
Abstract
Drought is one of the major challenges affecting crop productivity and yield. However, water stress responses are notoriously multigenic and quantitative with strong environmental effects on phenotypes. It is also clear that water stress often does not occur alone under field conditions but rather in conjunction with other abiotic stresses such as high temperature and high light intensities. A multidisciplinary approach with successful integration of a whole range of -omics technologies will not only define the system, but also provide new gene targets for both transgenic approaches and marker-assisted selection. Transcription factors are major players in water stress signaling and some constitute major hubs in the signaling webs. The main transcription factors in this network include MYB, bHLH, bZIP, ERF, NAC, and WRKY transcription factors. The role of WRKY transcription factors in abiotic stress signaling networks is just becoming apparent and systems biology approaches are starting to define their places in the signaling network. Using systems biology approaches, there are now many transcriptomic analyses and promoter analyses that concern WRKY transcription factors. In addition, reports on nuclear proteomics have identified WRKY proteins that are up-regulated at the protein level by water stress. Interactomics has started to identify different classes of WRKY-interacting proteins. What are often lacking are connections between metabolomics, WRKY transcription factors, promoters, biosynthetic pathways, fluxes and downstream responses. As more levels of the system are characterized, a more detailed understanding of the roles of WRKY transcription factors in drought responses in crops will be obtained.
Similar content being viewed by others


Introduction
A basic definition of omics is the cataloging of comprehensive sets of biological information from a given sample (Chow and Kay 2013). Today, there seems to be a new -omics field almost every week. In plants, the major -omics technologies include ionomics (the study of elemental accumulation), genomics (the study of genes and genomes), transcriptomics (the study of mRNA levels), regulomics (transcription factors and other molecules that regulate gene expression), proteomics (the study of proteins), phosphoproteomics (the study of phosphorylated proteins), secretomics (the study of secreted proteins), interactomics (the study of interactions), metabolomics (the study of metabolites), fluxomics (the study of fluxes through pathways), physiomics (the study of how physiological features are associated with genes, proteins and their networks), and phenomics (the study of phenotypes). All of these technologies together make up systems biology and the integration of these -omics technologies will facilitate an understanding of what the plant is doing at the whole system level. For example, regulomics will help explain the changes seen in transcriptomics and these changes, at least in part, will be reflected in proteomics analyses. Ultimately, changes in protein activities will affect the metabolome and so on.
Drought
Drought is one of the major events that cause huge losses in crop productivity and yield and it is therefore a major focus of research. Analyses of water stress responses at the systems biology levels therefore have great promise for the improvement of crop plants. A recent World Agricultural Supply and Demand Estimates report (October 11, 2012) shows that production of major crops like corn, soybean, sorghum, wheat, etc., was reduced by 27.6 % during the severe US drought in 2012 (http://www.ers.usda.gov/topics/in-the-news/us-drought-2012-farm-and-food-impacts.aspx) and this trend is increasing year by year (WASDE report 2012). Not only are crops affected, drought simultaneously affects livestock and farms with the same intensity.
Water deficiency has a major impact on ecological and agricultural systems and is a limiting factor during the initial phase of plant growth and establishment (Roschefort and Woodward 1992; Shao et al. 2009).
Water stress results in stomatal closure and reduced transpiration rates, decrease in water potential, decrease in photosynthesis, accumulation of compatible solutes, synthesis of new proteins and formation of ROS-scavenging compounds like ascorbate, glutathione, and alpha-tocopherol (Yordanov and Tsonev 2003). It is also necessary to understand how the system might be affected by the interplay of external factors (e.g., water availability) and internal factors (developmental triggers). Therefore, analyses of the system structures (e.g., gene interactions) and dynamics (e.g., metabolic fluxes) become important (Kitano 2002; Moore et al. 2009). Correlation of next-generation sequencing, genome-scale molecular analysis, modeling of physiological and molecular data to the physiology of the plant leads to new data about adaptability and improved traits which can ultimately be incorporated in crop plants to improve productivity under stress (Weckwerth 2011).
“Drought tolerance” can be considered as the tolerance of moderate dehydration down to a moisture content below which there is no bulk cytoplasmic water potential (Hoekstra et al. 2001). “Desiccation tolerance” generally refers to the tolerance of further dehydration, when the hydration shell of molecules is gradually lost (Hoekstra et al. 2001). The desiccation tolerance program can be switched on by dehydration and also by the plant hormone abscisic acid (ABA), and results in a decrease in cellular volume, which leads to accumulation of cytoplasmic components. This ultimately leads to protein denaturation and membrane fusion (Hoekstra et al. 2001). In response to cellular dehydration, many plants and microorganisms accumulate compatible solutes irrespective of whether the dehydration was brought on by drought, freezing or osmotic shock (Hoekstra et al. 2001). Partitioning of cytoplasmic amphiphiles into membranes during dehydration was also reported as one of the major strategies towards dehydration tolerance because it assists the automatic insertion of antioxidants or phospholipase inhibitors with amphiphilic properties and thus slows the aging process (Langridge et al. 2006). After severe water loss, hydrogen bonding and glass formation are the mechanisms by which membranes and proteins are structurally and functionally preserved (Yordanov and Tsonev 2003). Thus, compatible solutes, especially sugars play an important role in removal of the closely associated water from protein without leading to any conformational changes and loss of enzymatic function. Subsequently, downstream genes like late embryogenesis abundant proteins (LEA) and group-1 dehydrins are activated leading to plant protection during dehydration (Yordanov and Tsonev 2003). A small perturbation in partitioning-induced membranes causes impairment of the electron transport chain that leads to increased formation of ROS (Hoekstra et al. 2001). This leads to an accumulation of enzymes or other compounds that act in ROS scavenging.
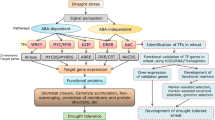
To understand drought responses in the field, it is important to have a comprehensive understanding of the signaling cascades at the molecular level and to identify the critical events that regulate different aspects of the signaling. To develop a drought-tolerant crop variety, it is necessary to elucidate the different molecular events along with their agronomical aspects, so that strategies to produce a better and more appropriate variety can be devised. New strategies to improve drought tolerance therefore often require multidisciplinary approaches that combine marker-assisted selection with a whole range of -omics technologies (Fig. 1). These -omics techniques not only define the system, but also provide new gene targets for both transgenic approaches and marker-assisted selection.

Simplified overview of successful integration of systems biology and molecular breeding approaches for crop improvement during drought. The forward genetics/breeding section of the diagram is modified from Fleury et al. (2010)
Transcription factors and drought
Environmental stresses affect physiological as well as biochemical aspects of the plant and hence influence the regulation of important proteins and genes. Plant adaptation to drought is dependent on molecular networks for drought perception, signal transduction, expression of a subset of genes and production of metabolites that protect and maintain the structure of cellular components (Umezawa et al. 2006a). Within the plant system, the processes of managing growth, developmental and stress responses are so interwoven that it is sometimes hard to separate these aspects from each other. Two major types of proteins are involved in the responses to environmental stress—‘effector proteins’ that, for example, hold water and control homeostasis and ‘regulatory proteins’, which are involved in signal perception and signal transduction. Regulatory proteins include transcription factors, protein kinases, and other signaling molecules. It is known that altering the activity of regulatory proteins might modulate the system because this class of proteins may regulate many downstream genes. This property makes them prime candidate target genes for improving responses to complex abiotic stresses like drought.
In general, the drought response pathways can be classified into two categories: one is dependent on the stress hormone ABA and the other is ABA independent (Umezawa et al. 2006b). ABA-responsive elements (ABREs) are one of the main cis-acting elements in ABA-dependent signaling of water stress responses (Shinozaki and Yamaguchi-Shinozaki 2007). It has been shown that this element directs ABA-dependent signaling of the Arabidopsis RD29B gene via bZIP transcription factors (Uno et al. 2000). The ABRE sequence CACGTG is also a potential binding site for both bHLH and NACs. Both bHLH and NAC TFS have been shown to play role in water stress responses. The cognate bZIP TFs have been named ABA-responsive element binding proteins (AREB)/ABA-responsive element binding factors (ABFs). The mode of activation appears to be via phosphorylation of the AREB/ABF proteins (Kagaya et al. 2002; Uno et al. 2000). Overexpression studies of ABF3 or ABREB2/ABF genes showed that the resulting ABA hypersensitivity led to an increase in drought tolerance in Arabidopsis (Kang et al. 2002).
Interestingly, Fujita et al. (2005) have shown that overexpression of the active form of AREB1 with N-terminal activation and bZIP DNA-binding domains (AREB1ΔQT) leads to plants displaying ABA hypersensitivity and enhanced drought tolerance. Yang et al. (2011) have shown that rice ABI5-Like1 (ABL1) regulates ABA and auxin responses through ABRE-containing WRKY genes. OsWRKY69 has an ABRE element in its promoter that leads to specific binding of the bZIP transcription factor (ABL1), which suppresses auxin signaling while enhancing ABA signaling and hence provides insights into ABA and auxin crosstalk (Yang et al. 2011). In another study, similar altered responses in auxin signaling and enhanced responses to ABA were observed when OsWRKY72 was ectopically overexpressed in Arabidopsis (Yu et al. 2010). Scanning 1 kb of upstream promoter region from OsWRKY72 through the PLACE database (Higo et al. 1998) shows an enrichment of ABRE-like elements. This suggests that this interaction of bZIP transcription factors with promoters from some WRKY gene promoters is crucial not only for ABA signaling but also for auxin signaling.
Transcription factors have been reported to play important roles at various levels in the signaling web to enable plants to cope with water-stress/drought. We now focus on drought/water-stress systems biology and WRKY TFs because the importance of WRKY TFs in abiotic stress signaling is just becoming clear.
WRKY transcription factors
The first reports about WRKY TFs came almost two decades ago and since then WRKY TFs have been reported as playing pivotal roles in defense signaling and regulating different growth and developmental processes in plants (Rushton et al. 2010). The initial reports of WRKY TFs defined the roles played in regulation of gene expression in sucrose inducibility (SPF1) (Ishiguro and Nakamura 1994) or during germination (ABF1 and ABF2) (Rushton et al. 1995). The history and nomenclature of this family of transcription factors have been thoroughly reviewed by Rushton et al. (2010). Studies now show that WRKYs are both positive and negative regulators of gene expression (Eulgem and Somssich 2007). Subsequently, progress has been made towards an understanding of the functionality of WRKY TFs not only in biotic stress conditions (where they are well studied) but also during seed germination, flower development, senescence and abiotic stresses as well.
WRKY transcription factors—domain structure and binding
The key-defining feature of WRKY TFs is their DNA-binding domain. This is called the WRKY domain because of the presence of the highly conserved WRKY amino acid sequence (WRKYGQK) also referred to as the “signature sequence” at the N-terminus (Maleck et al. 2000; Rushton et al. 1996). The WRKY domain is approximately 60 amino acid residues in length with the WRKY signature at the N-terminus and a zinc finger structure at the C-terminus. Based on the number of WRKY domains in the protein and structure of the zinc finger motif the whole family was divided into three different groups (Eulgem et al. 2000). Detailed phylogenetic analyses subsequently revealed a distribution of the WRKY family in higher plants into groups I, IIa+b, IIc, IId+e and III (Rushton et al. 2008a; Zhang and Wang 2005).
With regard to the structure of the WRKY domain, Rushton et al. (1995) initially showed that these proteins might contain a novel zinc finger structure. In the solution structure description by Yamasaki et al. (2005) it became clear that the WRKY domain constitutes a four-stranded β-sheet, with a zinc-binding pocket formed by the conserved Cys/His residues. The WRKY signature sequence enters the major groove of DNA and binds to its cognate DNA-binding site (TTGACC/T) known as the W box (Yamasaki et al. 2005). A few years later, a crystal structure description of the AtWRKY1 C terminal WRKY domain showed a similar structure (Duan et al. 2007). Recently, another major advance was made with the first reporting of the solution structure of a WRKY domain in complex with the W box binding site (Yamasaki et al. 2013). This revealed that a four-stranded β-sheet enters the major groove of the DNA in an atypical mode termed the β-wedge, so called because the sheet is nearly perpendicular to the DNA helical axis.
Recently, VQ proteins were reported to act as co-factors that positively or negatively regulate gene expression (Cheng et al. 2012). Also, chromatin remodeling has also started to emerge as an important mode whereby the regulatory mechanism modulates expression of target gene expression during stress responses (Trivedi et al. 2012; Wang et al. 2012a). Thus, mechanisms other than mere recognition of the core W box promoter elements are necessary to achieve the regulatory specificity of WRKY TFs (Chi et al. 2013). Therefore, the involvement and utilization of systems biology techniques (in this case interactomics) can enable us to decipher the behavior of WRKYs with their interacting partners and help characterize the resulting dynamic output (Chi et al. 2013).
Abiotic stress and WRKY transcription factors: altered expression and mutants
WRKY TFs play pivotal roles in regulating many stress reactions in plants but until recently unraveling their roles in abiotic stress responses has lagged behind that of biotic stresses (Rushton et al. 2012). Over the last 5 years, this situation has changed rapidly and there are now dozens of reports of WRKY TFs functioning in abiotic stress responses, including water stress (see Table 1 for examples). In rice, heat shock inducible HSP101 promoter driven overexpression of OsWRKY11 led to enhanced heat and drought tolerance (Wu et al. 2009). Similarly, overexpression of OsWRKY45 resulted in enhanced salt and drought tolerance in addition to increased disease resistance (Qiu and Yu 2009), again showing cross talk between these stress responses. These encouraging crop plant-based results are supplemented by others in the model plant Arabidopsis and further evidence came from altered plant responses to different abiotic stresses following the overexpression of three stress-inducible soybean WRKY genes in Arabidopsis. Zhou et al. (2008) revealed enhanced cold tolerance in comparison to wild type when GmWRKY21 was overexpressed. In comparison, GmWRKY54-overexpressing plants were shown to be more salt and drought tolerant and GmWRKY13 overexpression resulted in increased sensitivity to salt and mannitol stresses (Zhou et al. 2008).
Information regarding the different downstream target genes of WRKY TFs that regulate abiotic stress-induced responses is limited. Advances have, however, been made in a dehydration tolerance signaling pathway in the resurrection plant Boea hygrometrica (Wang et al. 2009). An important downstream target gene, Galactinol synthase 1 (BhGolS1), which plays a role in drought and cold tolerance (Teruaki et al. 2002) was found to be dehydration and ABA inducible. The BhGolS1 promoter contains four W boxes and chromatin immunoprecipitation showed that it is bound in vivo by the early dehydration and ABA-inducible BhWRKY1 TF (Wang et al. 2009). These observations provide direct insights into the role of a dehydration-inducible WRKY TF that interacts with a downstream target gene that plays an important role in drought responses. Recently, convincing evidence has been presented to show that AtWRKY8 functions antagonistically with its interacting partner VQ9 to modulate salinity tolerance (Hu et al. 2013). AtWRKY8 is highly up-regulated by salt and a wrky8 mutation rendered plants hypersensitive to salt. Conversely, a vq9 mutation enhanced tolerance to salt. Furthermore, chromatin immunoprecipitation assays showed that AtWRKY8 bound directly to the promoter of RD29A, suggesting that it is one of the AtWRKY8 target genes.
One recent and important discovery in WRKY TF research came from the T-DNA insertion mutant abo3. This mutation is in AtWRKY63 and leads to a hypersensitive response upon ABA treatment in both seedling establishment and seedling growth. Importantly, since stomatal closure was less sensitive to ABA, this mutant was also less drought tolerant than wild-type plants (Ren et al. 2010). A detailed analysis of the abo3 mutant illustrates some of the roles of this WRKY transcription factor by placing AtWRKY63 downstream of ABI1, ABI2 and ABI5 but upstream of ABF2, RD29A and COR4. Interestingly, AtWRKY40 appears to act upstream of the bZIP transcription factor ABI5 (Shang et al. 2010) whereas AtWRKY63 acts downstream of it and hence taken together, this shows that there is a cascade of transcription factors initiated by ABA with AtWRKY40 repressing ABI5 gene expression in the absence of ABA. Upon ABA perception, de-repression of ABI5 leads to activation of AtWRKY63 at the transcriptional level which further activates the target genes including RD29A and COR47 (Rushton et al. 2012). Importantly, other reports have shown that modulation of WRKY TF gene expression may lead to improved drought responses through changes, not in stomata but in root architecture (Song et al. 2010).
Up-regulation of multiple TFs by water stress may lead to enhanced expression of target genes. Babitha et al. (2013) showed that co-expression of AtWRKY28 and AtbHLH17 resulted in increased expression of different target genes in response to various abiotic stresses like drought, salt and oxidative stress in Arabidopsis (Babitha et al. 2013). Various downstream target genes like RD29A, rd22 and FSD2 were found to have W boxes in their promoter regions whereas KIN1 and ADH1 have G-box elements while P5C5 and LEA14 have both elements. All of them showed an enhanced expression in transgenic lines compared to wild-type plants when subjected to different osmoticum and stress treatments. However, there is not a simple relationship between the presence or absence of binding sites and expression levels, genes having binding site for both classes of transcription factors do not show higher expression when compared with the ones which have just one of the two binding sites alone. However, it is clear that co-expression of multiple transcription factors leads to up-regulation of more targets (Babitha et al. 2013). Hence, cooperative behavior of the different transcription factors appears to result in tighter regulation of the molecular responses to different stress conditions. In another study, Wang et al. (2012a, b) showed in Tamarix androssowii (Tamaricaceae), a member of a class of highly drought- and salt-tolerant trees or shrubs, that a conserved mechanism of transcriptional regulation was followed by WRKY and RAV (an AP2/ERF and B3 domain containing transcription factor) TFs which results in activation of TaeIF5A and confers tolerance to abiotic stresses (Wang et al. 2012b). Interestingly, the presence of a W box in the promoter region of TaeIF5A appeared to be the necessary element required for binding of the TaRAV and TaWRKY, as a mutation in the core W box motif results in loss of function. This specific interaction upstream of TaeIF5A regulates the expression in an ABA-dependent signal transduction pathway and leads to stress tolerance by various physiological pathways such as enhancing ROS scavenging, and preventing chlorophyll loss and membrane damage (Wang et al. 2012b).
As described earlier, reports have emerged from different crop plants where knowledge gathered from the model plant Arabidopsis has been successfully translated to crop species. In many cases, comparative genomics leads to new findings in agriculturally and economically important crops (Marchive et al. 2007; Mare et al. 2004; Petitot et al. 2013; Shekhawat et al. 2011). Recently, Luo et al. (2013) have described that in wild soybean, Glycine soja, GsWRKY20 promotes the expression of negative regulators of ABA signaling while repressing positive regulators. When overexpressed, GsWRKY20-overexpressing plants show higher tolerance to drought with a significant decline in water loss and stomatal density (Luo et al. 2013). This study shows that GsWRKY20 plays an important role in ABA-mediated stomatal closure, which ultimately leads to increased resistance to drought. In another study, tobacco plants overexpressing wheat TaWRKY10 show tolerance to multiple stresses including drought (Wang et al. 2013). WRKY TFs appear to play important roles in regulating water-stress/drought by modulating the osmotic balance, ROS scavenging and affecting expression of different stress-related genes (Agarwal et al. 2011).
These new insights show that some WRKY TFs represent major hubs in abiotic stress signaling as they take input signals from multiple stimuli (Rushton et al. 2012). This has major implications for the use of WRKY genes in crop improvement because it may make the manipulation of a single plant process difficult to improve using a single gene because several different stress responses are simultaneously affected. On the other hand, manipulation of a single WRKY gene may improve response to multiple stresses. A comprehensive systems biology approach would be a useful strategy to address such problems.
Transcriptomics
Transcriptomics, using oligo arrays and more recently RNA-seq, is the most widely used -omics technology. Until recently, WRKY TFs were well known as key regulators of plant responses to disease but they had barely been studied in regard to abiotic stresses like drought stress (Bartels and Sunkar 2005). Transcriptomics gave the first clues that WRKY TFs are regulators of abiotic stress responses. Transcriptome analyses in Arabidopsis using 7,000 genes revealed forty-three drought, cold or high-salinity stresses induced transcription factor genes. Among these were six DREBs, two ERFs, ten zinc finger-containing factors, three MYBs, two bHLHs, four bZIPs, five NACs and four WRKYs (Bartels and Sunkar 2005; Seki et al. 2002; Umezawa et al. 2006a). In addition, the expression patterns of the complete rice WRKY family under drought conditions in both the root and leaf was investigated using massively parallel signature sequencing (MPSS). At least seventeen rice WRKY genes (over 15 % of the gene family) were highly induced by drought stress. This provided missing evidence that WRKYs may play a role in drought responses (http://mpss.udel.edu/rice/). Since then, there has been a great deal of published transcriptomics data and a role of WRKY genes in water stress responses is no longer in doubt.
Transcriptome comparisons of water stress-inducible WRKY genes from three different families of plants
We have been able to investigate the water stress inducibility of WRKY genes from representative species from three plant families. Arabidopsis genes that are induced at least sevenfold by water stress at the mRNA level were identified using Genevestigator (Zimmermann et al. 2004). Soybean genes that are induced sevenfold by water stress were identified using our own transcriptome data set (Tripathi et al. unpublished). Tobacco genes that are induced sevenfold by water stress were also identified using our own transcriptome data set (Rabara et al. unpublished). A combined phylogenetic analysis proved revealing as there are not only similarities across all three species but also marked species-specific differences (Fig. 2). Similarities include the induction by water stress of genes from the I, IIa, IIc and III subfamilies. This includes notable hotspots of co-expressed genes in the group IIc (Fig. 3) and group IIa subfamilies. There are striking differences also between the plant species. Only IId genes from tobacco are induced by water stress (Fig. 2). Arabidopsis group IIb genes, unlike tobacco and soybean, do not appear to contain water stress-inducible members.

Combined phylogenetic tree of Arabidopsis, soybean and tobacco WRKY genes indicating water stress inducibility at the mRNA level. The complete WRKY gene family from Arabidopsis was taken from the TAIR web site. The tobacco genes were taken from the TOBFAC database (Rushton et al. 2008b). The soybean genes were taken from phytozome (Goodstein et al. 2012) and named arbitrarily except for previously published genes. The evolutionary history was inferred using the neighbor-joining method. The bootstrap consensus tree inferred from 1,000 replicates is taken to represent the evolutionary history of the taxa analyzed. The tree is drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree using MEGA5 (Tamura et al. 2011). The evolutionary distances were computed using the Poisson correction method and are in the units of the number of amino acid substitutions per site. Blue dots denote Arabidopsis genes that are induced at least sevenfold by water stress according to the transcriptome data sets in Genevestigator (Zimmermann et al. 2004). Red dots denote soybean and green dots denote tobacco genes that are induced sevenfold by water stress according to our transcriptome data sets

Hotspot of water stress-inducible WRKY genes in the group IIc subfamily. Blue dots denote Arabidopsis genes that are induced at least sevenfold by water stress according to the transcriptome data sets in Genevestigator (Zimmermann et al. 2004). Red dots denote soybean genes that are induced sevenfold by water stress according to our transcriptome data sets (Tripathi et al. unpublished, data not shown). Green dots denote tobacco genes that are induced sevenfold by water stress according to our transcriptome data sets (Rabara et al. unpublished, data not shown)
These initial analyses suggest that although some WRKY genes may form a core component of water stress deficit signaling, that others represent family or even species-specific responses. Further detailed analyses using more standardized experimental procedures will be necessary to shed further light on this, but it is tempting to speculate that the family/species-specific inducible WRKY genes may regulate family/species-specific processes such as parts of secondary metabolism.
Proteomics
Proteomics analyses of transcription factors, particularly shotgun proteomics, are hampered by the low concentration of transcription factors in the cell. Fortunately, most transcription factors are present in the nucleus and so one approach to circumvent this problem is subcellular fractionation. Therefore, nuclear proteomics may become the method of choice for WRKY proteins and other transcription factors. There are a very limited number of reports on the nuclear proteomics of water stress responses. Choudhary et al. (2009) reported on the dehydration-responsive nuclear proteome of rice. The proteome was established using nuclear enrichment followed by two-dimensional gel electrophoresis and LC–ESI–MS/MS. One hundred and fifty polypeptides showed changes in intensity of more than 2.5-fold. Ten percent of the identified proteins were involved in transcriptional regulation and chromatin remodeling. Among these proteins were a bZIP transcription factor, that showed up-regulation at 72–96 h of dehydration, and a WRKY transcription factor that displayed up-regulation across all the time points. This is a clear indication of the dehydration-induced increase in concentration of a WRKY TF in the nucleus and suggests that this WRKY is regulated by water stress (Choudhary et al. 2009). Excitingly, a similar study of the dehydration-responsive nuclear proteome of chickpea (Pandey et al. 2008) from the same group also identified a similar WRKY protein that is up-regulated by dehydration in chickpea. The chickpea WRKY protein appears to be an ortholog of the Arabidopsis group I protein, AtWRKY4. In each of the two studies, only a handful of transcription factors were identified as up-regulated and in each case a WRKY protein was one of these, suggesting that WRKY transcription factors are among the most important regulators of water stress responses.
Although nuclear proteomics is still in its infancy, such results together with transcriptomics, promoter analyses, mutational approaches, and metabolomics suggest that systems biology approaches will reveal more important roles for WRKY transcription factors in drought responses in crops.
Interactomics
Interactomics, in this case the study of the interacting partners of WRKY proteins, has made some significant advances over recent years. From their function as transcription factors, WRKY proteins must bind DNA and interact with other proteins to either activate or repress transcription, but how do they do this? WRKY proteins interact with a wide range of proteins (Chi et al. 2013) and the field of interactomics is allowing the elucidation of signaling webs that they form parts of. First, there is now considerable evidence that WRKY transcription factors form homo- and hetero-complexes. This includes interaction of group IIa WRKY proteins with each other via leucine zipper motifs (Cormack et al. 2002; Xu et al. 2006), interaction of group III WRKY TFs (Besseau et al. 2012) and heterodimerization of members of group IIb. In addition, there are reports of heterodimerization of WRKY TFs from different subfamilies such as OsWRKY71 and OsWRKY51 (groups IIa and IId, respectively) (Xie et al. 2006) and the group IIa TFs AtWRKY40 and AtWRKY60 interact with AtWRKY36 (IId) and AtWRKY38 (III) (Arabidopsis Interactome Mapping Consortium (2011)). This suggests a complex set of functional interactions that include cooperative and antagonistic binding in the “WRKY Wide Web of Signaling.”
It has also been shown that WRKY TFs interact with calmodulin, which binds to the C-motif (DxxVxKFKxVISLLxxxR) that is present in some group IId WRKY proteins (Park et al. 2005). This implicates group IId WRKY TFs in transducing Ca2+ signals. Also, interaction studies show that group I WRKY proteins are among the targets of MAP kinase signaling cascades, suggesting that phosphorylation plays a major role in WRKY activity. In Nicotiana benthamiana, NbWRKY8 phosphorylation by MAP kinases enhances its DNA-binding and transcription-activating activities (Ishihama et al. 2011). There are many other examples of WRKY-MAP kinase interactions (Chi et al. 2013). Other interacting partners of WRKY proteins include 14-3-3 proteins (Chang et al. 2009), chromatin remodeling proteins such as Arabidopsis histone deacetylase 19 (Kim et al. 2010), VQ proteins (Hu et al. 2013), CC-NBR-LRR-type R proteins (Shen et al. 2007), E3 ubiquitin ligases (Miao and Zentgraf 2010) and the chloroplast ⁄ plastid-localized ABA receptor, ABAR (Shang et al. 2010). These interacting partners of WRKY TFs are providing increasing information as to the roles and mechanisms involved in WRKY TF action at the systems level.
Metabolomics
Metabolomics analyses are also rare but correlations between knockdown/overexpression of specific WRKY genes and changes in metabolite profiles hold great promise for the establishment of areas of metabolism that are regulated by WRKY transcription factors. Reports from resurrection plants using parallel transcriptomic and metabolomic profiling have reported on WRKY TFs as regulating lysine biosynthesis (Urbanczyk-Wochniak et al. 2003), reprogramming normal growth to promote energy saving and promoting stress protective programs during water stress (Gechev et al. 2013). In their study, Urbanczyk-Wochniak et al. (2003) found that the nutritionally important metabolites such as ascorbate, tocopherol and lysine were closely related to the expression of various transcription factors: ascorbate was negatively correlated with a homologue of the clock gene CONSTANS and lysine was positively regulated by the transcription factor WRKY6. These observations provide several links between metabolites and the transcriptome. As mentioned earlier, research on a dehydration tolerance signaling pathway in the resurrection plant Boea hygrometrica links the WRKY transcription factor BhWRKY1 with the galactinol synthase gene (BhGolS1), and therefore with regulating the accumulation of osmolytes in situations of low water potential (Wang et al. 2009). The BhGolS1 promoter contains four W boxes and chromatin immunoprecipitation showed that it is bound in vivo by the early dehydration and ABA-inducible BhWRKY1 transcription factor (Wang et al. 2009). Gechev et al. (2013) produced a model where they explained that initial perception of the signaling events during drought is received by transcription factors including MADS box, NAC and WRKY proteins. This leads to production of ELIPs, LEAs, HSPs, PR proteins, regulation of ROS-scavenging proteins/metabolites, down-regulation of photosynthetic genes, accumulation of sucrose, and synthesis of GABA. These and other responses may contribute to desiccation tolerance and tolerance to salinity.
Conclusion and future perspectives
To have better insights into the mode of regulation of co-expressed genes, a comprehensive approach of parallel -omics profiling at the gene and protein level is a good approach. Such omics analyses can tell us who are major players and probably how they are receiving a stimulus. Various post-translational mechanisms are able to change the fate of proteins and direct them to act with potentially different function. To better understand the web of signaling involved in drought responses and the role of WRKY TFs in this web, functional and structural descriptions from transcriptomics and proteomics approaches are a good start. However, without the full description of the dynamics of fluxes, it is insufficient for an understanding of signaling cascades. Hence, it is important to integrate multiple -omics level analyses to understand signaling during water stress.
Water stress responses are notoriously multigenic and quantitative with strong environmental effects on phenotypes (Fleury et al. 2010). Looking to the future, the best approach to study water stress responses is therefore a multidisciplinary and integrated systems biology approach that has the potential to lead to crop improvement. Many different -omics technologies are required to give a clear view of the complete system at each level. For water stress, one of the possible approaches to define the system and find candidate genes for crop improvement is to first select candidate genes that are differentially regulated during the dehydration responses using transcriptome data. Subjecting these genes to promoter motif enrichment analysis can provide an overview of modules in signaling. In this way, target genes for TFs such as WRKY proteins can be tentatively assigned and signaling webs constructed. Further mutant, overexpression, and knockdown analyses can add functional data to reveal upstream and downstream signaling components of water stress. More elegantly, chromatin immunoprecipitation followed by next-generation sequencing (ChIP-seq) has the potential to provide a snap shot of the target promoters of a given TF in a genome wide manner. Other levels of the system can also be investigated. Metabolomics has the potential to establish connections between transcription factors, promoters, biosynthetic pathways, fluxes and downstream responses. Other approaches, such as phosphoproteomics and interactomics, have the capacity to establish signaling events that occur earlier in the signaling webs, such as kinase cascades or phosphorylation of TFs. Further exciting new advances can see the mutant, overexpression, knockout and molecular breeding approaches connected to large physiology datasets via high throughput phenotyping facilities. Taken together, these -omics technologies should provide new gene targets and power breeding programs. For transcription factors in plants, such as WRKY transcription factors, we have only just scratched the surface of what is becoming possible.
References
Agarwal P, Reddy MP, Chikara J (2011) WRKY: its structure, evolutionary relationship, DNA-binding selectivity, role in stress tolerance and development of plants. Mol Biol Rep 38:3883–3896
Arabidopsis Interactome Mapping Consortium (2011) Evidence for network evolution in an Arabidopsis interactome map. Science 333:601–607
Babitha KC, Ramu SV, Pruthvi V, Mahesh P, Nataraja KN, Udayakumar M (2013) Co-expression of AtbHLH17 and AtWRKY28 confers resistance to abiotic stress in Arabidopsis. Transgenic Res 22:327–341
Bartels D, Sunkar R (2005) Drought and salt tolerance in plants. Crit Rev Plant Sci 24:23–58
Berri S, Abbruscato P, Faivre-Rampant O, Brasileiro AC, Fumasoni I, Satoh K, Kikuchi S, Mizzi L, Morandini P, Pe ME, Piffanelli P (2009) Characterization of WRKY co-regulatory networks in rice and Arabidopsis. BMC Plant Biol 9:120
Besseau S, Li J, Palva ET (2012) WRKY54 and WRKY70 co-operate as negative regulators of leaf senescence in Arabidopsis thaliana. J Exp Bot 63:2667–2679
Chang IF, Curran A, Woolsey R, Quilici D, Cushman JC, Mittler R, Harmon A, Harper JF (2009) Proteomic profiling of tandem affinity purified 14-3-3 protein complexes in Arabidopsis thaliana. Proteomics 9:2967–2985
Cheng Y, Zhou Y, Yang Y, Chi YJ, Zhou J, Chen JY, Wang F, Fan B, Shi K, Zhou YH, Yu JQ, Chen Z (2012) Structural and functional analysis of VQ motif-containing proteins in Arabidopsis as interacting proteins of WRKY transcription factors. Plant Physiol 159:810–825
Chi Y, Yang Y, Zhou Y, Zhou J, Fan B, Yu JQ, Chen Z (2013) Protein–protein interactions in the regulation of WRKY transcription factors. Mol Plant 6:287–300
Choudhary MK, Basu D, Datta A, Chakraborty N, Chakraborty S (2009) Dehydration-responsive nuclear proteome of rice (Oryza sativa L.) illustrates protein network, novel regulators of cellular adaptation, and evolutionary perspective. Mol Cell Proteomics: MCP 8:1579–1598
Chow BY, Kay SA (2013) Global approaches for telling time: omics and the Arabidopsis circadian clock. Semin Cell Dev Biol 24:383–392
Cormack RS, Eulgem T, Rushton PJ, Kochner P, Hahlbrock K, Somssich IE (2002) Leucine zipper-containing WRKY proteins widen the spectrum of immediate early elicitor-induced WRKY transcription factors in parsley. Biochim Biophys Acta 1576:92–100
Duan MR, Nan J, Liang YH, Mao P, Lu L, Li L, Wei C, Lai L, Li Y, Su XD (2007) DNA binding mechanism revealed by high resolution crystal structure of Arabidopsis thaliana WRKY1 protein. Nucleic Acids Res 35:1145–1154
Eulgem T, Somssich IE (2007) Networks of WRKY transcription factors in defense signaling. Curr Opin Plant Biol 10:366–371
Eulgem T, Rushton PJ, Robatzek S, Somssich IE (2000) The WRKY superfamily of plant transcription factors. Trends Plant Sci 5:199–206
Fleury D, Jefferies S, Kuchel H, Langridge P (2010) Genetic and genomic tools to improve drought tolerance in wheat. J Exp Bot 61:3211–3222
Fujita Y, Fujita M, Satoh R, Maruyama K, Parvez MM, Seki M, Hiratsu K, Ohme-Takagi M, Shinozaki K, Yamaguchi-Shinozaki K (2005) AREB1 is a transcription activator of novel ABRE-dependent ABA signaling that enhances drought stress tolerance in Arabidopsis. Plant Cell 17(12):3470–3488
Gechev TS, Benina M, Obata T, Tohge T, Sujeeth N, Minkov I, Hille J, Temanni MR, Marriott AS, Bergstrom E, Thomas-Oates J, Antonio C, Mueller-Roeber B, Schippers JH, Fernie AR, Toneva V (2013) Molecular mechanisms of desiccation tolerance in the resurrection glacial relic Haberlea rhodopensis. Cell Mol Life Sci: CMLS 70:689–709
Goodstein DM, Shu S, Howson R, Neupane R, Hayes RD, Fazo J, Mitros T, Dirks W, Hellsten U, Putnam N, Rokhsar DS (2012) Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res 40(Database issue):D1178–D1186. doi:10.1093/nar/gkr944
Higo K, Ugawa Y, Iwamoto M, Higo H (1998) PLACE: a database of plant cis-acting regulatory DNA elements. Nucleic Acids Res 26:358–359
Hoekstra FA, Golovina EA, Buitink J (2001) Mechanisms of plant desiccation tolerance. Trends Plant Sci 6:431–438
Hu Y, Chen L, Wang H, Zhang L, Wang F, Yu D (2013) Arabidopsis transcription factor WRKY8 functions antagonistically with its interacting partner VQ9 to modulate salinity stress tolerance. Plant J 74:730–745
Huang T, Duman JG (2002) Cloning and characterization of a thermal hysteresis (antifreeze) protein with DNA-binding activity from winter bittersweet nightshade, Solanum dulcamara. Plant Mol Biol 48:339–350
Ishiguro S, Nakamura K (1994) Characterization of a cDNA encoding a novel DNA-binding protein, SPF1, that recognizes SP8 sequences in the 5′ upstream regions of genes coding for sporamin and beta-amylase from sweet potato. Mol Gen Genet 244:563–571
Ishihama N, Yamada R, Yoshioka M, Katou S, Yoshioka H (2011) Phosphorylation of the Nicotiana benthamiana WRKY8 transcription factor by MAPK functions in the defense response. Plant Cell 23:1153–1170
Jiang Y, Deyholos MK (2009) Functional characterization of Arabidopsis NaCl-inducible WRKY25 and WRKY33 transcription factors in abiotic stresses. Plant Mol Biol 69:91–105
Jiang Y, Liang G, Yu D (2012) Activated expression of WRKY57 confers drought tolerance in Arabidopsis. Mol Plant 5:1375–1388
Kagaya Y, Hobo T, Murata M, Ban A, Hattori T (2002) Abscisic acid-induced transcription is mediated by phosphorylation of an abscisic acid response element binding factor, TRAB1. Plant Cell 14:3177–3189
Kang JY, Choi HI, Im MY, Kim SY (2002) Arabidopsis basic leucine zipper proteins that mediate stress-responsive abscisic acid signaling. Plant Cell 14:343–357
Kim JM, To TK, Nishioka T, Seki M (2010) Chromatin regulation functions in plant abiotic stress responses. Plant Cell Environ 33:604–611
Kitano H (2002) Systems biology: a brief overview. Science 295:1662–1664
Langridge P, Paltridge N, Fincher G (2006) Functional genomics of abiotic stress tolerance in cereals. Brief Funct Genomic Proteomic 4:343–354
Li S, Fu Q, Huang W, Yu D (2009) Functional analysis of an Arabidopsis transcription factor WRKY25 in heat stress. Plant Cell Rep 28:683–693
Li S, Fu Q, Chen L, Huang W, Yu D (2011) Arabidopsis thaliana WRKY25, WRKY26, and WRKY33 coordinate induction of plant thermotolerance. Planta 233:1237–1252
Luo X, Bai X, Sun X, Zhu D, Liu B, Ji W, Cai H, Cao L, Wu J, Hu M, Liu X, Tang L, Zhu Y (2013) Expression of wild soybean WRKY20 in Arabidopsis enhances drought tolerance and regulates ABA signalling. J Exp Bot 64:2155–2169
Maleck K, Levine A, Eulgem T, Morgan A, Schmid J, Lawton KA, Dangl JL, Dietrich RA (2000) The transcriptome of Arabidopsis thaliana during systemic acquired resistance. Nat Genet 26:403–410
Marchive C, Mzid R, Deluc L, Barrieu F, Pirrello J, Gauthier A, Corio-Costet MF, Regad F, Cailleteau B, Hamdi S, Lauvergeat V (2007) Isolation and characterization of a Vitis vinifera transcription factor, VvWRKY1, and its effect on responses to fungal pathogens in transgenic tobacco plants. J Exp Bot 58:1999–2010
Mare C, Mazzucotelli E, Crosatti C, Francia E, Stanca AM, Cattivelli L (2004) Hv-WRKY38: a new transcription factor involved in cold- and drought-response in barley. Plant Mol Biol 55:399–416
Miao Y, Zentgraf U (2010) A HECT E3 ubiquitin ligase negatively regulates Arabidopsis leaf senescence through degradation of the transcription factor WRKY53. Plant J 63:179–188
Moore JP, Le NT, Brandt WF, Driouich A, Farrant JM (2009) Towards a systems-based understanding of plant desiccation tolerance. Trends Plant Sci 14:110–117
Niu CF, Wei W, Zhou QY, Tian AG, Hao YJ, Zhang WK, Ma B, Lin Q, Zhang ZB, Zhang JS, Chen SY (2012) Wheat WRKY genes TaWRKY2 and TaWRKY19 regulate abiotic stress tolerance in transgenic Arabidopsis plants. Plant Cell Environ 35:1156–1170
Pandey A, Chakraborty S, Datta A, Chakraborty N (2008) Proteomics approach to identify dehydration responsive nuclear proteins from chickpea (Cicer arietinum L.). Mol Cell Proteomics: MCP 7:88–107
Park CY, Lee JH, Yoo JH, Moon BC, Choi MS, Kang YH, Lee SM, Kim HS, Kang KY, Chung WS, Lim CO, Cho MJ (2005) WRKY group IId transcription factors interact with calmodulin. FEBS Lett 579:1545–1550
Petitot AS, Barsalobres-Cavallari C, Ramiro D, Albuquerque Freire E, Etienne H, Fernandez D (2013) Promoter analysis of the WRKY transcription factors CaWRKY1a and CaWRKY1b homoeologous genes in coffee (Coffea arabica). Plant Cell Rep 32:1263–1276
Qiu YP, Yu DQ (2009) Over-expression of the stress-induced OsWRKY45 enhances disease resistance and drought tolerance in Arabidopsis. Environ Exp Bot 65:35–47
Ramamoorthy R, Jiang SY, Kumar N, Venkatesh PN, Ramachandran S (2008) A comprehensive transcriptional profiling of the WRKY gene family in rice under various abiotic and phytohormone treatments. Plant Cell Physiol 49:865–879
Ren X, Chen Z, Liu Y, Zhang H, Zhang M, Liu Q, Hong X, Zhu JK, Gong Z (2010) ABO3, a WRKY transcription factor, mediates plant responses to abscisic acid and drought tolerance in Arabidopsis. Plant J 63:417–429
Roschefort L, Woodward FI (1992) Effect of climate change and a doubling of CO2 on climate diversity. J Exp Bot 43:1169–1180
Rushton PJ, Macdonald H, Huttly AK, Lazarus CM, Hooley R (1995) Members of a new family of DNA-binding proteins bind to a conserved cis-element in the promoters of alpha-Amy2 genes. Plant Mol Biol 29:691–702
Rushton PJ, Torres JT, Parniske M, Wernert P, Hahlbrock K, Somssich IE (1996) Interaction of elicitor-induced DNA-binding proteins with elicitor response elements in the promoters of parsley PR1 genes. EMBO J 15:5690–5700
Rushton PJ, Bokowiec MT, Han SC, Zhang HB, Brannock JF, Chen XF, Laudeman TW, Timko MP (2008a) Tobacco transcription factors: novel insights into transcriptional regulation in the Solanaceae. Plant Physiol 147:280–295
Rushton PJ, Bokowiec MT, Laudeman TW, Brannock JF, Chen X, Timko MP (2008b) TOBFAC: the database of tobacco transcription factors. BMC Bioinformatics 9:53
Rushton PJ, Somssich IE, Ringler P, Shen QJ (2010) WRKY transcription factors. Trends Plant Sci 15:247–258
Rushton DL, Tripathi P, Rabara RC, Lin J, Ringler P, Boken AK, Langum TJ, Smidt L, Boomsma DD, Emme NJ, Chen X, Finer JJ, Shen QJ, Rushton PJ (2012) WRKY transcription factors: key components in abscisic acid signalling. Plant Biotechnol J 10:2–11
Seki M, Ishida J, Narusaka M, Fujita M, Nanjo T, Umezawa T, Kamiya A, Nakajima M, Enju A, Sakurai T, Satou M, Akiyama K, Yamaguchi-Shinozaki K, Carninci P, Kawai J, Hayashizaki Y, Shinozaki K (2002) Monitoring the expression pattern of around 7,000 Arabidopsis genes under ABA treatments using a full-length cDNA microarray. Funct Integr Genomics 2:282–291
Shang Y, Yan L, Liu ZQ, Cao Z, Mei C, Xin Q, Wu FQ, Wang XF, Du SY, Jiang T, Zhang XF, Zhao R, Sun HL, Liu R, Yu YT, Zhang DP (2010) The Mg-chelatase H subunit of Arabidopsis antagonizes a group of WRKY transcription repressors to relieve ABA-responsive genes of inhibition. Plant Cell 22:1909–1935
Shao HB, Chu LY, Jaleel CA, Manivannan P, Panneerselvam R, Shao MA (2009) Understanding water deficit stress-induced changes in the basic metabolism of higher plants—biotechnologically and sustainably improving agriculture and the ecoenvironment in arid regions of the globe. Crit Rev Biotechnol 29:131–151
Shekhawat UK, Ganapathi TR, Srinivas L (2011) Cloning and characterization of a novel stress-responsive WRKY transcription factor gene (MusaWRKY71) from Musa spp. cv. Karibale Monthan (ABB group) using transformed banana cells. Mol Biol Rep 38:4023–4035
Shen QH, Saijo Y, Mauch S, Biskup C, Bieri S, Keller B, Seki H, Ulker B, Somssich IE, Schulze-Lefert P (2007) Nuclear activity of MLA immune receptors links isolate-specific and basal disease-resistance responses. Science 315:1098–1103
Shinozaki K, Yamaguchi-Shinozaki K (2007) Gene networks involved in drought stress response and tolerance. J Exp Bot 58:221–227
Song Y, Jing S, Yu D (2010) Overexpression of the stress-induced OsWRKY08 improves osmotic stress tolerance in Arabidopsis. Chin Sci Bull 54:4671–4678
Suzuki N, Rizhsky L, Liang H, Shuman J, Shulaev V, Mittler R (2005) Enhanced tolerance to environmental stress in transgenic plants expressing the transcriptional coactivator multiprotein bridging factor 1c. Plant Physiol 139:1313–1322
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739
Teruaki T, Chieko O, Satoshi I, Motoaki S, Mie K, Masatomo K, Kazuko Y-S, Kazuo S (2002) Important roles of drought- and cold-inducible genes for galactinol synthase in stress tolerance in Arabidopsis thaliana. Plant J 29:417–426
Trivedi I, Ranjan A, Sharma YK, Sawant S (2012) The histone H1 variant accumulates in response to water stress in the drought tolerant genotype of Gossypium herbaceum L. Protein J 31:477–486
Umezawa T, Fujita M, Fujita Y, Yamaguchi-Shinozaki K, Shinozaki K (2006a) Engineering drought tolerance in plants: discovering and tailoring genes to unlock the future. Curr Opin Biotechnol 17:113–122
Umezawa T, Okamoto M, Kushiro T, Nambara E, Oono Y, Seki M, Kobayashi M, Koshiba T, Kamiya Y, Shinozaki K (2006b) CYP707A3, a major ABA 8′-hydroxylase involved in dehydration and rehydration response in Arabidopsis thaliana. Plant J 46:171–182
Uno Y, Furihata T, Abe H, Yoshida R, Shinozaki K, Yamaguchi-Shinozaki K (2000) Arabidopsis basic leucine zipper transcription factors involved in an abscisic acid-dependent signal transduction pathway under drought and high-salinity conditions. Proc Natl Acad Sci USA 97:11632–11637
Urbanczyk-Wochniak E, Luedemann A, Kopka J, Selbig J, Roessner-Tunali U, Willmitzer L, Fernie AR (2003) Parallel analysis of transcript and metabolic profiles: a new approach in systems biology. EMBO Rep 4:989–993
Wang Z, Zhu Y, Wang LL, Liu X, Liu YX, Phillips J, Deng X (2009) A WRKY transcription factor participates in dehydration tolerance in Boea hygrometrica by binding to the W-box elements of the galactinol synthase (BhGolS1) promoter. Planta 230:1155–1166
Wang JN, Kuang JF, Shan W, Chen J, Xie H, Lu WJ, Chen JW, Chen JY (2012a) Expression profiles of a banana fruit linker histone H1 gene MaHIS1 and its interaction with a WRKY transcription factor. Plant Cell Rep 31:1485–1494
Wang L, Xu C, Wang C, Wang Y (2012b) Characterization of a eukaryotic translation initiation factor 5A homolog from Tamarix androssowii involved in plant abiotic stress tolerance. BMC Plant Biol 12:118
Wang C, Deng P, Chen L, Wang X, Ma H, Hu W, Yao N, Feng Y, Chai R, Yang G, He G (2013) A wheat WRKY transcription factor TaWRKY10 confers tolerance to multiple abiotic stresses in transgenic tobacco. PLoS One 8:e65120
Weckwerth W (2011) Green systems biology—from single genomes, proteomes and metabolomes to ecosystems research and biotechnology. J Proteomics 75:284–305
Wu X, Shiroto Y, Kishitani S, Ito Y, Toriyama K (2009) Enhanced heat and drought tolerance in transgenic rice seedlings overexpressing OsWRKY11 under the control of HSP101 promoter. Plant Cell Rep 28:21–30
Xie Z, Zhang ZL, Zou XL, Yang GX, Komatsu S, Shen QXJ (2006) Interactions of two abscisic-acid induced WRKY genes in repressing gibberellin signaling in aleurone cells. Plant J 46:231–242
Xiong X, James VA, Zhang H, Altpeter F (2010) Constitutive expression of the barley HvWRKY38 transcription factor enhances drought tolerance in turf and forage grass (Paspalum notatum Flugge). Mol Breed 25:419–432
Xu X, Chen C, Fan B, Chen Z (2006) Physical and functional interactions between pathogen-induced Arabidopsis WRKY18, WRKY40, and WRKY60 transcription factors. Plant Cell 18:1310–1326
Yamasaki K, Kigawa T, Inoue M, Tateno M, Yamasaki T, Yabuki T, Aoki M, Seki E, Matsuda T, Tomo Y, Hayami N, Terada T, Shirouzu M, Tanaka A, Seki M, Shinozaki K, Yokoyama S (2005) Solution structure of an arabidopsis WRKY DNA binding domain. Plant Cell 17:944–956
Yamasaki K, Kigawa T, Seki M, Shinozaki K, Yokoyama S (2013) DNA-binding domains of plant-specific transcription factors: structure, function, and evolution. Trends Plant Sci 18(5):267–276. doi:10.1016/j.tplants.2012.09.001
Yang X, Yang YN, Xue LJ, Zou MJ, Liu JY, Chen F, Xue HW (2011) Rice ABI5-Like1 regulates abscisic acid and auxin responses by affecting the expression of ABRE-containing genes. Plant Physiol 156:1397–1409
Yordanov IVV, Tsonev T (2003) Plant responses to drought and stress tolerance. Bulg J Plant Physiol 29:187–206
Yu S, Ligang C, Liping Z, Diqiu Y (2010) Overexpression of OsWRKY72 gene interferes in the abscisic acid signal and auxin transport pathway of Arabidopsis. J Biosci 35:459–471
Zhang Y, Wang L (2005) The WRKY transcription factor superfamily: its origin in eukaryotes and expansion in plants. BMC Evol Biol 5:1
Zhou QY, Tian AG, Zou HF, Xie ZM, Lei G, Huang J, Wang CM, Wang HW, Zhang JS, Chen SY (2008) Soybean WRKY-type transcription factor genes, GmWRKY13, GmWRKY21, and GmWRKY54, confer differential tolerance to abiotic stresses in transgenic Arabidopsis plants. Plant Biotechnol J 6:486–503
Zimmermann P, Hirsch-Hoffmann M, Hennig L, Gruissem W (2004) Genevestigator. Arabidopsis microarray database and analysis toolbox. Plant Physiol 136:2621–2632
Acknowledgments
Authors would like to thank Aravind Galla for the critical reading of the manuscript. This project was supported by National Research Initiative Grants 2008-35100-04519 and 2008-35100-05969 from the USDA National Institute of Food and Agriculture.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Tripathi, P., Rabara, R.C. & Rushton, P.J. A systems biology perspective on the role of WRKY transcription factors in drought responses in plants. Planta 239, 255–266 (2014). https://doi.org/10.1007/s00425-013-1985-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00425-013-1985-y