Surg 90:1016–1017. doi:10.1016/j.athoracsur.2010.01.083
Harrer WV, Patchefsky AS (1972) Malignant granular-cell myoblastoma of the posterior mediastinum. Chest 61:95–96
Hartman KR, Moncur JT, Minniti CP, Creamer KM (2009) Mediastinal Kaposiform hemangioendothelioma and Kasabach-Merritt phenomenon in an infant: treatment with interferon. J Pediatr Hematol Oncol 31:690–692. doi:10.1097/MPH.0b013e3181a1c291
Hayashi K, Yamamoto M, Nishimura H, Inou N (1994) Angiomyolipoma of the anterior mediastinum–a case report. Nihon Kyobu Geka Gakkai Zasshi 42:584–587
Herlitzka AJ, Gale JW (1958) Tumors and cysts of the mediastinum; survey of one hundred seventy-four mediastinal tumors treated surgically during the past eighteen years at the University of Wisconsin Hospitals. AMA Arch Surg 76:697–706
Hilliard RI, McKendry JB, Phillips MJ (1990) Congenital abnormalities of the lymphatic system: a new clinical classification. Pediatrics 86:988–994
Hiraiwa H, Hamazaki M, Tsuruta S, Hattori H, Mimaya J, Hasegawa S, Kohno S, Aoki K (1998) Infantile hemangioendothelioma of the thymus with massive pleural effusion and Kasabach--Merritt syndrome: histopathological, flow cytometrical analysis of the tumor. Acta Paediatr Jpn 40:604–607
Hirano H, Kizaki T, Sashikata T, Maeda T, Yoshii Y (2003) Leiomyosarcoma arising from soft tissue tumor of the mediastinum. Med Electron Microsc 36:52–58. doi:10.1007/s007950300007
Hirose T, Hasegawa T, Seki K, Yang P, Sano T, Morizumi H, Tsuyuguchi M (1996) Atypical glomus tumor in the mediastinum: a case report with immunohistochemical and ultrastructural studies. Ultrastruct Pathol 20:451–456
Hishida T, Yoshida J, Nishimura M, Ishii G, Nakao M, Nagai K (2009) Extraskeletal osteosarcoma arising in anterior mediastinum: brief report with a review of the literature. J Thorac Oncol 4:927–929. doi:10.1097/JTO.0b013e3181a52c63
Hoseok I, Jeong YJ, Choi KU, Kim YD (2008) Symptomatic posterior mediastinal angioleiomyoma. Yonsei Med J 49:666–668. doi:10.3349/ymj.2008.49.4.666
Ikeda T, Ishihara T, Yoshimatsu H, Kikuchi K, Murakami M (1974) Primary osteogenic sarcoma of the mediastinum. Thorax 29:582–588
Ishibashi H, Ota S, Hirose M, Tanio N, Fukuchi K, Muro H, Nakajima N (2010) Granular cell tumor in the mediastinum; report of a case. Kyobu Geka 63:496–499
Isowa N, Hasegawa S, Mino M, Morimoto K, Wada H (2002) Mediastinal epithelioid hemangioendothelioma resected by hemi-plastron window technique. Ann Thorac Surg 74:567–569
Iwami D, Shimaoka S, Mochizuki I, Sakuma T (2006) Kaposiform hemangioendothelioma of the mediastinum in a 7-month-old boy: a case report. J Pediatr Surg 41:1486–1488. doi:10.1016/j.jpedsurg.2006.04.013
Iwata T, Miura T, Inoue K, Hanada S, Inoue H, Miyamoto Y (2012) Primary leiomyosarcoma of the anterior mediastinum encasing the aortic arch, left common carotid and left subclavian arteries. Ann Thorac Cardiovasc Surg 18:140–143
Jeong SS, Choi PJ, Kim DW, Son C, Roh MS (2013) Primary extraskeletal mesenchymal chondrosarcoma of the anterior mediastinum. Korean J Pathol 47:492–494. doi:10.4132/KoreanJPathol.2013.47.5.492
Kalra B, Kingsley PA, Bedi HS, Kwatra KS, Negi P (2014) Malignant peripheral nerve sheath tumor of the anterior mediastinum: a rare presentation. Rare Tumors 6:5528. doi:10.4081/rt.2014.5528
Kardamakis D, Bouboulis N, Ravazoula P, Dimopoulos P, Dougenis D (1996) Primary hemangiosarcoma of the mediastinum. Lung Cancer 16:81–86
Katakura H, Fukuse T, Shiraishi I, Hayatsu E, Nishijo K, Toguchida J, Nakashima Y, Wada H (2009) Mediastinal synovial sarcoma. Thorac Cardiovasc Surg 57:183–185. doi:10.1055/s-2006-955886
Kim do Y, Jeon HW, Kim KS, Park JK, Sung SW (2014) A rare case of mediastinal granular cell tumor. Korean J Thorac Cardiovasc Surg 47:494–496. doi:10.5090/kjtcs.2014.47.5.494
Kim YH, Kwon NY, Myung NH, Kim EJ, Choi YH, Yoon SY, Choi EK, Park JS, Kim KY, Lee KY (2001) A case of mediastinal angiomyolipoma. Korean J Intern Med 16:277–280
Knight CS, Cerfolio RJ, Winokur TS (2008) Angiomyolipoma of the anterior mediastinum. Ann Diagn Pathol 12:293–295. doi:10.1016/j.anndiagpath.2006.12.007
Kocatas A, Dural AC, Sever N, Kankaya B, Dogan M, Yegen G, Gonenc M, Bilgic BM, Bedirhan MA, Alis H (2014) Minimal invasive endoscopic management of synchronous granular cell tumours in the colon and posterior mediastinum. J Minim Access Surg 10:34–36. doi:10.4103/0972-9941.124469
Koezuka S, Hata Y, Sato F, Otsuka H, Makino T, Tochigi N, Iyoda A (2014) Malignant peripheral nerve sheath tumor in the anterior mediastinum: a case report. Mol Clin Oncol 2:987–990. doi:10.3892/mco.2014.343
Koizumi K, Haraguchi S, Mikami I, Kubokura H, Okada D, Yamagishi S, Kinoshita H, Enomoto Y, Shimizu K, Maeda M (2005) Video-assisted thoracic surgery for Ewing’s sarcoma of the mediastinum in a 3-year-old girl. Ann Thorac Cardiovasc Surg 11:117–120
Kuschill-Dziurda J, Mastalerz L, Grzanka P, Nizankowska-Mogilnicka E (2009) Rhabdomyoma as a tumor of the posterior mediastinum. Pol Arch Med Wewn 119:599–602
Kuzucu A, Erkal HS, Soysal O, Serin M (2006) Extraskeletal Ewing’s sarcoma presenting with multifocal intrathoracic mass lesions associated with mediastinal shift. Ann Thorac Surg 81:1487–1488. doi:10.1016/j.athoracsur.2005.03.116
Lamovec J, Sobel HJ, Zidar A, Jerman J (1990) Epithelioid hemangioendothelioma of the anterior mediastinum with osteoclast-like giant cells. Light microscopic, immunohistochemical, and electron microscopic study. Am J Clin Pathol 93:813–817
Lamy AL, Fradet GJ, Luoma A, Nelems B (1994) Anterior and middle mediastinum paraganglioma: complete resection is the treatment of choice. Ann Thorac Surg 57:249–252
Lang-Lazdunski L, Pons F, Jancovici R (2003) Malignant “Triton” tumor of the posterior mediastinum: prolonged survival after staged resection. Ann Thorac Surg 75:1645–1648
Lazure T, Essamet W, Palazzo L, Epardeau B, Fabre M (2001) Cytological findings of a primary mediastino-pulmonary leiomyosarcoma. Report of a case diagnosed by endoscopic ultrasonography-guided fine needle aspiration. Cytopathology 12:410–413
Lee DH, Park CK, Keum DY, Kim JB, Hwang I (2012) Leiomyosarcoma of the posterior mediastinum extending into the adjacent spinal canal. Korean J Thorac Cardiovasc Surg 45:192–195. doi:10.5090/kjtcs.2012.45.3.192
Lee TJ, Reddy GP, Leung JW, Gotway MB (2008) Mediastinal glomangioma: CT and octreotide scintigraphy appearance, and review of the literature. J Thorac Imaging 23:289–291. doi:10.1097/RTI.0b013e31817eee70
Li XM, Lin XY, Xu HT, Yu JH, Wang L, Fan CF, Liu Y, Wang EH (2013) Mediastinal epithelioid hemangioendothelioma with abundant spindle cells and osteoclast-like giant cells mimicking malignant fibrous histiocytoma. Diagn Pathol 8:103. doi:10.1186/1746-1596-8-103
Lin J, Ho J, Chan A, Yeo W, Yip KM, Johnson PJ (1995) Extraosseous osteogenic sarcoma of the mediastinum occurring in a Chinese patient. Clin Oncol (R Coll Radiol) 7:200–201
Lincoln JC (1965) Leiomyosarcoma of the anterior mediastinum. Thorax 20:362–366
Linnoila RI, Tsokos M, Triche TJ, Marangos PJ, Chandra RS (1986) Evidence for neural origin and PAS-positive variants of the malignant small cell tumor of thoracopulmonary region (“Askin tumor”). Am J Surg Pathol 10:124–133
Lodding P, Kindblom LG, Angervall L, Stenman G (1990) Cellular schwannoma. A clinicopathologic study of 29 cases. Virchows Arch A Pathol Anat Histopathol 416:237–248
Lyons LL, North PE, Mac-Moune Lai F, Stoler MH, Folpe AL, Weiss SW (2004) Kaposiform hemangioendothelioma: a study of 33 cases emphasizing its pathologic, immunophenotypic, and biologic uniqueness from juvenile hemangioma. Am J Surg Pathol 28:559–568
Maamouri F, Ennaifer Jerbi E, Tounsi Guettiti H, Boussen H, Dellagi K, Boubaker S (2008) Multicentric epithelioid hemangioendothelioma involving lung, mediastinum and liver. Tunis Med 86:616–617
Macchiarini P, Ostertag H (2004) Uncommon primary mediastinal tumours. Lancet Oncol 5:107–118. doi:10.1016/S1470-2045(04)01385-3
Machida E, Haniuda M, Eguchi T, Kurai M, Yamanda T, Amano J, Ota H (2003) Granular cell tumor of the mediastinum. Intern Med 42:178–181
Malagon HD, Valdez AM, Moran CA, Suster S (2007) Germ cell tumors with sarcomatous components: a clinicopathologic and immunohistochemical study of 46 cases. Am J Surg Pathol 31:1356–1362. doi:10.1097/PAS.0b013e318033c7c4
Manduch M, Dexter DF, Ellis PM, Reid K, Isotalo PA (2008) Extraskeletal Ewing’s sarcoma/primitive neuroectodermal tumor of the posterior mediastinum with t(11;22)(q24;q12). Tumori 94:888–891
Manivel C, Wick MR, Abenoza P, Rosai J (1986) The occurrence of sarcomatous components in primary mediastinal germ cell tumors. Am J Surg Pathol 10:711–717
Mansour Z, Neuville A, Massard G (2010) Mediastinal epithelioid haemangioendothelioma: a rare mediastinal tumour. Interact Cardiovasc Thorac Surg 10:122–124. doi:10.1510/icvts.2009.216978
Marchevsky AM (1999) Mediastinal tumors of peripheral nervous system origin. Semin Diagn Pathol 16:65–78
Matsuoka H, Nishio W, Sakamoto T, Harada H, Sashikata T, Tsubota N (2002) Mediastinal angioleiomyoma. Ann Thorac Surg 73:1653–1654
McConnell YJ, Giacomantonio CA (2012) Malignant triton tumors—complete surgical resection and adjuvant radiotherapy associated with improved survival. J Surg Oncol 106:51–56. doi:10.1002/jso.23042
Michel M, Pratt JW (2004) Anterior mediastinal nonseminomatous germ cell tumor with malignant transformation: a case report. Curr Surg 61:576–579. doi:10.1016/j.cursur.2004.05.021
Miller R, Kurtz SM, Powers JM (1978) Mediastinal rhabdomyoma. Cancer 42:1983–1988
Mineo TC, Biancari F, Cristino B, D’Andrea V (1995) Benign vascular tumours of the mediastinum: presentation of three cases and review of the literature. Thorac Cardiovasc Surg 43:361–364. doi:10.1055/s-2007-1013811
Misugi K, Okajima H, Newton WA Jr, Kmetz DR, Delorimier AA (1965) Mediastinal origin of a melanotic progonoma or retinal anlage tumor: ultrastructural evidence for neural crest origin. Cancer 18:477–484
Moran CA, Koss MN (1993) Rhabdomyomatous thymoma. Am J Surg Pathol 17:633–636
Moran CA, Suster S (1995) Mediastinal hemangiomas: a study of 18 cases with emphasis on the spectrum of morphological features. Hum Pathol 26:416–421
Moran CA, Suster S (1997) Primary germ cell tumors of the mediastinum: I. Analysis of 322 cases with special emphasis on teratomatous lesions and a proposal for histopathologic classification and clinical staging. Cancer 80:681–690
Moran CA, Suster S, Fishback N, Koss MN (1993) Mediastinal paragangliomas. A clinicopathologic and immunohistochemical study of 16 cases. Cancer 72:2358–2364
Moran CA, Suster S, Perino G, Kaneko M, Koss MN (1994) Malignant smooth muscle tumors presenting as mediastinal soft tissue masses. A clinicopathologic study of 10 cases. Cancer 74:2251–2260
Morita K, Shida Y, Shinozaki K, Uehara S, Seto T, Sugio K, Ichinose Y, Nishiyama K, Matsuo Y, Hatakenaka M, Honda H (2012) Angiomyolipomas of the mediastinum and the lung. J Thorac Imaging 27:W21–W23. doi:10.1097/RTI.0b013e31823150c7
Murakawa T, Nakajima J, Fukami T, Tanaka M, Takeuchi E, Takamoto S (2001) Malignant fibrous histiocytoma in the anterior mediastinum. Jpn J Thorac Cardiovasc Surg 49:722–727
Nakamura M, Tachibana H, Mukaeyama T (1996) A case of leiomyoma of the posterior mediastinum. Nihon Kyobu Geka Gakkai Zasshi 44:2082–2086
Nakao M, Hishida T, Ishii G, Yoshida J, Nishimura M, Nagai K (2012) Malignant granular cell tumor of the posterior mediastinum with dissemination. Asian Cardiovasc Thorac Ann 20:71–73. doi:10.1177/0218492311421453
Nayak HK, Vangipuram DR, Sonika U, Kar P, Kumar N, Kapoor N (2011) Mediastinal mass—a rare presentation of desmoplastic small round cell tumour. BMJ Case Rep 2011. doi: 10.1136/bcr.10.2011.5042
Niwa H, Kondou K, Sekiguchi K (1988) A case of mediastinal leiomyoma associated with cystic change and calcification—a review of 7 cases in Japanese literatures. Nihon Kyobu Geka Gakkai Zasshi 36:2136–2139
O’Regan GM, Irvine AD, Yao N, O’Marcaigh A, Sheridan-Pereira M, Phelan E, McDermott MB, Twomey A, Russell J, Watson R (2009) Mediastinal and neck kaposiform hemangioendothelioma: report of three cases. Pediatr Dermatol 26:331–337. doi:10.1111/j.1525-1470.2009.00913.x
Odze R, Begin L (1990) Malignant paraganglioma of the posterior mediastinum. Cancer 65:667–694
Odze R, Begin LR (1990) Malignant paraganglioma of the posterior mediastinum. A case report and review of the literature. Cancer 65:564–569
Ohebsion J, O’Connor WN, Attili AK, Diaz-Guzman E (2010) A 30-year-old man with facial flushing and a mediastinal mass. Chest 138:746–749. doi:10.1378/chest.09-2847
Ohtsuka M, Satoh H, Inoue M, Yazawa T, Yamashita YT, Sekizawa K, Hasegawa S (2000) Disseminated metastasis of neuroblastomatous component in immature mediastinal teratoma: a case report. Anticancer Res 20:527–530
Okudela K, Nakamura N, Sano J, Ito T, Kitamura H (2001) Thymic carcinosarcoma consisting of squamous cell carcinomatous and embryonal rhabdomyosarcomatous components. Report of a case and review of the literature. Pathol Res Pract 197:205–210
Olson J, Sayler W (1978) Mediastinal paragangliomas (aortic body tumor): a report of four cases and a review of the literature. Cancer 41:2405–2412
Oshikiri T, Morikawa T, Jinushi E, Kawakami Y, Katoh H (2001) Five cases of the lymphangioma of the mediastinum in adult. Ann Thorac Cardiovasc Surg 7:103–105
Otani Y, Morishita Y, Yoshida I, Ishikawa S, Otaki A, Aihara T, Nakajima T (1996) A malignant Triton tumor in the anterior mediastinum requiring emergency surgery: report of a case. Surg Today 26:834–836
Ouadnouni Y, Achir A, Bekarsabein S, Bouchikh M, Smahi M, Msougar Y, Mahassini N, Benosman A (2009) Primary mediastinal leiomyoma: a case report. Cases J 2:8555. doi:10.4076/1757-1626-2-8555
Pachter MR (1963) Mediastinal nonchromaffin paraganglioma. A clinicopathologic study based on eight cases. J Thorac Cardiovasc Surg 45:152–160
Pachter MR, Lattes R (1963) Mesenchymal tumors of the mediastinum II. Tumors of blood vascular origin. Cancer 16:95–107
Panasuk DB, Bauer TL, Davies AL, Schneider C, Flynn C (2003) Common malignancies with uncommon sites of presentation: case 1. Anterior mediastinal rhabdomyosarcoma. J Clin Oncol 21:4455–4456. doi:10.1200/JCO.2003.03.149
Pandit S, Mukherjee S, Bhattacharya S, Dattachaudhuri A, Bhuniya S, Deb J, Bhanja P (2012) A rare mediastinal tumour in a young male mimicking massive pleural effusion. Lung India 29:66–69. doi:10.4103/0970-2113.92368
Papagiannopoulos K, Sheppard MN, Goldstraw P (2000) Thymic hemangioma presenting with recurrent pleural effusion. Ann Thorac Surg 70:297–299
Parham DM, Weeks DA, Beckwith JB (1994) The clinicopathologic spectrum of putative extrarenal rhabdoid tumors. An analysis of 42 cases studied with immunohistochemistry or electron microscopy. Am J Surg Pathol 18:1010–1029
Park JG, Aubry MC, Godfrey JA, Midthun DE (2006) Mediastinal lymphangioma: Mayo Clinic experience of 25 cases. Mayo Clin Proc 81:1197–1203. doi:10.4065/81.9.1197
Park JY, Park NJ, Kim SP, Kwon KY, Lee SS (2012) A soft tissue perineurioma and a hybrid tumor of perineurioma and schwannoma. Korean J Pathol 46:75–78. doi:10.4132/KoreanJPathol.2012.46.1.75
Perlman EJ, Ali SZ, Robinson R, Lindato R, Griffin CA (1998) Infantile extrarenal rhabdoid tumor. Pediatr Dev Pathol 1:149–152
Ponce FA, Killory BD, Wait SD, Theodore N, Dickman CA (2011) Endoscopic resection of intrathoracic tumors: experience with and long-term results for 26 patients. J Neurosurg Spine 14:377–381. doi:10.3171/2010.11.SPINE09718
Qi Y, Chang B, Pang L, Liu C, Li F (2011) Solid alveolar rhabdomyosarcoma with spindle-shaped cells and epithelial differentiation of the mediastinum in a 68-year-old man: a case report and literature review. J Cancer Res Ther 7:353–356. doi:10.4103/0973-1482.87009
Quan GM, Irons S, Scripcaru G, Tang SK (2014) Posterior mediastinal paraspinal angioleiomyoma causing thoracic radiculopathy. ANZ J Surg. doi:10.1111/ans.12639
Rajasingham AS, Cooray GH (1954) A large leiomyoma of the mediastinum. Br J Surg 41:446–447
Rasaretnam R, Panabokke RG (1975) Leiomyosarcoma of the mediastinum. Br J Dis Chest 69:63–69
Reali A, Mortellaro G, Allis S, Trevisiol E, Anglesio SM, Bartoncini S, Ruo Redda MG (2013) A case of primary mediastinal Ewing’s sarcoma/primitive neuroectodermal tumor presenting with initial compression of superior vena cava. Ann Thorac Med 8:121–123. doi:10.4103/1817-1737.109834
Reeder LB (2000) Neurogenic tumors of the mediastinum. Semin Thorac Cardiovasc Surg 12:261–267
Ren W, Xu X, Yan J, Qian X, Liu B (2014) Malignant triton tumor of the anterior mediastinum: a case report. Oncol Lett 7:807–810. doi:10.3892/ol.2014.1787
Ribet ME, Cardot GR (1994) Neurogenic tumors of the thorax. Ann Thorac Surg 58:1091–1095
Robinson JM, Knoll R, Henry DA (1988) Intrathoracic granular cell myoblastoma. South Med J 81:1453–1457
Romano R, Cesario A, Lococo F, Petrone G, Margaritora S, Granone P (2013) Successful multidisciplinary management of a primary mediastinal Ewing’s sarcoma. Minerva Chir 68:121–124
Rosenbloom PM, Barrows GH, Kmetz DR, Canty TG (1975) Granular cell myoblastoma arising from the thoracic sympathetic nerve chain. J Pediatr Surg 10:819–822
Saito A, Watanabe K, Kusakabe T, Abe M, Suzuki T (1998) Mediastinal mature teratoma with coexistence of angiosarcoma, granulocytic sarcoma and a hematopoietic region in the tumor: a rare case of association between hematological malignancy and mediastinal germ cell tumor. Pathol Int 48:749–753
Salah S, Al-Ibraheem A, Daboor A, Al-Hussaini M (2013) Synovial sarcoma presenting with huge mediastinal mass: a case report and review of the literature. BMC Res Notes 6:240
Salah S, Salem A (2014) Primary synovial sarcomas of the mediastinum: a systematic review and pooled analysis of the published literature. ISRN Oncol 2014:412527. doi:10.1155/2014/412527
Salih Deveci M, Ceyhan K, Deveci G, Finci R (2001) Pericardial rhabdomyomatous spindle cell thymoma with mucinous cystic degeneration. Histopathology 38:479–481
Sartelet H, Lantuejoul S, Armari-Alla C, Pin I, Delattre O, Brambilla E (1998) Solid alveolar rhabdomyosarcoma of the thorax in a child. Histopathology 32:165–171
Sayan M, Celik A, Ertunc O, Uluoglu O, Tastepe AI (2014) Mediastinal neurothekeoma: a rare tumor. Asian Cardiovasc Thorac Ann. doi:10.1177/0218492314552857
Schenning R, Vajtai P, Troxell M, Pollock J, Hopkins K (2013) Alveolar soft part sarcoma: unusual etiology of mediastinal mass in an adolescent. Clin Pract 3:e26. doi:10.4081/cp.2013.e26
Schulman H, Newman-Heinman N, Kurtzbart E, Maor E, Zirkin H, Laufer L (2000) Thoracoabdominal peripheral primitive neuroectodermal tumors in childhood: radiological features. Eur Radiol 10:1649–1652. doi:10.1007/s003300000367
Segawa M, Kusajima Y, Senda K, Saito K (2007) Mediastinal granular cell tumor arising from the left recurrent nerve; report of a case. Kyobu Geka 60:595–598
Shaffer K, Pugatch RD, Sugarbaker DJ (1990) Primary mediastinal leiomyoma. Ann Thorac Surg 50:301–302
Short M, Dramis A, Ramani P, Parikh DH (2008) Mediastinal and pulmonary infantile myofibromatosis: an unusual surgical presentation. J Pediatr Surg 43:e29–e31. doi:10.1016/j.jpedsurg.2008.06.046
Sidhu JS, Nicolas MM, Taylor W (2002) Mediastinal rhabdomyoma: a case report and review of the literature. Int J Surg Pathol 10:313–318
Siraj F, Dhawan S, Jain D (2011) Invasive thymoma with osseous metaplasia and cystic change in a case of myasthenia gravis: a rare presentation. Gen Thorac Cardiovasc Surg 59:583–586. doi:10.1007/s11748-010-0726-4
Smith AR, Gilbert CF, Strausbauch P, Silverman JF (1998) Fine needle aspiration cytology of a mediastinal granular cell tumor with histologic confirmation and ancillary studies. A case report. Acta Cytol 42:1011–1016
Soh WM, Yeong ML, Wong KP (2014) Malignant granular cell tumour of the mediastinum. Malays J Pathol 36:149–151
Stark P, Smith DC, Watkins GE, Chun KE (1990) Primary intrathoracic extraosseous osteogenic sarcoma: report of three cases. Radiology 174:725–726. doi:10.1148/radiology.174.3.2305056
Steen BC, Florez Martin S, Fernandez Fau L, Garcia Tirado J, Jareno Esteban J, Ancochea Bermudez J (1993) Mediastinal leiomyosarcoma. An Med Interna 10:83–85
Sunderrajan EV, Luger AM, Rosenholtz MJ, Maltby JD (1984) Leiomyosarcoma in the mediastinum presenting as superior vena cava syndrome. Cancer 53:2553–2556
Suster S, Moran CA (1997) Malignant cartilaginous tumors of the mediastinum: clinicopathological study of six cases presenting as extraskeletal soft tissue masses. Hum Pathol 28:588–594
Suster S, Moran CA (2005) Primary synovial sarcomas of the mediastinum: a clinicopathologic, immunohistochemical, and ultrastructural study of 15 cases. Am J Surg Pathol 29:569–578
Suster S, Moran CA, Koss MN (1994) Epithelioid hemangioendothelioma of the anterior mediastinum. Clinicopathologic, immunohistochemical, and ultrastructural analysis of 12 cases. Am J Surg Pathol 18:871–881
Suster S, Moran CA, Koss MN (1994) Rhabdomyosarcomas of the anterior mediastinum: report of four cases unassociated with germ cell, teratomatous, or thymic carcinomatous components. Hum Pathol 25:349–356
Tajima H, Tajima N, Yamamoto K, Maeda S, Koizumi K, Kumazaki T, Yoshida H, Egami K (1995) Anterior mediastinal schwannoma: a case report. Radiat Med 13:175–177
Tanaka Y, Yoshimasu T, Oura S, Hirai Y, Kawago M, Okamura Y (2014) Primary clear-cell sarcoma in the mediastinum. Case Rep Oncol 7:306–309. doi:10.1159/000363180
Tane S, Tanaka Y, Tauchi S, Uchino K, Nakai R, Yoshimura M (2011) Radically resected epithelioid angiosarcoma that originated in the mediastinum. Gen Thorac Cardiovasc Surg 59:503–506. doi:10.1007/s11748-010-0710-z
Tarr RW, Kerner T, McCook B, Page DL, Nance EP, Kaye JJ (1988) Primary extraosseous osteogenic sarcoma of the mediastinum: clinical, pathologic, and radiologic correlation. South Med J 81:1317–1319
Teh BM, Nickless D, Seevanayagam S (2011) Malignant ossifying fibromyxoid tumour: a large anterior mediastinal mass. Pathology 43:749–751. doi:10.1097/PAT.0b013e32834c72b1
Thomson TA, Klijanienko J, Couturier J, Brisse H, Pierron G, Freneaux P, Sastre-Garau X, Lagace R, Bourdeaut F (2011) Fine-needle aspiration of renal and extrarenal rhabdoid tumors: the experience of the Institut Curie regarding 20 tumors in 13 patients. Cancer Cytopathol 119:49–57. doi:10.1002/cncy.20121
Tirabosco R, Lang-Lazdunski L, Diss TC, Amary MF, Rodriguez-Justo M, Landau D, Lorenzi W, Flanagan AM (2009) Clear cell sarcoma of the mediastinum. Ann Diagn Pathol 13:197–200. doi:10.1016/j.anndiagpath.2008.02.014
Torigian DA, Kaiser LR, Soma LA, Tomaszewski JE, Kotloff R, Siegelman ES (2002) Symptomatic dysrhythmia caused by a posterior mediastinal angiomyolipoma. AJR Am J Roentgenol 178:93–96. doi:10.2214/ajr.178.1.1780093
Torres-Mora J, Dry S, Li X, Binder S, Amin M, Folpe AL (2014) Malignant melanotic schwannian tumor: a clinicopathologic, immunohistochemical, and gene expression profiling study of 40 cases, with a proposal for the reclassification of “melanotic schwannoma”. Am J Surg Pathol 38:94–105. doi:10.1097/PAS.0b013e3182a0a150
Toursarkissian B, O’Connor WN, Dillon ML (1990) Mediastinal epithelioid hemangioendothelioma. Ann Thorac Surg 49:680–685
Trupiano JK, Rice TW, Herzog K, Barr FG, Shipley J, Fisher C, Goldblum JR (2002) Mediastinal synovial sarcoma: report of two cases with molecular genetic analysis. Ann Thorac Surg 73:628–630
Ulusakarya A, Terrier P, Regnard JF, de Montpreville V, Munck JN (1999) Extraskeletal osteosarcoma of the mediastinum after treatment of a mediastinal germ-cell tumor. Am J Clin Oncol 22:609–614
Uno A, Sakurai M, Onuma K, Yamane Y, Kurita K, Hayashi I, Ikeda M, Hagiwara N, Tominaga K, Hakozaki H (1988) A case of giant mediastinal leiomyoma with long-term survival. Tohoku J Exp Med 156:1–6
Vaish AK, Verma SK, Shakya S, Goel MM (2012) Schwannoma in anterior mediastinum with massive pericardial effusion. BMJ Case Rep 2012. doi: 10.1136/bcr-2012-007867
Van Schil PE, Colpaert CG, Van Look R, Van Oosterom AT, EA V m, Reher SW, Schoofs EL (1993) Primary mediastinal leiomyosarcoma. Thorac Cardiovasc Surg 41:377–378. doi:10.1055/s-2007-1013895
Venuta F, Pescarmona EO, Rendina EA, Ciriaco P, De Giacomo T, Ricci C (1993) Primary osteogenic sarcoma of the posterior mediastinum. Case report. Scand J Thorac Cardiovasc Surg 27:169–173
Vrtik M, Larbalestier RI, Cameron D, Gupta A, Sinniah R (2004) Giant superior mediastinal angioleiomyoma. J Thorac Cardiovasc Surg 128:786–788. doi:10.1016/j.jtcvs.2004.03.035
Wallenstein MB, Hole MK, McCarthy C, Fijalkowski N, Jeng M, Wong WB (2014) Mediastinal Kaposiform hemangioendothelioma and Kasabach-Merritt phenomenon in a patient with no skin changes and a normal chest CT. Pediatr Hematol Oncol. doi:10.3109/08880018.2013.825356
Walsh MA, Carcao M, Pope E, Lee KJ (2008) Kaposiform hemangioendothelioma presenting antenatally with a pericardial effusion. J Pediatr Hematol Oncol 30:761–763. doi:10.1097/MPH.0b013e318175c244
Warth A, Herpel E, Schmahl A, Hoffmann H, Herth FJ, Schirmacher P, Dienemann H, Schnabel PA (2008) Mediastinal angiomyolipomas in a male patient affected by tuberous sclerosis. Eur Respir J 31:678–680. doi:10.1183/09031936.00021207
Watanabe S, Sato H, Tawaraya K, Tsubota M, Endo M, Seki M (1997) A case of mediastinal angiomyolipoma. Nihon Kyobu Geka Gakkai Zasshi 45:1889–1892
Watts KA, Watts JR Jr (2007) Incidental discovery of an anterior mediastinal angiomyolipoma. J Thorac Imaging 22:180–181. doi:10.1097/01.rti.0000213571.09280.f8
Weidner N (1991) Atypical tumor of the mediastinum: epithelioid hemangioendothelioma containing metaplastic bone and osteoclastlike giant cells. Ultrastruct Pathol 15:481–488
Weiss SW, Enzinger FM (1982) Epithelioid hemangioendothelioma: a vascular tumor often mistaken for a carcinoma. Cancer 50:970–981
Weissferdt A, Kalhor N, Suster S, Moran CA (2010) Primary angiosarcomas of the anterior mediastinum: a clinicopathologic and immunohistochemical study of 9 cases. Hum Pathol 41:1711–1717. doi:10.1016/j.humpath.2010.05.003
White W, Shiu MH, Rosenblum MK, Erlandson RA, Woodruff JM (1990) Cellular schwannoma. A clinicopathologic study of 57 patients and 58 tumors. Cancer 66:1266–1275
Wick MR, Rosai J (1991) Neuroendocrine neoplasms of the mediastinum. Semin Diagn Pathol 8:35–51
Wilken JJ, Meier FA, Kornstein MJ (2000) Kaposiform hemangioendothelioma of the thymus. Arch Pathol Lab Med 124:1542–1544. doi:10.1043/0003-9985(2000)124<1542:KHOTT>2.0.CO;2
Wilson H (1941) Extraskeletal ossifying tumors. Ann Surg 113:95–112
Wilson RW, Moran CA (1998) Primary ependymoma of the mediastinum: a clinicopathologic study of three cases. Ann Diagn Pathol 2:293–300
Woodruff JM, Godwin TA, Erlandson RA, Susin M, Martini N (1981) Cellular schwannoma: a variety of schwannoma sometimes mistaken for a malignant tumor. Am J Surg Pathol 5:733–744
Yanagawa N, Sasou S, Sato T, Shiono S, Kato H (2006) Mediastinal granular cell tumor: report of a case. Kyobu Geka 59:418–421
Yasuda A, Mizuno A, Mishima A, Sasaki S, Ochi N, Wakasugi T, Sawai H, Takeyama H, Manabe T (2007) Posterior mediastinal angioleiomyoma: report of a case. J Thorac Imaging 22:363–365. doi:10.1097/RTI.0b013e31813fabb5
Yasui N, Koh K, Kato M, Park MJ, Tomizawa D, Oshima K, Uchisaka N, Gocho Y, Arakawa A, Seki M, Oguma E, Kishimoto H, Watanabe S, Kikuchi A, Hanada R (2013) Kasabach-Merritt phenomenon: a report of 11 cases from a single institution. J Pediatr Hematol Oncol 35:554–558. doi:10.1097/MPH.0b013e318281558e
Yazici U, Gulhan E, Yazici U, Yaran P, Cakir E, Tastepe I (2011) A case of giant mediastinal leiomyoma. Turk J Gastroenterol 22:656–657
Younker D, Boozalis JE (1991) A thoracic leiomyosarcoma producing tracheal obstruction. J Clin Anesth 3:344–346
Yousem SA, Hochholzer L (1987) Unusual thoracic manifestations of epithelioid hemangioendothelioma. Arch Pathol Lab Med 111:459–463
Zehani A, Ayadi-Kaddour A, Daghfous H, Ridene I, Marghli A, Kilani T, El Mezni F (2011) Primary mediastinal sarcomas. Rev Mal Respir 28:14–24. doi:10.1016/j.rmr.2010.05.015
Zhang WD, Zhao LL, Huang XB, Cai PQ, Xu GX (2010) Computed tomography imaging of anterior and middle mediastinal Ewing sarcoma/primitive neuroectodermal tumors. J Thorac Imaging 25:168–172. doi:10.1097/RTI.0b013e3181a99117
Zisis C, Fragoulis S, Rontogianni D, Stratakos G, Bellenis I (2006) Malignant triton tumour of the anterior mediastinum as incidental finding. Monaldi Arch Chest Dis 65:222–224
Zolota V, Tzelepi V, Charoulis N, Apostolakis E, Dougenis D (2006) Mediastinal rhabdomyoma: case report and review of the literature. Virchows Arch 449:124–128. doi:10.1007/s00428-006-0211-8
Zukerberg LR, Nickoloff BJ, Weiss SW (1993) Kaposiform hemangioendothelioma of infancy and childhood. An aggressive neoplasm associated with Kasabach-Merritt syndrome and lymphangiomatosis. Am J Surg Pathol 17:321–328
Acknowledgments
The authors wish to thank Dr. Satoshi Kaneda (Department of Radiology, Saiseikai Central Hospital, Tokyo, Japan) for providing radiological images, Dr. Ieneke Hartmann (Department of Radiology, Maasstad Ziekenhuis, Rotterdam, The Netherlands) for reviewing the radiological images and for providing suggestions for the figure legends. The molecular analysis of the epithelioid hemangioendothelioma (Fig. 2) was kindly performed by Dr. Uta Flucke (Department of Pathology, Radboud University Medical Center, Nijmegen, The Netherlands).
Compliance with Ethical Standards
ᅟ
Conflict of interest
The authors declare that they have no competing interests.
Human and Animal Rights and Informed Consent
There was no research involving human participants and/or animals performed for this manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
den Bakker, M.A., Marx, A., Mukai, K. et al. Mesenchymal tumours of the mediastinum—part II. Virchows Arch 467, 501–517 (2015). https://doi.org/10.1007/s00428-015-1832-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00428-015-1832-6