
Abstract
Hutchinson–Gilford progeria syndrome (HGPS) is an extremely rare premature aging disease presenting many features resembling the normal aging process. HGPS patients die before the age of 20 years due to cardiovascular problems and heart failure. HGPS is linked to mutations in the LMNA gene encoding the intermediate filament protein lamin A. Lamin A is a major component of the nuclear lamina, a scaffold structure at the nuclear envelope that defines mechanochemical properties of the nucleus and is involved in chromatin organization and epigenetic regulation. Lamin A is also present in the nuclear interior where it fulfills lamina-independent functions in cell signaling and gene regulation. The most common LMNA mutation linked to HGPS leads to mis-splicing of the LMNA mRNA and produces a mutant lamin A protein called progerin that tightly associates with the inner nuclear membrane and affects the dynamic properties of lamins. Progerin expression impairs many important cellular processes providing insight into potential disease mechanisms. These include changes in mechanosignaling, altered chromatin organization and impaired genome stability, and changes in signaling pathways, leading to impaired regulation of adult stem cells, defective extracellular matrix production and premature cell senescence. In this review, we discuss these pathways and their potential contribution to the disease pathologies as well as therapeutic approaches used in preclinical and clinical tests.
Similar content being viewed by others


Introduction
Aging is a universal process in biological organisms that is characterized by a time-dependent progressive decline in cellular and tissue function. At the molecular and cellular level, nine hallmarks have been proposed to contribute to the extremely complex, multifactorial process of aging: genomic instability and defects in nuclear architecture, telomere attrition, epigenetic alterations and chromatin remodeling, loss of proteostasis, deregulated nutrient sensing, mitochondrial dysfunction, cellular senescence, stem cell exhaustion and altered intercellular communication (Lopez-Otin et al. 2013). Aging represents a major risk factor for the development of several diseases including cancer and cardiovascular and neurodegenerative diseases (Campisi et al. 2011; Niccoli and Partridge 2012).
Several premature aging-like syndromes have been described in humans, presenting many features that resemble normal aging (Navarro et al. 2006). Progeroid syndromes represent a group of rare genetic disorders with features of premature aging (Ghosh and Zhou 2014; Pereira et al. 2008). They are “segmental disorders” that affect multiple organs and tissues and display some but not all symptoms observed in physiological aging (Ghosh and Zhou 2014; Sahin and Depinho 2010; Navarro et al. 2006). Most of these syndromes have been well characterized, and a number of associated genes and causative mutations have been identified in recent years (Kudlow et al. 2007; Pereira et al. 2008). Several heritable premature aging syndromes have been linked to mutations in genes encoding DNA repair proteins such as Werner syndrome (WS), Cockayne syndrome (CS), Bloom syndrome (BS), ataxia-telangiectasia (A-T), xeroderma pigmentosum (XP) and Rothmund–Thomson syndrome (RTS), suggesting that the maintenance of genome integrity has a central role in human aging (Navarro et al. 2006; Pereira et al. 2008). In addition, mutations in the mitochondrial DNA (mtDNA) and impairment of mitochondrial pathways were shown to lead to the development of progeroid phenotypes in the mtDNA mutator premature aging mouse model (Trifunovic et al. 2004; Bratic and Larsson 2013).
A different group of genetic premature aging disorders is linked to mutations in the genes encoding A-type lamins or lamin-processing enzymes, including Hutchinson–Gilford progeria syndrome (HGPS) and restrictive dermopathy (RD) (Navarro et al. 2005; De Sandre-Giovannoli et al. 2003). HGPS has attracted much attention not only because of the severity of the disease, but also due to the hypothesis that the expression of the disease-causing lamin A variant called progerin may also be linked to the normal aging process. While in the most common form of HGPS a silent point mutation in the LMNA gene affects splicing of prelamin A mRNA and leads to the production of the disease-causing lamin A variant progerin (De Sandre-Giovannoli et al. 2003; Eriksson et al. 2003), the sporadic use of the same cryptic splice site in wild-type LMNA can lead to the production of mis-spliced prelamin A mRNA and progerin also in cells and tissues of aged healthy individuals (Scaffidi and Misteli 2006; McClintock et al. 2007). Furthermore, HGPS and normal aging share many cellular phenotypes, such as abnormal nuclear shape, loss of epigenetic marks and increased DNA damage, as well as tissue pathologies including reduced bone density and cardiovascular disease (Burtner and Kennedy 2010). Thus, better understanding of the molecular pathogenesis underlying progeroid syndromes can lead to a better understanding of the normal human aging process. In this review, we summarize the genetic cause of HGPS and consequences for posttranslational lamin processing and lamin functions. We also describe potential causative disease mechanisms and how they may contribute to the cellular, tissue and organismal phenotypes. Finally, we briefly summarize potential strategies for treatment of HGPS.
Nuclear lamins: biochemistry, functions and link to disease
Lamins are type V intermediate filament proteins expressed in all metazoan cells. They are the major building blocks of the nuclear lamina, a complex filamentous meshwork underneath the inner nuclear membrane (INM) (Dechat et al. 2010a; Gruenbaum and Foisner 2015). Lamins share with their cytoskeletal counterparts the domain organization, encompassing a ~45-nm-long central α-helical rod domain flanked by two globular domains (Coulombe et al. 2001; Herrmann et al. 2007), but they contain additional lamin-specific motifs and domains in the C-terminus, such as a nuclear localization signal, a highly conserved immunoglobulin (Ig)-like fold and in most cases a CaaX-box (C = cysteine, a = aliphatic residue, X = any amino acid) (Dechat et al. 2010a; Gruenbaum and Foisner 2015). Based on their sequence similarities, biochemical and structural properties and their expression patterns during development, lamins are classified into A- and B-types. B-type lamins are expressed throughout development, whereas A-type lamins are weakly or not at all expressed in early embryonic stages and in embryonic stem cells (Eckersley-Maslin et al. 2013), but they are upregulated at later stages during development (Gruenbaum and Foisner 2015). In mammals, LMNB1 and LMNB2 encode the two major B-type lamins, lamin B1 and B2, respectively, and LMNB2 encodes an additional smaller germ cell-specific isoform (lamin B3). A-type lamins are derived from a single LMNA gene by alternative splicing, which gives rise to the two major A-type isoforms (lamin A and the smaller splice variant lamin C) and two less abundant isoforms, the germ cell-specific lamin C2 and lamin AΔ10 (Broers et al. 2006; Dechat et al. 2010a). Lamin B1 and B2 and lamin A are expressed as prelamins and undergo several steps of posttranslational processing at their C-terminal–CaaX sequence (Young et al. 2005). The first three processing steps are common to B-type lamins and lamin A and include the addition of a farnesyl group to the C-terminal cysteine residue by farnesyltransferase (FTase) followed by cleavage of the -aaX tripeptide by FACE1/ZMPSTE24 or FACE2/Rce1 proteases, and carboxymethylation of the farnesylated cysteine residue by the isoprenyl-cysteine-carboxy-methylatransferase (ICMT) (Rusinol and Sinensky 2006). The processing of B-type lamins stops at this step, resulting in mature lamin B with a C-terminal farnesyl- and carboxymethyl group. The hydrophobic farnesyl group mediates strong interaction with the INM, leading to the predominant localization of B-type lamins at the nuclear periphery. In contrast, farnesylated prelamin A is further processed by FACE1/ZMPSTE24, which removes the 15 C-terminal amino acids including the farnesylated and carboxymethylated cysteine residue (Pendas et al. 2002). As a consequence, mature lamin A as well as lamin C, which lacks a CaaX-box and never becomes farnesylated, lack the hydrophobic farnesyl group and are therefore not only found at the peripheral lamina associated with the INM, but they can also localize to the nuclear interior (Dechat et al. 2010b; Kolb et al. 2011; Moir et al. 2000; Naetar et al. 2008). In proliferating cells, the nucleoplasmic pool of lamin A/C accounts for 10–15 % of total lamin A/C and is highly mobile compared to lamin A/C at the periphery (Moir et al. 2000).
Lamins have long been known as structural components providing mechanical support for the nucleus (Gruenbaum and Foisner 2015), and recent reports showed that lamins define the mechanochemical properties of the nucleus (Osmanagic-Myers et al. 2015); lamin A is responsible for nuclear stiffness, and B-type lamins for nuclear elasticity (Buxboim et al. 2014; Swift et al. 2013). Besides their mechanochemical role, lamins have a multitude of additional functions, including chromatin organization, gene regulation, DNA repair, and (mechano-) signaling (Amendola and van Steensel 2014; Andres and Gonzalez 2009; Dechat et al. 2010a; Dittmer and Misteli 2011; Gruenbaum and Foisner 2015; Ho and Lammerding 2012).
The multitude of functions of nuclear lamins can, at least in part, be explained by their interactions with a plethora of lamin-binding proteins at the nuclear envelope (Brachner and Foisner 2011; Gruenbaum and Foisner 2015; Wilson and Foisner 2010). It is generally assumed that ubiquitously expressed lamins together with their differentially expressed binding partners form “functional units” at the nuclear envelope responsible for diverse, tissue-specific roles of lamins (Korfali et al. 2012; Worman and Schirmer 2015). In contrast to the huge number of lamin-binding proteins at the nuclear envelope, only a few proteins are known to interact with the mobile nucleoplasmic pool of lamin A/C to form “functional units” in the nuclear interior. The best-characterized interaction partner of A-type lamins in the nuclear interior is lamina-associated polypeptide (LAP) 2α (Dechat et al. 2000; Markiewicz et al. 2002; Dorner et al. 2006; Naetar et al. 2008), a unique isoform of the LAP2 family. Unlike the other LAP2 isoforms, which are integral membrane proteins of the INM, LAP2α lacks a transmembrane domain and localizes in the nuclear interior, where it interacts with and stabilizes nucleoplasmic lamin A/C (Dechat et al. 2000; Naetar et al. 2008). Nucleoplasmic lamin A/C–LAP2α complexes have been implicated in the retinoblastoma protein-mediated regulation of cell proliferation and differentiation of tissue progenitor cells (Dorner et al. 2006; Markiewicz et al. 2002; Naetar et al. 2008) and in chromatin organization (Bronshtein et al. 2015; Zhang et al. 2013).
Given the multitude of functions of the lamins, it is not surprising that mutations in lamins and lamin-binding proteins are associated with a variety of human diseases exhibiting complex patterns of tissue-specific pathologies (Broers et al. 2006; Worman 2012). The majority of diseases are caused by mutations in the LMNA gene and are collectively termed laminopathies. Until today more than 500 mutations have been described in LMNA (www.umd.be/LMNA/) that give rise to four major groups of diseases with overlapping pathologies, including striated muscle diseases, lipodystrophic syndromes, peripheral neuropathy and accelerated aging disorders (Worman 2012).
Hutchinson–Gilford progeria syndrome: genetics and cellular and clinical phenotypes
Hutchinson–Gilford progeria syndrome (HGPS) is an extremely rare sporadic autosomal-dominant genetic disorder affecting 1 in 4–8 million newborns and displays phenotypic features of premature aging (Ghosh and Zhou 2014; Gordon et al. 2014). Children with HGPS appear normal at birth but start to exhibit many distinctive clinical features within the first year of life. Classical progeria symptoms include severe growth retardation, loss of hair and subcutaneous fat, prominent eyes and scalp veins, aged-looking skin, joint stiffness and reduced bone density. As children get older they suffer from osteoporosis, atherosclerosis and cardiovascular diseases as the most severe aspect of the disease. HGPS patients die at an average age of 14 years due to myocardial infarction, heart failure or progressive atherosclerosis (Cau et al. 2014; Kieran et al. 2007; Muchir and Worman 2010).
Classical HGPS is caused by a de novo heterozygous mutation (1824C>T, p.G608G) in exon 11 of LMNA (De Sandre-Giovannoli et al. 2003; Eriksson et al. 2003), which activates a cryptic splice donor site, resulting in the production of a prelamin A mRNA that contains an internal deletion of 150 base pairs. This transcript is translated into a mutant lamin A protein termed progerin, which harbors a deletion of 50 amino acids within its C-terminus including the FACE1/ZMSPTE24 cleavage site (Eriksson et al. 2003). As a consequence, progerin cannot undergo the final proteolytic processing step and permanently retains the C-terminal farnesyl group, leading to its stable association with the INM and predominant localization at the nuclear periphery (Dechat et al. 2007; Davies et al. 2009).
Progerin is expressed in multiple tissues, mostly of mesenchymal origin including skin, bone, skeletal muscle, adipose tissue, heart and large and small arteries (Gordon et al. 2014). Expression of progerin induces various cellular defects in a dominant-negative manner, including highly lobulated nuclei with thickened lamina, loss of peripheral heterochromatin, accumulation of DNA damage, telomere aberrations and mitochondrial dysfunction, leading to differentiation defects and premature cellular senescence (Bridger and Kill 2004; Brunauer and Kennedy 2015; Goldman et al. 2004; Gonzalo and Kreienkamp 2015; McCord et al. 2013; Scaffidi and Misteli 2008; Shumaker et al. 2006; Vidak et al. 2015). In addition, progerin expression leads to decreased expression levels of lamin B1, heterochromatin protein 1 α (HP1α) and LAP2α, and loss of nucleoplasmic lamins (Fig. 1) (Cenni et al. 2011; Scaffidi and Misteli 2005; Miller et al. 2013; Scaffidi and Misteli 2008; Vidak et al. 2015). These changes together with the abnormal nuclear morphology are often used as cellular disease markers to test therapeutic strategies in cell and mouse models (Cao et al. 2011b; Capell et al. 2005; Fong et al. 2006; Scaffidi and Misteli 2005).

Progerin expression causes loss of nucleoplasmic lamin A/C and LAP2α in primary HGPS fibroblasts. Immunofluorescence analysis of wild-type (WT) and HGPS primary human fibroblasts using anti-progerin (red), anti-lamin A/C (green) and anti-LAP2α (green) antibodies shows significant decrease in the nucleoplasmic pool of A-type lamins and LAP2α levels upon progerin expression. Scale bar 10 µm
In addition to the classical 1824C>T HGPS mutation, other heterozygous, homozygous or compound heterozygous mutations in LMNA have been reported, such as T10I, A57P, L59R, R133L, L140R, S143F, E145K, V169fsX176, D300N, E578V and R644C and c.412G>A (Kirschner et al. 2005; Csoka et al. 2004; Chen et al. 2003; Caux et al. 2003; Jacob et al. 2005; Mory et al. 2008; McPherson et al. 2009; Doh et al. 2009; Renard et al. 2009; Doubaj et al. 2012; Kane et al. 2013), all causing atypical progeroid syndromes (APS). APS mutations affect similar tissues (bone, skin, hair and body fat) and cause similar pathologies (growth retardation, alopecia, tight skin and beaked nose) as classical HGPS, but the course and severity of the symptoms vary greatly (Garg et al. 2009; Doubaj et al. 2012). Compound heterozygous mutations in FACE, leading to the complete loss of function of ZMPSTE24 protease and accumulation of farnesylated prelamin A causes autosomal recessive restrictive dermopathy (RD), a progeroid syndrome associated with neonatal death (Navarro et al. 2005). In 2011, a new autosomal recessive syndrome was identified and named Nestor–Guillermo progeria syndrome (NGPS) (Cabanillas et al. 2011). Patients with NGPS display several pathologies similar to HGPS but with a slow clinical course and relatively long survival. NGPS is caused by a homozygous missense mutation in BANF1 that encodes barrier-to-autointegration factor (BAF), a chromatin protein, which directly interacts with lamins and lamin-binding proteins and has been implicated in chromatin organization (Margalit et al. 2007).
Mouse models of HGPS
In the search for molecular disease mechanisms of HGPS, several progeroid mouse models have been created. One of the first of these mouse models was generated by knocking-in a mutant Lmna allele (Lmna HG) that produces exclusively progerin but no wild-type lamin A and lamin C (Yang et al. 2005). This knock-in mouse model displays phenotypes similar to HGPS children including loss of hair (alopecia) and subcutaneous fat, osteoporosis and premature death, but no cardiovascular defects were reported. In contrast, a transgenic mouse model that carries the mutated G608G human LMNA allele on a bacterial artificial chromosome (G608G BAC) develops progressive loss of vascular smooth muscle cells (VSMCs), a feature described also in HGPS patients, but did not show most of the other pathologies (Varga et al. 2006). Another mouse model, in which a point mutation in Lmna caused loss of exon 9 (Lmna L530P/L530P, also known as Lmna∆9), also displayed phenotypes overlapping with HGPS (Mounkes et al. 2003; Hernandez et al. 2010), but the mechanism is still unclear and may differ from that of the classical HGPS.
These mouse models phenocopy some of the phenotypes observed in the HGPS patients, but they do not recreate the exact molecular changes occurring at the LMNA locus in HGPS patients. Therefore, Osorio et al. created a mouse knock-in strain that carries a HGPS mutation in the mouse Lmna gene (Lmna G609G; 1827C>T; Gly609Gly) and produces progerin due to abnormal splicing of the endogenous Lmna mRNA like in HGPS patients (Osorio et al. 2011). These mice phenocopy the main clinical manifestations of human HGPS and open new avenues towards investigating the splicing defect in HGPS and identification of drugs that may correct faulty splicing of prelamin A mRNA in HGPS.
In addition to these transgenic mice expressing progerin ubiquitously, several mouse models with tissue-specific progerin expression have been generated. A transgenic mouse model that contains a human G608G LMNA minigene mutant under the control of a tet-operon (tetopLAG608G) allowed tissue-specific and inducible expression of progerin by crossing them with transgenic mice expressing the transactivator in specific tissues (Sagelius et al. 2008). Expression of progerin in keratin 5-expressing cells in the epidermis caused loss of subcutaneous fat, fibrosis of the dermis, incomplete development of sebaceous glands, dental problems, hair thinning, decreased stem cell population in epidermal tissues and impaired wound healing ability, supporting the hypothesis that an impaired regeneration capacity of epidermal stem cells may contribute to the HGPS phenotype (Rosengardten et al. 2011). The osteoblast-specific expression of progerin reduced bone density and caused spontaneous fractures most likely due to an abnormal osteoblast differentiation (Schmidt et al. 2012). In contrast, expression of progerin in the aged brain, which usually expresses low levels of lamin A/C, caused some structural nuclear abnormalities but no signs of impaired brain function were detected (Baek et al. 2015).
Deletion of Zmpste24, the metalloproteinase involved in the posttranslational maturation of prelamin A, results in the expression of farnesylated prelamin A and produces various progeroid phenotypes in mice. Zmpste24 −/− mice are normal at birth, but within 4–6 weeks of age they start to display many of the progeroid phenotypes such as growth retardation, alopecia, loss of adipose tissue, multiple spontaneous bone fractures, abnormal nuclear morphology and premature death, as well as muscular dystrophy and dilated cardiomyopathy (Bergo et al. 2002; Pendas et al. 2002). The latter phenotypes may also be linked to the fact that no mature lamin A is produced in these mice, while HGPS phenotypes may be caused by the accumulation of farnesylated prelamin A. In support of this hypothesis, blocking prelamin A farnesylation in these mice by farnesyltransferase inhibitors (FTIs) improves some of the HGPS phenotypes (Fong et al. 2006). Furthermore, a knock-in mouse model that exclusively expresses non-farnesylated prelamin A (Lmna nPLAO/nPLAO) shows no progeroid phenotypes but develops severe cardiomyopathy (Davies et al. 2010). In contrast, observations in Lmna HG/+ mice that FTI treatment ameliorates but not completely abolishes progeria phenotypes suggested that the non-farnesylated form of progerin may retain some toxic features relevant for HGPS. To test this hypothesis, a mouse model exclusively expressing the non-farnesylated form of progerin (Lmna nHG) was generated by replacing the cysteine in the CaaX motif in the Lmna HG allele with a serine (CSIM → SSIM). Interestingly, homozygote and heterozygote Lmna nHG mice showed similar, but less severe progeroid phenotypes than those described for Lmna HG mice (Yang et al. 2008). To exclude that the cysteine-to-serine substitution may itself be toxic, an alternative non-farnesylated progerin-expressing mouse model was generated by deleting the isoleucine in the CSIM motif to create a protein ending in CSM rather than SSIM (Lmna csmHG). Surprisingly, Lmna csmHG mice showed no noticeable progeroid defects, suggesting that cysteine-to-serine substitution in the Lmna nHG mouse model may be toxic, although the authors do not exclude the possibility that toxicity of progerin is linked to the 50 amino acid deletion in its C-terminus independent of farnesylation and the deletion of the isoleucine in CSIM may neutralize the toxicity of the protein (Yang et al. 2011b).
Molecular and cellular mechanisms contributing to the HGPS pathologies
HGPS-causing mutations have been shown to affect many fundamental cellular processes, but how these contribute to the described pathologies is not completely understood. In most cases, we do not even know the detailed molecular mechanisms how progerin expression affects the molecular processes found to be impaired in HGPS. However, based on what is known about lamin A/C and progerin biochemistry, function and interactions (Kubben et al. 2010a, b), one may hypothesize about potential pathways and mechanisms (Fig. 2) discussed below.

Cellular functions affected by the expression of the lamin A mutant protein progerin. Progerin accumulates at the nuclear membrane/lamina leading to reduced levels of nucleoplasmic lamin A/C–LAP2α. These changes in lamin dynamics affect mechanical properties and mechanosignaling, lead to dissociation of heterochromatic lamina-associated domains (LADs) in the genome from the lamina, and impair signaling pathways and gene expression, all contributing to defects in self-renewal and differentiation of adult stem cells, the production of a faulty extracellular matrix (ECM) and cell senescence
Mechanical defects in HGPS
The presence of the farnesyl group in progerin is thought to be a predominant toxic feature in the pathogenesis of the disease. In healthy cells, A- and B-type lamins form distinct homopolymers and micro-domains at the nuclear periphery (Shimi et al. 2008), but this segregation may be lost in HGPS cells (Delbarre et al. 2006) due to the stable association of permanently farnesylated progerin with the membrane (Dechat et al. 2007). Progressive progerin accumulation at the INM during cellular aging of HGPS cells (Eriksson et al. 2003; Fong et al. 2006; Goldman et al. 2004; Varela et al. 2005) leads to immobilization of wild-type lamin A at the lamina, thickening and increased stiffness of the lamina, prominent lobulation of the nuclear envelope, and clustering of nuclear pores (Dahl et al. 2006; Goldman et al. 2004). Overall, these alterations disrupt the structural and functional integrity of the nuclear lamina and may render cells more susceptible to damage through physical stress (Verstraeten et al. 2007; Zhang et al. 2011). While in wild-type cells nuclei respond to shear stress by up-regulation and re-distribution of A-type lamins (Buxboim et al. 2014; Philip and Dahl 2008; Swift et al. 2013), this rearrangement does not work properly in HGPS cells (Dahl et al. 2006). This defect is of particular importance in tissues that are exposed to mechanical stress such as vasculature, bone and joints, which also present some of the most prominent pathologies in HGPS (Gordon et al. 2014; Zhang et al. 2011). In accordance, HGPS skin fibroblasts and VSMCs display increased mechanosensitivity under biomechanical strain (Verstraeten et al. 2008), which may then lead to the observed severe depletion of arterial VSMCs in progeria patients and some progeria mouse models (Stehbens et al. 1999; Varga et al. 2006).
Altered adaptation of progerin-expressing cells to high sheer stress may also be caused by progerin’s negative effect on the expression of many proteins involved in mechanotransduction and cytoskeletal organization, as well as extracellular matrix proteins (ECM) (Song et al. 2014; Brassard et al. 2015). In accordance with this, accumulation of progerin in the ascending aorta of Lmna G608G knock-in mice causes reduced expression of the cytoskeleatal protein vimentin (Song et al. 2014). As vimentin has a major role in maintaining cellular integrity and affects apoptotic pathways (Song et al. 2014; Moisan and Girard 2006; Barker et al. 2010), the impaired vimentin expression may lead to defects in mechanotransduction and mechanosignaling in progerin-expressing VSMCs (Song et al. 2014; Brassard et al. 2015). Furthermore, progerin expression was shown to impair linker of the nucleoskeleton and cytoskeleton (LINC) complexes at the nuclear envelope through the stabilization and accumulation of SUN1, a LINC component in the INM, and reducing SUN1 mobility (Chen et al. 2014). As the LINC complex is essential for nucleocytoskeletal coupling and mechanotransduction (Osmanagic-Myers et al. 2015), an impaired LINC complex may further contribute to impaired shear stress response in the vasculature of HGS patients.
Impaired chromatin organization
A-type lamins can directly interact with DNA and histones (Gruenbaum and Foisner 2015), and together with a number of lamin A-binding chromatin proteins, such as members of the LEM domain protein family (Brachner and Foisner 2011; Solovei et al. 2013), they have been implicated in higher-order chromatin organization, heterochromatin formation and epigenetic regulation (Amendola and van Steensel 2014; Dechat et al. 2009; Gonzalez-Suarez and Gonzalo 2010). A-type lamins contribute to the tethering of heterochromatic genomic regions, termed lamina-associated domains (LADs) to the nuclear lamina (Meuleman et al. 2013; Peric-Hupkes et al. 2010) but they also interact with promoter regions of genes, thereby affecting gene expression during cell differentiation (Lund et al. 2013; Ronningen et al. 2015).
It is therefore not surprising that progerin-expressing HGPS nuclei display significant changes in chromatin, such as loss of peripheral heterochromatin, a decrease in the repressive histone marks H3K9me3 and H3K27me3 and an increase in H4K20me3 (Columbaro et al. 2005; Kubben et al. 2012; McCord et al. 2013; Scaffidi and Misteli 2005; Shumaker et al. 2006). It is currently unknown how progerin affects chromatin organization mechanistically, but several reports showing an effect of progerin on epigenetic modifier and chromatin regulator proteins may provide clues towards potential mechanisms. For example, progerin expression causes upregulation of the methyltransferase Suv39h1, while Suv39h1 depletion delays senescence of HGPS cells and prolongs lifespan of Zmpste24 −/− mice (Liu et al. 2013). Furthermore, progerin-expressing cells have reduced levels of heterochromatin protein 1 (HP1) α (Scaffidi and Misteli 2008) and of several proteins of the nucleosome remodeling NuRD complex (Pegoraro et al. 2009; Meshorer and Gruenbaum 2009; Prokocimer et al. 2013). Interestingly, inactivation of the NuRD complex in wild-type cells can also induce aging-associated chromatin defects resembling those observed in HGPS patients. Changes in chromatin organization and epigenetic regulation in progeria cells may in turn have a profound impact on gene expression and genome stability, thereby contributing to many disease phenotypes (Prokocimer et al. 2013).
Most of the lamin A/C chromatin interaction studies so far have focused on the nuclear lamina, but, given the dual location of A-type lamins at the nuclear lamina and in the nuclear interior, it is tempting to speculate that nucleoplasmic lamins together with their binding partner LAP2α may interact with and regulate chromatin throughout the nucleus (Gesson et al. 2014). In support of this, a recent study demonstrated that only 30 % of all cellular LADs in the genome associate with the nuclear envelope in a given cell, while the rest localizes in the nuclear interior, but LADs are stochastically reshuffled to the nuclear lamina in each cell cycle (Kind et al. 2013). This indicates that lamin A–LAD interaction may also occur in the nuclear interior. In line with this model, lamin A/C-deficient cells show increased mobility of chromatin in the nuclear interior (Bronshtein et al. 2015), indicating that lamin A may cross-link chromatin fibers. Moreover, several recent findings indicate that A-type lamins interact also with genomic regions outside of the LADs (Lund et al. 2013, 2015; Ronningen et al. 2015). Furthermore, LAP2α has been found to interact with chromatin at a genome-wide level and to affect chromatin interaction of high-mobility group N protein 5 (HMGN5), a non-histone protein involved in higher-order chromatin organization (Zhang et al. 2013). As progerin expression reduces levels of nucleoplasmic lamin A/C and LAP2α (Dahl et al. 2006; Scaffidi and Misteli 2005; Vidak et al. 2015), it is conceivable that chromatin organization in the nuclear interior is particularly affected in HGPS. In support of this, re-expression of LAP2α in HGPS cells was found to rescue proliferation of the cells, most likely due to the rescue of the impaired chromatin organization at telomeres (Chojnowski et al. 2015) and at genomic regions harboring ECM genes (Vidak et al. 2015).
Genome instability
Progerin expression may also affect genome stability by negatively affecting DNA damage repair pathways (Liu et al. 2005). HGPS cells and cells derived from Zmpste24 −/− mice display impaired recruitment of the DNA double-strand break (DSB) repair factors p53-binding protein 1 (53BP1) and Rad50 and Rad51 to the sites of DNA damage (Liu et al. 2005; Manju et al. 2006), resulting in the accumulation of DSBs (Musich and Zou 2011; Richards et al. 2011). Progerin expression also affects the expression and localization of the nucleotide excision repair protein XPA (Xeroderma Pigmentosum complementation group A) at DNA lesions, resulting in persistent activation of DNA damage response checkpoint kinases ataxia-telangiectasia-mutated (ATM) and ATM and Rad3-related (ATR) protein (Manju et al. 2006; Gonzalo and Kreienkamp 2015). Persistent DNA damage in turn activates tumor suppressor p53 and promotes senescence, one of the phenotypes described as hallmarks of HGPS (Collado et al. 2007).
Deficiencies in DSB repair and telomere dysfunction are major contributors to genome instability in aging cells (Hoeijmakers 2009; Blasco 2005). Dysfunctional telomeres are recognized as DSBs and activate the non-homologous end joining (NHEJ) repair pathway (di Fagagna et al. 2003; Gonzalo and Kreienkamp 2015). Persistent telomere dysfunction and shortening of telomeres below a critical length cause permanent growth arrest known as replicative senescence (Gonzalo and Kreienkamp 2015). HGPS cells were found to undergo accelerated telomere shortening when grown in culture (Decker et al. 2009), and ectopic expression of progerin in wild-type fibroblasts leads to accumulation of DNA damage at telomeres (Benson et al. 2010; Cao et al. 2011a), both resulting in proliferation arrest and senescence. Accordingly, re-expression of telomerase improves proliferation and extends HGPS cellular lifespan (Kudlow et al. 2007; Benson et al. 2010; Chojnowski et al. 2015), suggesting that telomere dysfunction underlies genomic instability and premature senescence in progerin-expressing cells (Gonzalo and Kreienkamp 2015). The molecular mechanisms leading to telomere dysfunction upon progerin expression are poorly understood. Recent studies suggest a role of LAP2α in stabilizing telomere and chromatin structure by increasing the epigenetic H3K27me3 histone mark, preventing progerin-associated DNA damage and rescuing premature senescence (Chojnowski et al. 2015).
Altered regulation of signaling pathways
Lamins serve as scaffolds for various signaling molecules and transcription factors, thereby regulating their activity (Gruenbaum and Foisner 2015; Osmanagic-Myers et al. 2015). This scaffolding function has a dual role. On the one hand, lamins serve as a platform enabling efficient activation of signaling molecules, such as the extracellular-signal regulated kinase (ERK)-dependent activation of transcription factor c-fos at the lamina (Gonzalez et al. 2008). On the other hand, they sequester various transcription factors and their regulating molecules to the nuclear periphery attenuating their function on target genes (Ivorra et al. 2006; Scaffidi and Misteli 2008). Therefore, it is not surprising that progerin expression causes misregulation of various signaling pathways (Prokocimer et al. 2013) as shown in numerous examples; the molecular mechanisms, however, remain mostly obscure. Fibroblasts from the Lmna Δ9 progeria mouse model have reduced Wnt/β-catenin signaling causing defects in the expression of ECM genes (Hernandez et al. 2010). Similarly, hair follicle stem cells in Zmpste24 −/− mice have reduced levels of active β-catenin (Espada et al. 2008). As wnt signaling is important for cartilage and bone development, an impaired wnt signaling can contribute to the bone phenotype in HGPS patients. Notch signaling, another important pathway for the regulation of cell fate and stem cell differentiation during osteogenesis and adipogenesis (Hori et al. 2013), is also affected in progerin expressing cells (Scaffidi and Misteli 2008). Here, some insight into the potential molecular mechanism of progerin’s effect on Notch-signaling has been provided: Sequestration of the Notch co-activator SKIP (Ski-interacting protein) by wild-type lamins is impaired by progerin, leading to increased activation of major Notch downstream effectors (HES1, HES5, HEY1 and TLE1) (Scaffidi and Misteli 2008). NF-κB, an important transcription factor activated as a response to damage, stress and inflammation (Ghosh and Hayden 2008) and during aging (Tilstra et al. 2012), was found to be hyperactivated in progeroid mice (Zmpste24 −/− and LmnaG609G knock-in mice) (Osorio et al. 2012). Crossing Zmpste24 −/− mice with transgenic mice displaying reduced NF-κB signaling (haploinsufficient for the p65 (RelA) NF-κB subunit, RelA +/−) or treatment of mice with the NF-κB inhibitor sodium salicylate extended life span and rescued skin and immunological phenotypes (Kawahara et al. 2009; Osorio et al. 2012).
Complexes of nucleoplasmic A-type lamins and LAP2α interact directly with retinoblastoma protein (pRb) a major regulator of cell proliferation (Markiewicz et al. 2002; Dorner et al. 2006), thereby affecting pRb localization and stability (Johnson et al. 2004; Andres and Gonzalez 2009; Nitta et al. 2007) and expression of pRb target genes (Dorner et al. 2006; Naetar et al. 2008). Genome-wide expression studies in primary HGPS-derived dermal fibroblasts revealed an impaired pRb signaling network (Marji et al. 2010). Thus, the loss of nucleoplasmic lamin A/C and LAP2α in HGPS cells (Vidak et al. 2015) may be linked to a deregulation of the pRb pathway in HGPS, leading to impaired regulation of tissue stem cells (Naetar et al. 2008). An impairment of pRb signaling may also be linked to the reduced levels of heterochromatic histone marks in HGPS in view of the described role of pRb in stabilizing histone methylation (Gonzalo et al. 2005).
Altered regulation of adult stem cells
Adult stem cells constantly replace non-functional and dying cells in many tissues and an age-related decline in their regenerative capacity is an important factor in biological aging (Brassard et al. 2015). Several lines of evidence suggest that A-type lamins may be involved in the regulation of proliferation and differentiation of mesenchymal stem cells (MSCs) as well as tissue progenitor cells (Gotzman and Foisner 2006; Pekovic et al. 2007). MSCs are adult stem cells important for the regeneration of many tissues profoundly affected in HGPS such as bone, skin, muscle and adipose tissue (Scaffidi and Misteli 2008; Dreesen and Stewart 2011; Halaschek-Wiener and Brooks-Wilson 2007). Naive MSCs derived from HGPS patients express low progerin levels in vivo, but accumulate significant amounts of progerin with increasing passages in vitro (Wenzel et al. 2012). Both, adult stem cell self-renewal and differentiation may be affected in HGPS through impaired signaling and chromatin organization as described above. In line with this, Zmpste 24 −/− and tetop-Lmna G608G;K5tTA+ progeria mouse models (see in chapter mouse models) display decreased numbers and altered proliferative capacity of epidermal stem cells (Espada et al. 2008; Rosengardten et al. 2011) and muscle-derived stem/progenitor cells (MDPSCs) (Lavasani et al. 2012). Interestingly, intraperitoneal administration of MDPSCs from young wild-type mice to progeroid mice leads to significant extension of life span and health span (Lavasani et al. 2012), further supporting the hypothesis that an impaired stem cell regulation contributes to premature aging. Postnatal, but not embryonic fibroblasts, derived from the Lmna∆9 progeria mouse model, show proliferative arrest and premature senescence (Hernandez et al. 2010). Why this phenotype is detectable only in postnatal cells remains unknown, but it is likely linked to an impaired extracellular matrix (ECM) production (Hernandez et al. 2010).
Based on the emerging findings in HGPS, it is tempting to speculate that a misbalance in stem cell self-renewal and differentiation coupled with increased mechanical sensitivity could lead to stem cell exhaustion and inefficient repair of the damaged tissues, contributing to many of the phenotypes in HGPS (Gotzman and Foisner 2006).
ECM-mediated HGPS mechanisms
Several recent findings led to the newly emerging concept that progerin expression may lead to impaired expression of ECM components and formation of a faulty ECM, which in turn may be causative for many of the cellular phenotypes observed in HGPS. The ECM is well known to have a major role in cell proliferation, differentiation, cell adhesion and migration, and cell survival (Gattazzo et al. 2014; Humphrey et al. 2014), and ECM production is also compromised during physiological aging (Yang et al. 2011a). Abnormal ECM production and rearrangement during development and tissue homeostasis result in many pathological processes including tissue fibrosis and cancer.
Gene expression analyses in HGPS patient fibroblasts showed a profound deregulation of ECM components (Csoka et al. 2004), including decreased levels of ECM remodeling enzymes such as metaloproteinases (MMPs) (Harten et al. 2011; Vidak et al. 2015) and increased levels of type IV and VI collagen and fibronectin (Colige et al. 1991; Maquart et al. 1988; Song et al. 2014). The importance of a faulty ECM in the development of HGPS phenotypes is supported by several observations: Proliferation of human cells expressing progerin (Vidak et al. 2015) and of mouse adult fibroblasts derived from Lmna∆9 progeria mice (Hernandez et al. 2010) is rescued upon growth on ECM derived from wild-type cells. The most convincing data supporting the role of the ECM in disease development in vivo comes from Zmpste24 mosaic mice containing similar proportion of Zmpste24-deficient (prelamin A-accumulating) and Zmpste24-proficient (mature lamin A-containing) cells in tissues (de la Rosa et al. 2013). Surprisingly, these mice develop normally and maintain the same proportion of mutant versus wild-type cells in their tissues throughout life, indicating that progeroid Zmpste24 −/− cells develop normally in a background providing normal ECM and possibly other extrinsic factors.
Overall, a deregulation of ECM production and remodeling could account for both an impaired proliferation and differentiation of osteoblasts and chondrocytes during cartilage development (Muchir and Worman 2010), as well as an excessive ECM deposition in the vascular system causing increased arterial stiffness in HGPS (Olive et al. 2010).
Therapeutic approaches in progeria
Therapeutic approaches for HGPS treatment can be envisaged to work at different levels from genes to tissues, including approaches to correct protein function, RNA splicing and mutations in the LMNA gene, to cell replacement strategies and treatments to reverse the cellular phenotypes (Gordon et al. 2014). The first therapy developed for HGPS and already tested in clinical trials (Gordon et al. 2012) aimed at correcting the mutant protein. As the presence of the farnesyl group in progerin was proposed to be the predominant deleterious and toxic component, efforts were put into treatments blocking progerin farnesylation through pharmacologically targeting the isoprenoid biosynthesis pathway (Young et al. 2005; Cau et al. 2014). The initial treatment used drugs called farnesyltransferase inhibitors (FTIs), previously developed as potential anti-cancer drugs, which improve some disease parameters in cultured cells (e.g., nuclear shape and proliferation) and progeria mouse models (Fong et al. 2006; Toth et al. 2005; Yang et al. 2005). Based on these preclinical tests a prospective clinical trial was commenced at Boston Children’s Hospital from 2007 to 2009 including 26 HGPS patients (Gordon et al. 2012). Administration of the FTI drug lonafarnib improved weight gain, bone structure and vascular stiffness, with some of the HGPS patients developing mild drug-related side effects (Gordon et al. 2012). After the initiation of the first clinical trial, it was demonstrated that an alternative prenylation pathway called geranyl geranylation may be activated in the presence of FTIs, offering possible explanations for the only moderate efficiency of the FTI treatment in preclinical trials (Yang et al. 2008). Therefore, the pharmacological strategy was adjusted and tested in the Zmpste 24 −/− progeria mouse model, using a combined aminobisphosphonate (zoledronic acid) and statin (pravastatin) treatment, inhibiting farnesyl-pyrophosphatase synthase and the HMG-CoA reductase, respectively (Varela et al. 2008). Based on promising results in preclinical trials using the Zmpste 24 −/− progeria mouse model (Varela et al. 2008), a second clinical trial combining statins and aminobisphosphonates was initiated from 2008 to 2013 in Marseille’ La Timone Children’s Hospital, but the results of the study are not published yet (Cau et al. 2014).
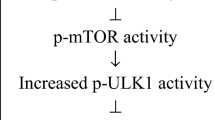
Another approach for treatment aims at reducing progerin protein levels rather than blocking progerin farnesylation. Two major cellular mechanisms are involved in removing misfolded, mutant or aggregated proteins, proteasomal degradation and autophagy. Although a detailed study on potential pathways involved in progerin degradation has not been done so far, several observations suggest that progerin may be removed by activating macroautophagy. Autophagy is a cytoplasmic degradation machinery that targets damaged proteins and organelles to lysosomal degradation and is upregulated in times of stress such as starvation to provide a source of amino acids (Tanida et al. 2008). Treatment with rapamycin, an inhibitor of the mammalian target of rapamycin (mTOR) pathway, upregulates autophagy and extends life span from yeast to mammals (Johnson et al. 2013; Jung et al. 2010; Madeo et al. 2010). Rapamycin treatment of cultured HGPS cells increases progerin clearance by macroautophagy-related pathways and reduces some of the disease phenotypes, such as lobulated nuclei, LAP2α levels and DNA damage (Cao et al. 2011b; Cenni et al. 2011). Interestingly, it has been suggested that FTIs may indirectly affect mTOR by inhibiting the farnesylation of Rheb GTPase, an upstream activator of mTOR (Hanker et al. 2010).
Sulforaphane, an antioxidant derived from cruciferous vegetables, which stimulates proteasome activity and autophagy in normal and HGPS fibroblast cultures, was also found to enhance progerin clearance by autophagy and to reverse the cellular hallmarks of HGPS (Gabriel et al. 2015). Furthermore, two recent studies suggested that retinoids alone (Kubben et al. 2015) or in a combination with rapamycin (Pellegrini et al. 2015) lower the amount of progerin and rescue progeroid phenotypes in cultured cells. These findings together suggest that compounds acting by decreasing progerin levels in the cell could represent a potent tool for new treatments. Autophagy-activating drugs could be particularly beneficial in progeria treatment, but careful in vivo analyses have to be conducted before including them in clinical trials.
An RNA-targeting therapeutic strategy aims at eliminating or inhibiting alternative splicing of progerin pre-mRNA. Introduction of a short (25-mer) antisense morpholino oligonucleotide that can sterically block the cryptic splice site in exon 11 of progerin pre-mRNA resulted in a concentration-dependent decrease in progerin mRNA and protein levels and in the reversion of cellular phenotypes in cultured HGPS cells (Scaffidi and Misteli 2005). A similar strategy was successfully tested in vivo in Zmpste24 −/− and Lmna G609G/G609G progeria mice, resulting in improved body weight, extended lifespan and improvement of several HGPS phenotypes (Osorio et al. 2011). This finding, together with the increasing evidence that the use of oligonucleotides for correction of splicing defects has growing therapeutic applications, initiated a set up of a new clinical trial that is currently under design (Cau et al. 2014).
Finally, a few approaches have been described to target cellular HGPS phenotypes. Resveratrol, an activator of SIRT1, a deacetylase involved in many cellular processes (Lavu et al. 2008), was found beneficial in the treatment of Zmpste24 −/− mice (Liu et al. 2012). Resveratrol treatment was previously shown to increase life span in yeast, worms, and flies and to enhance health span in rodents (Baur et al. 2006; Howitz et al. 2003; Milne et al. 2007; Wood et al. 2004). The mechanism of the beneficial effect of resveratol in HGPS mice is unclear, but it has been shown that SIRT1 interacts with lamin A in wild-type cells (Liu et al. 2012). In the presence of progerin or prelamin A, SIRT1 exhibits reduced association with the nuclear matrix and decreased deacetylase activity, leading to rapid depletion of adult stem cells in Zmpste24 −/− mice. Resveratrol enhances the binding of SIRT1 to A-type lamins, increases SIRT1 deacetylase activity and slows down body weight loss and significantly extends the life span of Zmpste24 −/− mice (Liu et al. 2012). However, another study conducted with an osteoblast and osteocyte-specific progerin-expressing mouse model (Schmidt et al. 2012) did not reveal a beneficial effect of resveratrol (Strandgren et al. 2015). Thus, more detailed studies are needed to find out whether resveratrol is a potentially promising drug for treatment of HGPS.
Concluding remarks
The number of molecular biological studies aiming at the identification of lamin-mediated molecular disease mechanisms involved in HGPS increased tremendously following the surprising discovery that LMNA is causally linked to the premature aging disease HGPS in 2003. Despite numerous cellular pathways that were identified to be affected by the expression of the mutant lamin A protein (Fig. 2), the mechanistic details behind these effects are still unclear in most cases. Knowledge based on what was already known on lamin biology before the protein was linked to HGPS and findings on novel roles of lamins in diverse pathways in recent years allowed the launch of translational studies and the efficient search for drug targets and therapeutic approaches within a short time period. The results of the first clinical trials taught us that some improvements of the disease phenotypes can be achieved by FTI treatment, but they also made clear that we need a much better understanding of the underlying disease mechanisms to be able to tackle specific aspects of the disease in a more focused approach. It will also be important to elucidate which of the numerous pathways found to be impaired in HGPS are most relevant for and causally involved in the pathologies, and which ones are just bystanders.
References
Amendola M, van Steensel B (2014) Mechanisms and dynamics of nuclear lamina–genome interactions. Curr Opin Cell Biol 28:61–68. doi:10.1016/j.ceb.2014.03.003
Andres V, Gonzalez JM (2009) Role of A-type lamins in signaling, transcription, and chromatin organization. J Cell Biol 187(7):945–957. doi:10.1083/jcb.200904124
Baek JH, Schmidt E, Viceconte N, Strandgren C, Pernold K, Richard TJ, Van Leeuwen FW, Dantuma NP, Damberg P, Hultenby K, Ulfhake B, Mugnaini E, Rozell B, Eriksson M (2015) Expression of progerin in aging mouse brains reveals structural nuclear abnormalities without detectible significant alterations in gene expression, hippocampal stem cells or behavior. Hum Mol Genet 24(5):1305–1321. doi:10.1093/hmg/ddu541
Barker AJ, Lanning C, Shandas R (2010) Quantification of hemodynamic wall shear stress in patients with bicuspid aortic valve using phase-contrast MRI. Ann Biomed Eng 38(3):788–800. doi:10.1007/s10439-009-9854-3
Baur JA, Pearson KJ, Price NL, Jamieson HA, Lerin C, Kalra A, Prabhu VV, Allard JS, Lopez-Lluch G, Lewis K, Pistell PJ, Poosala S, Becker KG, Boss O, Gwinn D, Wang M, Ramaswamy S, Fishbein KW, Spencer RG, Lakatta EG, Le Couteur D, Shaw RJ, Navas P, Puigserver P, Ingram DK, de Cabo R, Sinclair DA (2006) Resveratrol improves health and survival of mice on a high-calorie diet. Nature 444(7117):337–342. doi:10.1038/nature05354
Benson EK, Lee SW, Aaronson SA (2010) Role of progerin-induced telomere dysfunction in HGPS premature cellular senescence. J Cell Sci 123(15):2605–2612. doi:10.1242/jcs.067306
Bergo MO, Gavino B, Ross J, Schmidt WK, Hong C, Kendall LV, Mohr A, Meta M, Genant H, Jiang Y, Wisner ER, Van Bruggen N, Carano RA, Michaelis S, Griffey SM, Young SG (2002) Zmpste24 deficiency in mice causes spontaneous bone fractures, muscle weakness, and a prelamin A processing defect. Proc Natl Acad Sci USA 99(20):13049–13054. doi:10.1073/pnas.192460799
Blasco MA (2005) Telomeres and human disease: ageing, cancer and beyond. Nat Rev Genet 6(8):611–622. doi:10.1038/nrg1656
Brachner A, Foisner R (2011) Evolvement of LEM proteins as chromatin tethers at the nuclear periphery. Biochem Soc Trans 39(6):1735–1741. doi:10.1042/BST20110724
Brassard JA, Fekete N, Garnier A, Hoesli CA (2015) Hutchinson–Gilford progeria syndrome as a model for vascular aging. Biogerontology. doi:10.1007/s10522-015-9602-z
Bratic A, Larsson NG (2013) The role of mitochondria in aging. J Clin Invest 123(3):951–957. doi:10.1172/JCI64125
Bridger JM, Kill IR (2004) Aging of Hutchinson–Gilford progeria syndrome fibroblasts is characterised by hyperproliferation and increased apoptosis. Exp Gerontol 39(5):717–724
Broers JL, Ramaekers FC, Bonne G, Yaou RB, Hutchison CJ (2006) Nuclear lamins: laminopathies and their role in premature ageing. Physiol Rev 86(3):967–1008. doi:10.1152/physrev.00047.2005
Bronshtein I, Kepten E, Kanter I, Berezin S, Lindner M, Redwood AB, Mai S, Gonzalo S, Foisner R, Shav-Tal Y, Garini Y (2015) Loss of lamin A function increases chromatin dynamics in the nuclear interior. Nat Commun 6:8044. doi:10.1038/ncomms9044
Brunauer R, Kennedy BK (2015) Medicine. Progeria accelerates adult stem cell aging. Science 348(6239):1093–1094. doi:10.1126/science.aac4214
Burtner CR, Kennedy BK (2010) Progeria syndromes and ageing: what is the connection? Nat Rev Mol Cell Biol 11(8):567–578. doi:10.1038/nrm2944
Buxboim A, Swift J, Irianto J, Spinler KR, Dingal PC, Athirasala A, Kao YR, Cho S, Harada T, Shin JW, Discher DE (2014) Matrix elasticity regulates lamin-A, C phosphorylation and turnover with feedback to actomyosin. Curr Biol 24(16):1909–1917. doi:10.1016/j.cub.2014.07.001
Cabanillas R, Cadinanos J, Villameytide JA, Perez M, Longo J, Richard JM, Alvarez R, Duran NS, Illan R, Gonzalez DJ, Lopez-Otin C (2011) Nestor–Guillermo progeria syndrome: a novel premature aging condition with early onset and chronic development caused by BANF1 mutations. Am J Med Genet A 155A(11):2617–2625. doi:10.1002/ajmg.a.34249
Campisi J, Andersen JK, Kapahi P, Melov S (2011) Cellular senescence: a link between cancer and age-related degenerative disease? Semin Cancer Biol 21(6):354–359. doi:10.1016/j.semcancer.2011.09.001
Cao K, Blair CD, Faddah DA, Kieckhaefer JE, Olive M, Erdos MR, Nabel EG, Collins FS (2011a) Progerin and telomere dysfunction collaborate to trigger cellular senescence in normal human fibroblasts. J Clin Invest 121(7):2833–2844. doi:10.1172/JCI43578
Cao K, Graziotto JJ, Blair CD, Mazzulli JR, Erdos MR, Krainc D, Collins FS (2011b) Rapamycin reverses cellular phenotypes and enhances mutant protein clearance in Hutchinson–Gilford progeria syndrome cells. Sci Transl Med. doi:10.1126/scitranslmed.3002346
Capell BC, Erdos MR, Madigan JP, Fiordalisi JJ, Varga R, Conneely KN, Gordon LB, Der CJ, Cox AD, Collins FS (2005) Inhibiting farnesylation of progerin prevents the characteristic nuclear blebbing of Hutchinson–Gilford progeria syndrome. Proc Natl Acad Sci USA 102(36):12879–12884. doi:10.1073/pnas.0506001102
Cau P, Navarro C, Harhouri K, Roll P, Sigaudy S, Kaspi E, Perrin S, De Sandre-Giovannoli A, Levy N (2014) Nuclear matrix, nuclear envelope and premature aging syndromes in a translational research perspective. Semin Cell Dev Biol. doi:10.1016/j.semcdb.2014.03.022
Caux F, Dubosclard E, Lascols O, Buendia B, Chazouilleres O, Cohen A, Courvalin JC, Laroche L, Capeau J, Vigouroux C, Christin-Maitre S (2003) A new clinical condition linked to a novel mutation in lamins A and C with generalized lipoatrophy, insulin-resistant diabetes, disseminated leukomelanodermic papules, liver steatosis, and cardiomyopathy. J Clin Endocrinol Metab 88(3):1006–1013. doi:10.1210/jc.2002-021506
Cenni V, Capanni C, Columbaro M, Ortolani M, D’Apice MR, Novelli G, Fini M, Marmiroli S, Scarano E, Maraldi NM, Squarzoni S, Prencipe S, Lattanzi G (2011) Autophagic degradation of farnesylated prelamin A as a therapeutic approach to lamin-linked progeria. Eur J Histochem 55(4):200–205. doi:10.4081/ejh.2011.e36
Chen L, Lee L, Kudlow BA, Dos Santos HG, Sletvold O, Shafeghati Y, Botha EG, Garg A, Hanson NB, Martin GM, Mian IS, Kennedy BK, Oshima J (2003) LMNA mutations in atypical Werner’s syndrome. Lancet 362(9382):440–445. doi:10.1016/S0140-6736(03)14069-X
Chen ZJ, Wang WP, Chen YC, Wang JY, Lin WH, Tai LA, Liou GG, Yang CS, Chi YH (2014) Dysregulated interactions between lamin A and SUN1 induce abnormalities in the nuclear envelope and endoplasmic reticulum in progeric laminopathies. J Cell Sci 127(Pt 8):1792–1804. doi:10.1242/jcs.139683
Chojnowski A, Ong PF, Wong ES, Lim JS, Mutalif RA, Navasankari R, Dutta B, Yang H, Liow YY, Sze SK, Boudier T, Wright GD, Colman A, Burke B, Stewart CL, Dreesen O (2015) Progerin reduces LAP2α-telomere association in Hutchinson–Gilford progeria. Elife. doi:10.7554/eLife.07759
Colige A, Nusgens B, Lapiere CM (1991) Altered response of progeria fibroblasts to epidermal growth factor. J Cell Sci 100(Pt 3):649–655
Collado M, Blasco MA, Serrano M (2007) Cellular senescence in cancer and aging. Cell 130(2):223–233. doi:10.1016/j.cell.2007.07.003
Columbaro M, Capanni C, Mattioli E, Novelli G, Parnaik VK, Squarzoni S, Maraldi NM, Lattanzi G (2005) Rescue of heterochromatin organization in Hutchinson–Gilford progeria by drug treatment. Cell Mol Life Sci 62(22):2669–2678. doi:10.1007/s00018-005-5318-6
Coulombe PA, Ma L, Yamada S, Wawersik M (2001) Intermediate filaments at a glance. J Cell Sci 114(Pt 24):4345–4347
Csoka AB, Cao H, Sammak PJ, Constantinescu D, Schatten GP, Hegele RA (2004) Novel lamin A/C gene (LMNA) mutations in atypical progeroid syndromes. J Med Genet 41(4):304–308
Dahl KN, Scaffidi P, Islam MF, Yodh AG, Wilson KL, Misteli T (2006) Distinct structural and mechanical properties of the nuclear lamina in Hutchinson–Gilford progeria syndrome. Proc Natl Acad Sci USA 103(27):10271–10276. doi:10.1073/pnas.0601058103
Davies BSJ, Fong LG, Yang SH, Coffinier C, Young SG (2009) The posttranslational processing of prelamin A and disease. Annu Rev Genomics Hum Genet 10:153–174. doi:10.1146/annurev-genom-082908-150150
Davies BS, Barnes RH 2nd, Tu Y, Ren S, Andres DA, Spielmann HP, Lammerding J, Wang Y, Young SG, Fong LG (2010) An accumulation of non-farnesylated prelamin A causes cardiomyopathy but not progeria. Hum Mol Genet 19(13):2682–2694. doi:10.1093/hmg/ddq158
de la Rosa J, Freije JMP, Cabanillas R, Osorio FG, Fraga MF, Fernandez-Garcia MS, Rad R, Fanjul V, Ugalde AP, Liang Q, Prosser HM, Bradley A, Cadinanos J, Lopez-Otin C (2013) Prelamin A causes progeria through cell-extrinsic mechanisms and prevents cancer invasion. Nat Commun. doi:10.1038/Ncomms3268
De Sandre-Giovannoli A, Bernard R, Cau P, Navarro C, Amiel J, Boccaccio I, Lyonnet S, Stewart CL, Munnich A, Le Merrer M, Levy N (2003) Lamin a truncation in Hutchinson–Gilford progeria. Science 300(5628):2055. doi:10.1126/science.1084125
Dechat T, Korbei B, Vaughan OA, Vlcek S, Hutchison CJ, Foisner R (2000) Lamina-associated polypeptide 2α binds intranuclear A-type lamins. J Cell Sci 113(Pt 19):3473–3484
Dechat T, Shimi T, Adam SA, Rusinol AE, Andres DA, Spielmann HP, Sinensky MS, Goldman RD (2007) Alterations in mitosis and cell cycle progression caused by a mutant lamin A known to accelerate human aging. Proc Natl Acad Sci USA 104(12):4955–4960. doi:10.1073/pnas.0700854104
Dechat T, Adam SA, Goldman RD (2009) Nuclear lamins and chromatin: when structure meets function. Adv Enzyme Regul 49(1):157–166. doi:10.1016/j.advenzreg.2008.12.003
Dechat T, Adam SA, Taimen P, Shimi T, Goldman RD (2010a) Nuclear lamins. Cold Spring Harb Perspect Biol 2(11):a000547. doi:10.1101/cshperspect.a000547
Dechat T, Gesson K, Foisner R (2010b) Lamina-independent lamins in the nuclear interior serve important functions. Cold Spring Harb Symp Quant Biol 75:533–543. doi:10.1101/sqb.2010.75.018
Decker ML, Chavez E, Vulto I, Lansdorp PM (2009) Telomere length in Hutchinson–Gilford Progeria Syndrome. Mech Ageing Dev 130(6):377–383. doi:10.1016/j.mad.2009.03.001
Delbarre E, Tramier M, Coppey-Moisan M, Gaillard C, Courvalin JC, Buendia B (2006) The truncated prelamin A in Hutchinson–Gilford progeria syndrome alters segregation of A-type and B-type lamin homopolymers. Hum Mol Genet 15(7):1113–1122. doi:10.1093/hmg/ddl026
di Fagagna FD, Reaper PM, Clay-Farrace L, Fiegler H, Carr P, von Zglinicki T, Saretzki G, Carter NP, Jackson SP (2003) A DNA damage checkpoint response in telomere-initiated senescence. Nature 426(6963):194–198. doi:10.1038/nature02118
Dittmer T, Misteli T (2011) The lamin protein family. Genome Biol. doi:10.1186/Gb-2011-12-5-222
Doh YJ, Kim HK, Jung ED, Choi SH, Kim JG, Kim BW, Lee IK (2009) Novel LMNA gene mutation in a patient with Atypical Werner’s Syndrome. Korean J Intern Med 24(1):68–72. doi:10.3904/kjim.2009.24.1.68
Dorner D, Vlcek S, Foeger N, Gajewski A, Makolm C, Gotzmann J, Hutchison CJ, Foisner R (2006) Lamina-associated polypeptide 2α regulates cell cycle progression and differentiation via the retinoblastoma-E2F pathway. J Cell Biol 173(1):83–93. doi:10.1083/jcb.200511149
Doubaj Y, De Sandre-Giovannoli A, Vera EV, Navarro CL, Elalaoui SC, Tajir M, Levy N, Sefiani A (2012) An inherited LMNA gene mutation in atypical progeria syndrome. Am J Med Genet A 158A(11):2881–2887. doi:10.1002/ajmg.a.35557
Dreesen O, Stewart CL (2011) Accelerated aging syndromes, are they relevant to normal human aging? Aging (Albany NY) 3(9):889–895
Eckersley-Maslin MA, Bergmann JH, Lazar Z, Spector DL (2013) Lamin A/C is expressed in pluripotent mouse embryonic stem cells. Nucleus 4(1):53–60. doi:10.4161/nucl.23384
Eriksson M, Brown WT, Gordon LB, Glynn MW, Singer J, Scott L, Erdos MR, Robbins CM, Moses TY, Berglund P, Dutra A, Pak E, Durkin S, Csoka AB, Boehnke M, Glover TW, Collins FS (2003) Recurrent de novo point mutations in lamin A cause Hutchinson–Gilford progeria syndrome. Nature 423(6937):293–298. doi:10.1038/nature01629
Espada J, Varela I, Flores I, Ugalde AP, Cadinanos J, Pendas AM, Stewart CL, Tryggvason K, Blasco MA, Freije JM, Lopez-Otin C (2008) Nuclear envelope defects cause stem cell dysfunction in premature-aging mice. J Cell Biol 181(1):27–35. doi:10.1083/jcb.200801096
Fong LG, Frost D, Meta M, Qiao X, Yang SH, Coffinier C, Young SG (2006) A protein farnesyltransferase inhibitor ameliorates disease in a mouse model of progeria. Science 311(5767):1621–1623. doi:10.1126/science.1124875
Gabriel D, Roedl D, Gordon LB, Djabali K (2015) Sulforaphane enhances progerin clearance in Hutchinson–Gilford progeria fibroblasts. Aging Cell 14(1):78–91. doi:10.1111/acel.12300
Garg A, Subramanyam L, Agarwal AK, Simha V, Levine B, D’Apice MR, Novelli G, Crow Y (2009) Atypical progeroid syndrome due to heterozygous missense LMNA mutations. J Clin Endocrinol Metab 94(12):4971–4983. doi:10.1210/jc.2009-0472
Gattazzo F, Urciuolo A, Bonaldo P (2014) Extracellular matrix: a dynamic microenvironment for stem cell niche. Biochim Biophys Acta 1840(8):2506–2519. doi:10.1016/j.bbagen.2014.01.010
Gesson K, Vidak S, Foisner R (2014) Lamina-associated polypeptide (LAP)2α and nucleoplasmic lamins in adult stem cell regulation and disease. Semin Cell Dev Biol 29:116–124. doi:10.1016/j.semcdb.2013.12.009
Ghosh S, Hayden MS (2008) New regulators of NF-κB in inflammation. Nat Rev Immunol 8(11):837–848. doi:10.1038/nri2423
Ghosh S, Zhou Z (2014) Genetics of aging, progeria and lamin disorders. Curr Opin Genet Dev 26:41–46. doi:10.1016/j.gde.2014.05.003
Goldman RD, Shumaker DK, Erdos MR, Eriksson M, Goldman AE, Gordon LB, Gruenbaum Y, Khuon S, Mendez M, Varga R, Collins FS (2004) Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson–Gilford progeria syndrome. Proc Natl Acad Sci USA 101(24):8963–8968. doi:10.1073/pnas.0402943101
Gonzalez JM, Navarro-Puche A, Casar B, Crespo P, Andres V (2008) Fast regulation of AP-1 activity through interaction of lamin A/C, ERK1/2, and c-Fos at the nuclear envelope. J Cell Biol 183(4):653–666. doi:10.1083/jcb.200805049
Gonzalez-Suarez I, Gonzalo S (2010) Nurturing the genome: A-type lamins preserve genomic stability. Nucleus 1(2):129–135. doi:10.4161/nucl.1.2.10797
Gonzalo S, Kreienkamp R (2015) DNA repair defects and genome instability in Hutchinson–Gilford progeria syndrome. Curr Opin Cell Biol 34:75–83. doi:10.1016/j.ceb.2015.05.007
Gonzalo S, Garcia-Cao M, Fraga MF, Schotta G, Peters AH, Cotter SE, Eguia R, Dean DC, Esteller M, Jenuwein T, Blasco MA (2005) Role of the RB1 family in stabilizing histone methylation at constitutive heterochromatin. Nat Cell Biol 7(4):420–428. doi:10.1038/ncb1235
Gordon LB, Kleinman ME, Miller DT, Neuberg DS, Giobbie-Hurder A, Gerhard-Herman M, Smoot LB, Gordon CM, Cleveland R, Snyder BD, Fligor B, Bishop WR, Statkevich P, Regen A, Sonis A, Riley S, Ploski C, Correia A, Quinn N, Ullrich NJ, Nazarian A, Liang MG, Huh SY, Schwartzman A, Kieran MW (2012) Clinical trial of a farnesyltransferase inhibitor in children with Hutchinson–Gilford progeria syndrome. Proc Natl Acad Sci USA 109(41):16666–16671. doi:10.1073/pnas.1202529109
Gordon LB, Rothman FG, Lopez-Otin C, Misteli T (2014) Progeria: A paradigm for translational medicine. Cell 156(3):400–407. doi:10.1016/j.cell.2013.12.028
Gotzman J, Foisner R (2006) Lamins and emerin in muscular distrophy. In: Winder SJ (ed) Molecular Mechanisms of Muscular Distrophy. Eureka/Landes Bioscience, New Yoork, pp 142–159
Gruenbaum Y, Foisner R (2015) Lamins: nuclear intermediate filament proteins with fundamental functions in nuclear mechanics and genome regulation. Annu Rev Biochem 84:131–164. doi:10.1146/annurev-biochem-060614-034115
Halaschek-Wiener J, Brooks-Wilson A (2007) Progeria of stem cells: stem cell exhaustion in Hutchinson–Gilford progeria syndrome. J Gerontol A Biol Sci Med Sci 62(1):3–8
Hanker AB, Mitin N, Wilder RS, Henske EP, Tamanoi F, Cox AD, Der CJ (2010) Differential requirement of CAAX-mediated posttranslational processing for Rheb localization and signaling. Oncogene 29(3):380–391. doi:10.1038/onc.2009.336
Harten IA, Zahr RS, Lemire JM, Machan JT, Moses MA, Doiron RJ, Curatolo AS, Rothman FG, Wight TN, Toole BP, Gordon LB (2011) Age-dependent loss of MMP-3 in Hutchinson–Gilford progeria syndrome. J Gerontol A Biol Sci Med Sci 66(11):1201–1207. doi:10.1093/gerona/glr137
Hernandez L, Roux KJ, Wong ES, Mounkes LC, Mutalif R, Navasankari R, Rai B, Cool S, Jeong JW, Wang H, Lee HS, Kozlov S, Grunert M, Keeble T, Jones CM, Meta MD, Young SG, Daar IO, Burke B, Perantoni AO, Stewart CL (2010) Functional coupling between the extracellular matrix and nuclear lamina by Wnt signaling in progeria. Dev Cell 19(3):413–425. doi:10.1016/j.devcel.2010.08.013
Herrmann H, Bar H, Kreplak L, Strelkov SV, Aebi U (2007) Intermediate filaments: from cell architecture to nanomechanics. Nat Rev Mol Cell Biol 8(7):562–573. doi:10.1038/nrm2197
Ho CY, Lammerding J (2012) Lamins at a glance. J Cell Sci 125(9):2087–2093. doi:10.1242/Jcs.087288
Hoeijmakers JH (2009) DNA damage, aging, and cancer. N Engl J Med 361(15):1475–1485. doi:10.1056/NEJMra0804615
Hori K, Sen A, Artavanis-Tsakonas S (2013) Notch signaling at a glance. J Cell Sci 126(Pt 10):2135–2140. doi:10.1242/jcs.127308
Howitz KT, Bitterman KJ, Cohen HY, Lamming DW, Lavu S, Wood JG, Zipkin RE, Chung P, Kisielewski A, Zhang LL, Scherer B, Sinclair DA (2003) Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature 425(6954):191–196. doi:10.1038/nature01960
Humphrey JD, Dufresne ER, Schwartz MA (2014) Mechanotransduction and extracellular matrix homeostasis. Nat Rev Mol Cell Biol 15(12):802–812. doi:10.1038/nrm3896
Ivorra C, Kubicek M, Gonzalez JM, Sanz-Gonzalez SM, Alvarez-Barrientos A, O’Connor JE, Burke B, Andres V (2006) A mechanism of AP-1 suppression through interaction of c-Fos with lamin A/C. Genes Dev 20(6):747 (vol 20, p 307
Jacob KN, Baptista F, dos Santos HG, Oshima J, Agarwal AK, Garg A (2005) Phenotypic heterogeneity in body fat distribution in patients with atypical Werner’s Syndrome due to heterozygous Arg133Leu lamin A/C mutation. J Clin Endocrinol Metab 90(12):6699–6706. doi:10.1210/jc.2005-0939
Johnson BR, Nitta RT, Frock RL, Mounkes L, Barbie DA, Stewart CL, Harlow E, Kennedy BK (2004) A-type lamins regulate retinoblastoma protein function by promoting subnuclear localization and preventing proteasomal degradation. Proc Natl Acad Sci USA 101(26):9677–9682. doi:10.1073/pnas.0403250101
Johnson SC, Rabinovitch PS, Kaeberlein M (2013) mTOR is a key modulator of ageing and age-related disease. Nature 493(7432):338–345. doi:10.1038/nature11861
Jung CH, Ro SH, Cao J, Otto NM, Kim DH (2010) mTOR regulation of autophagy. FEBS Lett 584(7):1287–1295. doi:10.1016/j.febslet.2010.01.017
Kane MS, Lindsay ME, Judge DP, Barrowman J, Ap Rhys C, Simonson L, Dietz HC, Michaelis S (2013) LMNA-associated cardiocutaneous progeria: an inherited autosomal dominant premature aging syndrome with late onset. Am J Med Genet A 161A(7):1599–1611. doi:10.1002/ajmg.a.35971
Kawahara TL, Michishita E, Adler AS, Damian M, Berber E, Lin M, McCord RA, Ongaigui KC, Boxer LD, Chang HY, Chua KF (2009) SIRT6 links histone H3 lysine 9 deacetylation to NF-κB-dependent gene expression and organismal life span. Cell 136(1):62–74. doi:10.1016/j.cell.2008.10.052
Kieran MW, Gordon L, Kleinman M (2007) New approaches to progeria. Pediatrics 120(4):834–841. doi:10.1542/peds.2007-1356
Kind J, Pagie L, Ortabozkoyun H, Boyle S, de Vries SS, Janssen H, Amendola M, Nolen LD, Bickmore WA, van Steensel B (2013) Single-cell dynamics of genome-nuclear lamina interactions. Cell 153(1):178–192. doi:10.1016/j.cell.2013.02.028
Kirschner J, Brune T, Wehnert M, Denecke J, Wasner C, Feuer A, Marquardt T, Ketelsen UP, Wieacker P, Bonnemann CG, Korinthenberg R (2005) p. S143F mutation in lamin A/C: a new phenotype combining myopathy and progeria. Ann Neurol 57(1):148–151. doi:10.1002/ana.20359
Kolb T, Maass K, Hergt M, Aebi U, Herrmann H (2011) Lamin A and lamin C form homodimers and coexist in higher complex forms both in the nucleoplasmic fraction and in the lamina of cultured human cells. Nucleus 2(5):425–433. doi:10.4161/nucl.2.5.17765
Korfali N, Wilkie GS, Swanson SK, Srsen V, de Las Heras J, Batrakou DG, Malik P, Zuleger N, Kerr AR, Florens L, Schirmer EC (2012) The nuclear envelope proteome differs notably between tissues. Nucleus 3(6):552–564. doi:10.4161/nucl.22257
Kubben N, Voncken JW, Demmers J, Calis C, van Almen G, Pinto Y, Misteli T (2010a) Identification of differential protein interactors of lamin A and progerin. Nucleus 1(6):513–525. doi:10.4161/nucl.1.6.13512
Kubben N, Voncken JW, Misteli T (2010b) Mapping of protein- and chromatin-interactions at the nuclear lamina. Nucleus 1(6):460–471. doi:10.4161/nucl.1.6.13513
Kubben N, Adriaens M, Meuleman W, Voncken JW, van Steensel B, Misteli T (2012) Mapping of lamin A- and progerin-interacting genome regions. Chromosoma 121(5):447–464. doi:10.1007/s00412-012-0376-7
Kubben N, Brimacombe KR, Donegan M, Li Z, Misteli T (2015) A high-content imaging-based screening pipeline for the systematic identification of anti-progeroid compounds. Methods. doi:10.1016/j.ymeth.2015.08.024
Kudlow BA, Kennedy BK, Monnat RJ Jr (2007) Werner and Hutchinson–Gilford progeria syndromes: mechanistic basis of human progeroid diseases. Nat Rev Mol Cell Biol 8(5):394–404. doi:10.1038/nrm2161
Lavasani M, Robinson AR, Lu A, Song M, Feduska JM, Ahani B, Tilstra JS, Feldman CH, Robbins PD, Niedernhofer LJ, Huard J (2012) Muscle-derived stem/progenitor cell dysfunction limits healthspan and lifespan in a murine progeria model. Nat Commun 3:608. doi:10.1038/ncomms1611
Lavu S, Boss O, Elliott PJ, Lambert PD (2008) Sirtuins–novel therapeutic targets to treat age-associated diseases. Nat Rev Drug Discov 7(10):841–853. doi:10.1038/nrd2665
Liu BH, Wang JM, Chan KM, Tjia WM, Deng W, Guan XY, Huang JD, Li KM, Chau PY, Chen DJ, Pei DQ, Pendas AM, Cadinanos J, Lopez-Otin C, Tse HF, Hutchison C, Chen JJ, Cao YH, Cheah KSE, Tryggvason K, Zhou ZJ (2005) Genomic instability in laminopathy-based premature aging. Nat Med 11(7):780–785. doi:10.1038/nm1266
Liu B, Ghosh S, Yang X, Zheng H, Liu X, Wang Z, Jin G, Zheng B, Kennedy BK, Suh Y, Kaeberlein M, Tryggvason K, Zhou Z (2012) Resveratrol rescues SIRT1-dependent adult stem cell decline and alleviates progeroid features in laminopathy-based progeria. Cell Metab 16(6):738–750. doi:10.1016/j.cmet.2012.11.007
Liu B, Wang Z, Zhang L, Ghosh S, Zheng H, Zhou Z (2013) Depleting the methyltransferase Suv39h1 improves DNA repair and extends lifespan in a progeria mouse model. Nat Commun 4:1868. doi:10.1038/ncomms2885
Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G (2013) The hallmarks of aging. Cell 153(6):1194–1217. doi:10.1016/j.cell.2013.05.039
Lund E, Oldenburg AR, Delbarre E, Freberg CT, Duband-Goulet I, Eskeland R, Buendia B, Collas P (2013) Lamin A/C-promoter interactions specify chromatin state-dependent transcription outcomes. Genome Res 23(10):1580–1589. doi:10.1101/gr.159400.113
Lund EG, Duband-Goulet I, Oldenburg A, Buendia B, Collas P (2015) Distinct features of lamin A-interacting chromatin domains mapped by ChIP-sequencing from sonicated or micrococcal nuclease-digested chromatin. Nucleus 6(1):30–39. doi:10.4161/19491034.2014.990855
Madeo F, Tavernarakis N, Kroemer G (2010) Can autophagy promote longevity? Nat Cell Biol 12(9):842–846. doi:10.1038/ncb0910-842
Manju K, Muralikrishna B, Parnaik VK (2006) Expression of disease-causing lamin A mutants impairs the formation of DNA repair foci. J Cell Sci 119(13):2704–2714. doi:10.1242/jcs.03009
Maquart FX, Bellon G, Gillery P, Borel JP, Labeille B, Risbourg B, Denoeux JP (1988) Increased secretion of fibronectin and collagen by progeria (Hutchinson–Gilford) fibroblasts. Eur J Pediatr 147(4):442
Margalit A, Brachner A, Gotzmann J, Foisner R, Gruenbaum Y (2007) Barrier-to-autointegration factor—a BAFfling little protein. Trends Cell Biol 17(4):202–208
Marji J, O’Donoghue SI, McClintock D, Satagopam VP, Schneider R, Ratner D, Worman HJ, Gordon LB, Djabali K (2010) Defective lamin A-Rb signaling in Hutchinson–Gilford progeria syndrome and reversal by farnesyltransferase inhibition. PLoS ONE 5(6):e11132. doi:10.1371/journal.pone.0011132
Markiewicz E, Dechat T, Foisner R, Quinlan RA, Hutchison CJ (2002) Lamin A/C binding protein LAP2α is required for nuclear anchorage of retinoblastoma protein. Mol Biol Cell 13(12):4401–4413. doi:10.1091/mbc.E02-07-0450
McClintock D, Ratner D, Lokuge M, Owens DM, Gordon LB, Collins FS, Djabali K (2007) The mutant form of Lamin A that causes Hutchinson–Gilford progeria is a biomarker of cellular aging in human skin. PLoS ONE. doi:10.1371/journal.pone.0001269
McCord RP, Nazario-Toole A, Zhang H, Chines PS, Zhan Y, Erdos MR, Collins FS, Dekker J, Cao K (2013) Correlated alterations in genome organization, histone methylation, and DNA-lamin A/C interactions in Hutchinson–Gilford progeria syndrome. Genome Res 23(2):260–269. doi:10.1101/gr.138032.112
McPherson E, Turner L, Zador I, Reynolds K, Macgregor D, Giampietro PF (2009) Ovarian failure and dilated cardiomyopathy due to a novel lamin mutation. Am J Med Genet A 149A(4):567–572. doi:10.1002/ajmg.a.32627
Meshorer E, Gruenbaum Y (2009) NURD keeps chromatin young. Nat Cell Biol 11(10):1176–1177. doi:10.1038/ncb1009-1176
Meuleman W, Peric-Hupkes D, Kind J, Beaudry JB, Pagie L, Kellis M, Reinders M, Wessels L, van Steensel B (2013) Constitutive nuclear lamina–genome interactions are highly conserved and associated with A/T-rich sequence. Genome Res 23(2):270–280. doi:10.1101/gr.141028.112
Miller JD, Ganat YM, Kishinevsky S, Bowman RL, Liu B, Tu EY, Mandal PK, Vera E, Shim JW, Kriks S, Taldone T, Fusaki N, Tomishima MJ, Krainc D, Milner TA, Rossi DJ, Studer L (2013) Human iPSC-based modeling of late-onset disease via progerin-induced aging. Cell Stem Cell 13(6):691–705. doi:10.1016/j.stem.2013.11.006
Milne JC, Lambert PD, Schenk S, Carney DP, Smith JJ, Gagne DJ, Jin L, Boss O, Perni RB, Vu CB, Bemis JE, Xie R, Disch JS, Ng PY, Nunes JJ, Lynch AV, Yang HY, Galonek H, Israelian K, Choy W, Iffland A, Lavu S, Medvedik O, Sinclair DA, Olefsky JM, Jirousek MR, Elliott PJ, Westphal CH (2007) Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature 450(7170):712–716. doi:10.1038/nature06261
Moir RD, Yoon M, Khuon S, Goldman RD (2000) Nuclear lamins A and B1: different pathways of assembly during nuclear envelope formation in living cells. J Cell Biol 151(6):1155–1168
Moisan E, Girard D (2006) Cell surface expression of intermediate filament proteins vimentin and lamin B1 in human neutrophil spontaneous apoptosis. J Leukoc Biol 79(3):489–498. doi:10.1189/jlb.0405190
Mory PB, Crispim F, Kasamatsu T, Gabbay MAL, Dib SA, Moises RS (2008) Atypical generalized lipoatrophy and severe insulin resistance due to a heterozygous LMNA p.T101 mutation. Arq Bras Endocrinol Metabol 52(8):1252–1256
Mounkes LC, Kozlov S, Hernandez L, Sullivan T, Stewart CL (2003) A progeroid syndrome in mice is caused by defects in A-type lamins. Nature 423(6937):298–301. doi:10.1038/nature01631
Muchir A, Worman HJ (2010) Signaling defects and the nuclear envelope in progeria. Dev Cell 19(3):355–356. doi:10.1016/j.devcel.2010.08.019
Musich PR, Zou Y (2011) DNA-damage accumulation and replicative arrest in Hutchinson–Gilford progeria syndrome. Biochem Soc Trans 39:1764–1769. doi:10.1042/Bst20110687
Naetar N, Korbei B, Kozlov S, Kerenyi MA, Dorner D, Kral R, Gotic I, Fuchs P, Cohen TV, Bittner R, Stewart CL, Foisner R (2008) Loss of nucleoplasmic LAP2α—lamin A complexes causes erythroid and epidermal progenitor hyperproliferation. Nat Cell Biol 10(11):1341–1348. doi:10.1038/ncb1793
Navarro CL, Cadinanos J, Sandre-Giovannoli AD, Bernard R, Courrier S, Boccaccio I, Boyer A, Kleijer WJ, Wagner A, Giuliano F, Beemer FA, Freije JM, Cau P, Hennekam RCM, Lopez-Otin C, Badens C, Levy N (2005) Loss of ZMPSTE24 (FACE-1) causes autosomal recessive restrictive dermopathy and accumulation of Lamin A precursors. Hum Mol Genet 14(11):1503–1513. doi:10.1093/hmg/ddi159
Navarro CL, Cau P, Levy N (2006) Molecular bases of progeroid syndromes. Hum Mol Genet 15(2):R151–R161. doi:10.1093/hmg/ddl214
Niccoli T, Partridge L (2012) Ageing as a risk factor for disease. Curr Biol 22(17):R741–R752. doi:10.1016/j.cub.2012.07.024
Nitta RT, Smith CL, Kennedy BK (2007) Evidence that proteasome-dependent degradation of the retinoblastoma protein in cells lacking A-type lamins occurs independently of gankyrin and MDM2. PLoS ONE. doi:10.1371/journal.pone.0000963
Olive M, Harten I, Mitchell R, Beers JK, Djabali K, Cao K, Erdos MR, Blair C, Funke B, Smoot L, Gerhard-Herman M, Machan JT, Kutys R, Virmani R, Collins FS, Wight TN, Nabel EG, Gordon LB (2010) Cardiovascular pathology in Hutchinson–Gilford progeria: correlation with the vascular pathology of aging. Arterioscler Thromb Vasc Biol 30(11):2301–2309. doi:10.1161/ATVBAHA.110.209460
Osmanagic-Myers S, Dechat T, Foisner R (2015) Lamins at the crossroads of mechanosignaling. Genes Dev 29(3):225–237. doi:10.1101/gad.255968.114
Osorio FG, Navarro CL, Cadinanos J, Lopez-Mejia IC, Quiros PM, Bartoli C, Rivera J, Tazi J, Guzman G, Varela I, Depetris D, de Carlos F, Cobo J, Andres V, De Sandre-Giovannoli A, Freije JM, Levy N, Lopez-Otin C (2011) Splicing-directed therapy in a new mouse model of human accelerated aging. Sci Transl Med 3(106):106ra107. doi:10.1126/scitranslmed.3002847
Osorio FG, Lopez-Otin C, Freije JM (2012) NF-kB in premature aging. Aging (Albany NY) 4(11):726–727
Pegoraro G, Kubben N, Wickert U, Gohler H, Hoffmann K, Misteli T (2009) Ageing-related chromatin defects through loss of the NURD complex. Nat Cell Biol 11(10):1261–1267. doi:10.1038/ncb1971
Pekovic V, Harborth J, Broers JL, Ramaekers FC, van Engelen B, Lammens M, von Zglinicki T, Foisner R, Hutchison C, Markiewicz E (2007) Nucleoplasmic LAP2α—lamin A complexes are required to maintain a proliferative state in human fibroblasts. J Cell Biol 176(2):163–172. doi:10.1083/jcb.200606139
Pellegrini C, Columbaro M, Capanni C, D’Apice MR, Cavallo C, Murdocca M, Lattanzi G, Squarzoni S (2015) All-trans retinoic acid and rapamycin normalize Hutchinson–Gilford progeria fibroblast phenotype. Oncotarget 6(30):29914–29928. doi:10.18632/oncotarget.4939
Pendas AM, Zhou Z, Cadinanos J, Freije JM, Wang J, Hultenby K, Astudillo A, Wernerson A, Rodriguez F, Tryggvason K, Lopez-Otin C (2002) Defective prelamin A processing and muscular and adipocyte alterations in Zmpste24 metalloproteinase-deficient mice. Nat Genet 31(1):94–99. doi:10.1038/ng871
Pereira S, Bourgeois P, Navarro C, Esteves-Vieira V, Cau P, De Sandre-Giovannoli A, Levy N (2008) HGPS and related premature aging disorders: from genomic identification to the first therapeutic approaches. Mech Ageing Dev 129(7–8):449–459. doi:10.1016/j.mad.2008.04.003
Peric-Hupkes D, Meuleman W, Pagie L, Bruggeman SW, Solovei I, Brugman W, Graf S, Flicek P, Kerkhoven RM, van Lohuizen M, Reinders M, Wessels L, van Steensel B (2010) Molecular maps of the reorganization of genome-nuclear lamina interactions during differentiation. Mol Cell 38(4):603–613. doi:10.1016/j.molcel.2010.03.016
Philip JT, Dahl KN (2008) Nuclear mechanotransduction: response of the lamina to extracellular stress with implications in aging. J Biomech 41(15):3164–3170. doi:10.1016/j.jbiomech.2008.08.024
Prokocimer M, Barkan R, Gruenbaum Y (2013) Hutchinson–Gilford progeria syndrome through the lens of transcription. Aging Cell 12(4):533–543. doi:10.1111/acel.12070
Renard D, Fourcade G, Milhaud D, Bessis D, Esteves-Vieira V, Boyer A, Roll P, Bourgeois P, Levy N, De Sandre-Giovannoli A (2009) Novel LMNA mutation in atypical Werner syndrome presenting with ischemic disease. Stroke 40(2):e11–e14. doi:10.1161/STROKEAHA.108.531780
Richards SA, Muter J, Ritchie P, Lattanzi G, Hutchison CJ (2011) The accumulation of un-repairable DNA damage in laminopathy progeria fibroblasts is caused by ROS generation and is prevented by treatment with N-acetyl cysteine. Hum Mol Genet 20(20):3997–4004. doi:10.1093/hmg/ddr327
Ronningen T, Shah A, Oldenburg AR, Vekterud K, Delbarre E, Moskaug JO, Collas P (2015) Prepatterning of differentiation-driven nuclear lamin A/C-associated chromatin domains by GlcNAcylated histone H2B. Genome Res 25(12):1825–1835. doi:10.1101/gr.193748.115
Rosengardten Y, McKenna T, Grochova D, Eriksson M (2011) Stem cell depletion in Hutchinson–Gilford progeria syndrome. Aging Cell 10(6):1011–1020. doi:10.1111/j.1474-9726.2011.00743.x
Rusinol AE, Sinensky MS (2006) Farnesylated lamins, progeroid syndromes and farnesyl transferase inhibitors. J Cell Sci 119(Pt 16):3265–3272. doi:10.1242/jcs.03156
Sagelius H, Rosengardten Y, Hanif M, Erdos MR, Rozell B, Collins FS, Eriksson M (2008) Targeted transgenic expression of the mutation causing Hutchinson–Gilford progeria syndrome leads to proliferative and degenerative epidermal disease. J Cell Sci 121(Pt 7):969–978. doi:10.1242/jcs.022913
Sahin E, Depinho RA (2010) Linking functional decline of telomeres, mitochondria and stem cells during ageing. Nature 464(7288):520–528. doi:10.1038/nature08982
Scaffidi P, Misteli T (2005) (R)eversal of the cellular phenotype in the premature aging disease Hutchinson–Gilford progeria syndrome. Nat Med 11(4):440–445. doi:10.1038/nm1204
Scaffidi P, Misteli T (2006) Lamin A-dependent nuclear defects in human aging. Science 312(5776):1059–1063. doi:10.1126/science.1127168
Scaffidi P, Misteli T (2008) Lamin A-dependent misregulation of adult stem cells associated with accelerated ageing. Nat Cell Biol 10(4):452–459. doi:10.1038/ncb1708
Schmidt E, Nilsson O, Koskela A, Tuukkanen J, Ohlsson C, Rozell B, Eriksson M (2012) Expression of the Hutchinson–Gilford progeria mutation during osteoblast development results in loss of osteocytes, irregular mineralization, and poor biomechanical properties. J Biol Chem 287(40):33512–33522. doi:10.1074/jbc.M112.366450
Shimi T, Pfleghaar K, Kojima S, Pack CG, Solovei I, Goldman AE, Adam SA, Shumaker DK, Kinjo M, Cremer T, Goldman RD (2008) The A- and B-type nuclear lamin networks: microdomains involved in chromatin organization and transcription. Genes Dev 22(24):3409–3421. doi:10.1101/gad.1735208
Shumaker DK, Dechat T, Kohlmaier A, Adam SA, Bozovsky MR, Erdos MR, Eriksson M, Goldman AE, Khuon S, Collins FS, Jenuwein T, Goldman RD (2006) Mutant nuclear lamin A leads to progressive alterations of epigenetic control in premature aging. Proc Natl Acad Sci USA 103(23):8703–8708. doi:10.1073/pnas.0602569103
Solovei I, Wang AS, Thanisch K, Schmidt CS, Krebs S, Zwerger M, Cohen TV, Devys D, Foisner R, Peichl L, Herrmann H, Blum H, Engelkamp D, Stewart CL, Leonhardt H, Joffe B (2013) LBR and lamin A/C sequentially tether peripheral heterochromatin and inversely regulate differentiation. Cell 152(3):584–598. doi:10.1016/j.cell.2013.01.009
Song M, San H, Anderson SA, Cannon RO 3rd, Orlic D (2014) Shear stress-induced mechanotransduction protein deregulation and vasculopathy in a mouse model of progeria. Stem Cell Res Ther 5(2):41. doi:10.1186/scrt429
Stehbens WE, Wakefield SJ, Gilbert-Barness E, Olson RE, Ackerman J (1999) Histological and ultrastructural features of atherosclerosis in progeria. Cardiovasc Pathol 8(1):29–39
Strandgren C, Nasser HA, McKenna T, Koskela A, Tuukkanen J, Ohlsson C, Rozell B, Eriksson M (2015) Transgene silencing of the Hutchinson–Gilford progeria syndrome mutation results in a reversible bone phenotype, whereas resveratrol treatment does not show overall beneficial effects. FASEB J 29(8):3193–3205. doi:10.1096/fj.14-269217
Swift J, Harada T, Buxboim A, Shin JW, Tang HY, Speicher DW, Discher DE (2013) Label-free mass spectrometry exploits dozens of detected peptides to quantify lamins in wildtype and knockdown cells. Nucleus 4(6):450–459. doi:10.4161/nucl.27413
Tanida I, Ueno T, Kominami E (2008) LC3 and autophagy. Methods Mol Biol 445:77–88. doi:10.1007/978-1-59745-157-4_4
Tilstra JS, Robinson AR, Wang J, Gregg SQ, Clauson CL, Reay DP, Nasto LA, St Croix CM, Usas A, Vo N, Huard J, Clemens PR, Stolz DB, Guttridge DC, Watkins SC, Garinis GA, Wang Y, Niedernhofer LJ, Robbins PD (2012) NF-κB inhibition delays DNA damage-induced senescence and aging in mice. J Clin Invest 122(7):2601–2612. doi:10.1172/JCI45785
Toth JI, Yang SH, Qiao X, Beigneux AP, Gelb MH, Moulson CL, Miner JH, Young SG, Fong LG (2005) Blocking protein farnesyltransferase improves nuclear shape in fibroblasts from humans with progeroid syndromes. Proc Natl Acad Sci USA 102(36):12873–12878. doi:10.1073/pnas.0505767102
Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE, Bohlooly-Y M, Gidlof S, Oldfors A, Wibom R, Tornell J, Jacobs HT, Larsson NG (2004) Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 429(6990):417–423. doi:10.1038/nature02517
Varela I, Cadinanos J, Pendas AM, Gutierrez-Fernandez A, Folgueras AR, Sanchez LM, Zhou Z, Rodriguez FJ, Stewart CL, Vega JA, Tryggvason K, Freije JM, Lopez-Otin C (2005) Accelerated ageing in mice deficient in Zmpste24 protease is linked to p53 signalling activation. Nature 437(7058):564–568. doi:10.1038/nature04019
Varela I, Pereira S, Ugalde AP, Navarro CL, Suarez MF, Cau P, Cadinanos J, Osorio FG, Foray N, Cobo J, de Carlos F, Levy N, Freije JMP, Lopez-Otin C (2008) Combined treatment with statins and aminobisphosphonates extends longevity in a mouse model of human premature aging. Nat Med 14(7):767–772. doi:10.1038/nm1786
Varga R, Eriksson M, Erdos MR, Olive M, Harten I, Kolodgie F, Capell BC, Cheng J, Faddah D, Perkins S, Avallone H, San H, Qu X, Ganesh S, Gordon LB, Virmani R, Wight TN, Nabel EG, Collins FS (2006) Progressive vascular smooth muscle cell defects in a mouse model of Hutchinson–Gilford progeria syndrome. Proc Natl Acad Sci USA 103(9):3250–3255. doi:10.1073/pnas.0600012103
Verstraeten VLRM, Broers JLV, Ramaekers FCS, van Steensel MAM (2007) The nuclear envelope, a key structure in cellular integrity and gene expression. Curr Med Chem 14(11):1231–1248
Verstraeten VL, Ji JY, Cummings KS, Lee RT, Lammerding J (2008) Increased mechanosensitivity and nuclear stiffness in Hutchinson–Gilford progeria cells: effects of farnesyltransferase inhibitors. Aging Cell 7(3):383–393. doi:10.1111/j.1474-9726.2008.00382.x
Vidak S, Kubben N, Dechat T, Foisner R (2015) Proliferation of progeria cells is enhanced by lamina-associated polypeptide 2α (LAP2α) through expression of extracellular matrix proteins. Genes Dev 29(19):2022–2036. doi:10.1101/gad.263939.115
Wenzel V, Roedl D, Gabriel D, Gordon LB, Herlyn M, Schneider R, Ring J, Djabali K (2012) Naive adult stem cells from patients with Hutchinson–Gilford progeria syndrome express low levels of progerin in vivo. Biol Open 1(6):516–526. doi:10.1242/bio.20121149
Wilson KL, Foisner R (2010) Lamin-binding proteins. Cold Spring Harb Perspect Biol 2(4):a000554. doi:10.1101/cshperspect.a000554
Wood JG, Rogina B, Lavu S, Howitz K, Helfand SL, Tatar M, Sinclair D (2004) Sirtuin activators mimic caloric restriction and delay ageing in metazoans. Nature 431(7004):107. doi:10.1038/nature02941 (vol 430, p 686, 2004)
Worman HJ (2012) Nuclear lamins and laminopathies. J Pathol 226(2):316–325. doi:10.1002/path.2999
Worman HJ, Schirmer EC (2015) Nuclear membrane diversity: underlying tissue-specific pathologies in disease? Curr Opin Cell Biol 34:101–112. doi:10.1016/j.ceb.2015.06.003
Yang SH, Bergo MO, Toth JI, Qiao X, Hu Y, Sandoval S, Meta M, Bendale P, Gelb MH, Young SG, Fong LG (2005) Blocking protein farnesyltransferase improves nuclear blebbing in mouse fibroblasts with a targeted Hutchinson-Gilford progeria syndrome mutation. Proc Natl Acad Sci USA 102(29):10291–10296. doi:10.1073/pnas.0504641102
Yang SH, Andres DA, Spielmann HP, Young SG, Fong LG (2008) Progerin elicits disease phenotypes of progeria in mice whether or not it is farnesylated. J Clin Invest 118(10):3291–3300. doi:10.1172/JCI35876
Yang KE, Kwon J, Rhim JH, Choi JS, Kim SI, Lee SH, Park J, Jang IS (2011a) Differential expression of extracellular matrix proteins in senescent and young human fibroblasts: a comparative proteomics and microarray study. Mol Cells 32(1):99–106. doi:10.1007/s10059-011-0064-0
Yang SH, Chang SY, Ren S, Wang Y, Andres DA, Spielmann HP, Fong LG, Young SG (2011b) Absence of progeria-like disease phenotypes in knock-in mice expressing a non-farnesylated version of progerin. Hum Mol Genet 20(3):436–444. doi:10.1093/hmg/ddq490
Young SG, Fong LG, Michaelis S (2005) Prelamin A, Zmpste24, misshapen cell nuclei, and progeria–new evidence suggesting that protein farnesylation could be important for disease pathogenesis. J Lipid Res 46(12):2531–2558. doi:10.1194/jlr.R500011-JLR200
Zhang J, Lian Q, Zhu G, Zhou F, Sui L, Tan C, Mutalif RA, Navasankari R, Zhang Y, Tse HF, Stewart CL, Colman A (2011) A human iPSC model of Hutchinson–Gilford progeria reveals vascular smooth muscle and mesenchymal stem cell defects. Cell Stem Cell 8(1):31–45. doi:10.1016/j.stem.2010.12.002
Zhang S, Schones DE, Malicet C, Rochman M, Zhou M, Foisner R, Bustin M (2013) High mobility group protein N5 (HMGN5) and lamina-associated polypeptide 2α (LAP2α) interact and reciprocally affect their genome-wide chromatin organization. J Biol Chem 288(25):18104–18109. doi:10.1074/jbc.C113.469544
Acknowledgments
Work in the authors laboratory was supported by Grants from the Austrian Science Fund (FWF Grant P26492-B20), the Progeria Research Foundation (Innovator Award, PRF2011-37) and the doctoral program “Molecular mechanisms of Cell Signalling” funded by Austrian Science Fund (FWF, DK W1220).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Vidak, S., Foisner, R. Molecular insights into the premature aging disease progeria. Histochem Cell Biol 145, 401–417 (2016). https://doi.org/10.1007/s00418-016-1411-1
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00418-016-1411-1