
Abstract
Desmosomes are patch-like intercellular adhering junctions (“maculae adherentes”), which, in concert with the related adherens junctions, provide the mechanical strength to intercellular adhesion. Therefore, it is not surprising that desmosomes are abundant in tissues subjected to significant mechanical stress such as stratified epithelia and myocardium. Desmosomal adhesion is based on the Ca2+-dependent, homo- and heterophilic transinteraction of cadherin-type adhesion molecules. Desmosomal cadherins are anchored to the intermediate filament cytoskeleton by adaptor proteins of the armadillo and plakin families. Desmosomes are dynamic structures subjected to regulation and are therefore targets of signalling pathways, which control their molecular composition and adhesive properties. Moreover, evidence is emerging that desmosomal components themselves take part in outside-in signalling under physiologic and pathologic conditions. Disturbed desmosomal adhesion contributes to the pathogenesis of a number of diseases such as pemphigus, which is caused by autoantibodies against desmosomal cadherins. Beside pemphigus, desmosome-associated diseases are caused by other mechanisms such as genetic defects or bacterial toxins. Because most of these diseases affect the skin, desmosomes are interesting not only for cell biologists who are inspired by their complex structure and molecular composition, but also for clinical physicians who are confronted with patients suffering from severe blistering skin diseases such as pemphigus. To develop disease-specific therapeutic approaches, more insights into the molecular composition and regulation of desmosomes are required.
Similar content being viewed by others


Introduction
Desmosomes are intercellular adhering junctions serving to attach neighbouring cells to each other. They are most numerous in tissues subjected to significant mechanical stress such as the stratified squamous epithelia of the skin (Bizzozero 1864) and of mucous membranes (Farquhar and Palade 1963) as well as the myocardium (Fawcett and Selby 1958). Moreover, desmosomes are found in simple epithelia and in non-epithelial cells such as the meningeal cells of the arachnoidea (Gusek 1962) and the follicular dendritic cells of lymph follicles (Swartzendruber 1965).
Desmosomes were discovered as cell contacts in the middle of the nineteenth century (Calkins and Setzer 2007). By the means of light microscopy, desmosomes were first described in the epidermis by the Italian pathologist Bizzozero in (1864). In his histology text book, the anatomist Josef Schaffer from Vienna introduced the term “desmosome”, by combining the greek words “desmos” (bond) and “soma” (body) although, to that time, he, like most others in the field believed that desmosomes were cytoplasm-filled intercellular bridges (Schaffer 1920). It took almost another century until Keith Porter, using electron microscopy, was able to confirm the basic observation of Bizzozero that desmosomes rather are contacts between adjacent cells and to allow the first description on desmosome ultrastructure (Porter 1956). With these new technical advances at hand, several studies were performed in the following years on the distribution and organization of desmosomes in various tissues. In addition, starting in the 1970s, biochemical approaches and molecular cloning techniques were applied to identify the desmosomal components and to characterize their interactions (Drochmans et al. 1978; Moll et al. 1986; Moll and Franke 1982; Schwarz et al. 1990; Skerrow and Matoltsy 1974).
Significant insights into the regulation of desmosomal adhesion also came from the field of dermatology since it was demonstrated that autoantibodies in patients suffering from the autoimmune blistering skin diseases pemphigus vulgaris (PV), and pemphigus foliaceus (PF), are directed to Ca2+-sensitive cell surface proteins within desmosomes (Eyre and Stanley 1987, 1988; Jones et al. 1986b; Karpati et al. 1993), which were identified as the desmosomal cadherins desmoglein 1 (Dsg 1) and Dsg 3 (Amagai et al. 1991; Koulu et al. 1984). The term “pemphigus” comes from the greek word “pemphix” (blister) and is being used in dermatology since 1791 (Schmidt et al. 2000), long before it was found that pemphigus is associated with autoantibodies against keratinocyte surface antigens (Beutner and Jordon 1964) and that these antibodies are sufficient to cause acantholysis, i.e. loss of cell–cell adhesion, in human skin in vivo and in vitro (Anhalt et al. 1982; Schiltz and Michel 1976). The final break-through was the finding that autoantibodies against the extracellular domains of Dsg 3 and Dsg 1 in PV and in PF are pathogenic (Amagai et al. 1995, 1994a, 1992). Therefore, autoantibodies from pemphigus patients have been used to characterize the mechanisms involved in the regulation of desmosomal adhesion. Except from pemphigus, other diseases in which desmosomal adhesion is altered by mutations or bacterial toxins helped to elucidate the functional role of the different desmosomal components.
During the last past several years, a number of comprehensive reviews have been published on both desmosome structure and function (Dusek et al. 2007b; Garrod et al. 2002; Getsios et al. 2004b; Green and Simpson 2007; Holthofer et al. 2007; Kitajima 2002; Kottke et al. 2006; Muller et al. 2008a; Yin and Green 2004) and/or on the mechanisms involved in pemphigus pathogenesis (Amagai 2003; Hashimoto 2003; Lanza et al. 2006; Payne et al. 2004; Sharma et al. 2007; Sitaru and Zillikens 2005; Stanley and Amagai 2006), which indicates that the perspective of the existing model of the desmosome and its role in pemphigus pathogenesis are constantly reshaped. Moreover, because even textbook knowledge such as on the molecular composition of myocardial intercalated discs needs revision (Borrmann et al. 2006; Franke et al. 2006), it becomes obvious that after almost 150 years of desmosome research, our knowledge is still far from complete. This article focuses on the mechanisms regulating desmosomal adhesion, which are compromised in diseases such as pemphigus.
The ultrastructure and composition of desmosomes
The first detailed analysis of desmosome ultrastructure was provided by Odland (1958). Desmosomes are discoid junctions with a diameter of about 0.2–0.5 μm and are composed of two electron-dense plaques in each of the two cells which are separated by an intercellular cleft of 24–30 nm (Figs. 1, 2) (Farquhar and Palade 1963; Odland 1958). Within the plaques, an outer dense plaque can be separated from a less dense inner plaque, the latter of which is linked to loops of intermediate filament bundles (Kelly 1966). Desmosomes contain members of at least three protein families. Desmosomal cadherins form the intercellular adhesive interface, whereas armadillo and plakin family proteins built up the plaques. It is believed that the cytoplasmic tail of Dsgs and Dscs interact with plakoglobin which in turn binds to desmoplakin (Fig. 1). Desmoplakin finally is anchored to the intermediate filament cytoskeleton (Green and Simpson 2007). These interactions seem to be stabilized laterally by plakophilin (Hatzfeld 2007).

Molecular model of the desmosome. The desmosomal cadherins desmoglein and desmocollin undergo homophilic and heterophilic binding via interaction with the amino-terminal extracellular (EC) 1 domain of partner molecules on the same (cis) as well as on the neighbouring cell (trans). The cytoplasmic domains are largely embedded in the outer dense plaque (ODP) where they are associated with plakoglobin and plakophilin. In the inner dense plaque (IDP), desmoplakin links these adaptor molecules to the intermediate filament cytoskeleton

Ultrastructure of the desmosome. The electron micrograph of a keratinocyte desmosome shows the desmosomal plaque with inserting cytokeratin intermediate filaments as well as some fuzzy material within the extracellular space likely reflecting the extracellular domains of desmosomal cadherins
Desmosomes and desmosome-like junctions
Adhering junctions are divided into two main forms: (1) desmosomes, which serve as anchoring structures for intermediate filaments to desmosomal cadherins, and (2) adherens junctions, which contain cell-type specific adhesion molecules from the cadherin super-family that are linked to the actin cytoskeleton. Both, desmosomes and adherens junctions can be found as constituents of more elaborated cell contact complexes. Moreover, chimeric cell contacts exist which share features of both adherens junctions and desmosomes.
Junctional complexes
Polarized epithelial cells display junctional complexes located at the uppermost section of the baso-lateral membrane. In apico-basal direction, the complex is composed of the zonula occludens (tight junction), the zonula adherens and a desmosome (macula adherens) (Farquhar and Palade 1963). Accompanied by a line of separated desmosomes, the zonula occludens and the zonula adherens span the entire cell by forming continuous junction belts. These junctional complexes are regarded as hallmarks of polarized epithelial cells but differ in terms of size and ultrastructure in cell type-specific manner.
Area composita
The intercalated discs of the myocardium also consist of three types of cell junctions, i.e. adherens junctions, desmosomes and gap junctions (Fig. 3). Although “transitional forms” between adherens junctions and desmosomes were described in the very beginning (Fawcett and Selby 1958), most morphological studies regarded the intercalated discs to be composed of separated desmosomes and adherens junctions, the former linked to the desmin type intermediate filament cytoskeleton and the latter to actin filaments of the myofibrills (Fig. 3) (Fawcett and McNutt 1969; McNutt and Fawcett 1969; Shimada et al. 2004). This view seemed to be supported by immuno-localization studies, which showed that desmoplakin as a desmosomal marker and the myocardial adherens junction protein N-cadherin displayed mutually exclusive spatial distribution patterns (Angst et al. 1997; Gutstein et al. 2003). However, a recent comprehensive set of studies unequivocally demonstrated that the desmosomal components desmoglein 2 (Dsg 2), desmocollin 2 (Dsc 2), desmoplakin and plakophilin-2 as well as the adherens junction components N-cadherin, cadherin-11, α-catenin and β-catenin, afadin, vinculin and ZO-1 are present in all parts of intercalated discs irrespective of whether their ultrastructure resembles more closely typical desmosomes or adherens junctions (Borrmann et al. 2006; Franke et al. 2006). It seems that the two types of junctions coalesce within the first-year postpartum and that plakophilin 2 is of special importance for junction integrity (Pieperhoff and Franke 2007; Pieperhoff et al. 2008). Therefore, the intercalated discs were now reclassified as “area composita”, a mixed type of adhering junctions.

Ultrastructure of the area composita of a myocardial intercalated disc. The electron micrograph shows an intercalated disc containing a gap junction (GJ) in its longitudinal section as well as an adhering junction with an extensive electron-dense plaque in the section perpendicular to the cellular axis. Note that insertion of actin filaments, which is typical for adherens junctions, is present in some parts of the junction (asterisk) but not in others (hash key). Based on the recent finding that all parts of these adhering junctions contain the same set of desmosomal components, they are now defined as area composita
Complexus adhaerentes and meningeal junctions
Lymphatic endothelial cells in certain lymphatic vessels and in the sinus of lymph nodes form a kind of chimeric cell contact, which contains components from desmosomes (desmoplakin, plakoglobin), adherens junctions (VE-cadherin, α-catenin, β-catenin, afadin) as well as from tight junctions (claudin-5 and ZO-1) (Hammerling et al. 2006; Schmelz and Franke 1993; Schmelz et al. 1994). The ultrastructure of complexus adhaerentes shares features of both adherens junctions and desmosomes. Complexus adhearentes form continuous belt-like junctions similar to adherens junctions of vascular endothelial cells, whereas their junctional plaques are more similar to desmosomes. It was proposed that plakoglobin in these contacts is responsible for recruitment of desmoplakin (Kowalczyk et al. 1998). Recently, in meningeoma cells, a new type of adhering junctions was discovered in which adherens junctions also contained the desmosomal plaque protein plakophilin 2 (Akat et al. 2008).
The desmosomal components
Desmosomal cadherins
The desmosomal members of the cadherin superfamily, desmogleins (Dsg 1–4) and desmocollins (Dsc 1–3), are single-pass transmembrane glycoproteins, which mediate adhesion in Ca2+-dependent manner (Buxton et al. 1993; Garrod et al. 2002; Getsios et al. 2004b; Nollet et al. 2000). Desmosomal adhesion molecules have been first isolated from desmosomal intercellular regions (Gorbsky and Steinberg 1981). Using specific antibodies to localize proteins at the cell surface and to inhibit desmosome formation, Dsc 1 (130 kDa) and Dsc 2 (115 kDa) were shown to be directly involved in cell–cell adhesion (Cowin et al. 1984) and later on were identified to be cadherin family proteins (Collins et al. 1991; Koch et al. 1991b). Similarly, Dsg 1 (165 kDa), Dsg 2 (116 kDa), Dsg 3 (130 kDa) and Dsc 3 (110 kDa) were characterized (Amagai et al. 1991; Arnemann et al. 1991; Jones et al. 1986a; King et al. 1995; Koch et al. 1991a, 1990; Schafer et al. 1994; Schmelz et al. 1986a, b). More recently, Dsg 4 (108 kDa) was found to be the principal Dsg expressed in hair follicles (Kljuic et al. 2003; Whittock and Bower 2003). The genes encoding desmosomal cadherins, which share an amino acid identity of approximately 30–50%, both with each other and with classical cadherins, are all located on chromosome 18 in humans (Cowley et al. 1997). Mouse models revealed that Dsg 2 and Dsc 3 are the most important desmosomal cadherin members because deficiency caused embryonic lethality (Table 1) (Den et al. 2006; Eshkind et al. 2002). Because lethality induced by Dsc 3 deficiency occurred before mature desmosmes were formed, and because Dsg 2 was observed to be also localized outside of desmosomes in embryonal stem cells, non-desmosmal functions of Dsc 3 and Dsg 2 seemed to be responsible in this context. In contrast, ablation of cadherins with more restrictive expression patterns such as Dsc 1 led to localized superficial epidermal acantholysis or mucosal and deep epidermal splitting in traumatized skin accompanied by hair loss in the case of Dsg 3 (Chidgey et al. 2001; Koch et al. 1998). Dsg 4 mutations are followed primarily by defective hair formation (Kljuic et al. 2003).
The structure of desmosomal cadherins
Desmosomal cadherins are type I integral membrane proteins. The amino-terminal extracellular domain of desmosomal cadherins consists of four cadherin repeats (EC1–4) of about 110 amino acids followed by a less related membrane-proximal domain (EC 5) (Dusek et al. 2007b). Based on the crystal structure of classical cadherins (Boggon et al. 2002; Overduin et al. 1995; Shapiro et al. 1995), the EC 1–4 domains are thought to be connected via flexible linkers which are rigidified by binding of up to three Ca2+ ions each (Pertz et al. 1999). In the cytoplasmic domain, a juxtamembranuous anchor (IA) region is located which, at least in the case of Dsc 1a, contains a desmoplakin-binding element (Troyanovsky et al. 1994b) and maybe be involved in binding and trafficking of p120catenin similar to its role in E-cadherin (Miranda et al. 2003). The following cadherin-typical sequence (ICS) is required for binding to plakoglobin (Mathur et al. 1994; Troyanovsky et al. 1994a). For desmocollins, in which a long and short “a” and “b” isoform is generated by alternative splicing (Collins et al. 1991), it has been shown that the ICS domain is lacking in the b isoform which therefore is unable to bind plakoglobin but instead associates with plakophilin 3 (Bonne et al. 2003; Troyanovsky et al. 1993). The functions of the C-terminal proline-rich linker (L), the Dsg-specific repeated unit domains (RUDs) and the desmoglein terminal domain (DTD), which are all only present in desmogleins, are not clear (Dusek et al. 2007b).
The Ca2+-dependency of desmosomal cadherin-mediated binding
It is well established that binding of desmosomal and classical cadherins is strictly Ca2+-dependent (Chitaev and Troyanovsky 1997; Heupel et al. 2007; Pertz et al. 1999; Waschke et al. 2007). For Dsg 1, Ca2+-dependency of homophilic binding has been characterized in more detail. It was found that the Ca2+ concentration for half-maximal binding activity of Dsg 1 is 0.8 mM Ca2+ and that binding is highly cooperative with the Hill coefficient being ≥5 (Waschke et al. 2007). This indicates that Dsg 1 binding is strong only at extracellular Ca2+concentrations higher than 0.8 mM. Although the exact extracellular Ca2+concentration within the epidermis is unknown, it has been shown that a gradient exists with low Ca2+concentrations in the basal layers and high concentrations in the superficial epidermis (Elias et al. 2002; Menon and Elias 1991). Therefore, if homophilic binding of Dsg 1 occurs in vivo, it has to be considered that it may contribute to effective intercellular adhesion only in the superficial epidermis.
Transinteraction mechanisms of desmosmal cadherins
At present, it is still a matter of debate how desmosmal cadherins interact with each other in vivo. However, several lines of evidence indicate that the N-terminal EC 1 domain is important. Similar to classical cadherins, desmosomal cadherins contain a cell adhesion recognition (CAR) site containing a central alanine residue (Blaschuk et al. 1990). However, instead of the conserved tripeptide HAV sequence of classical cadherins, the sequence is YAT for Dsc 1 and RAL for Dsg 1, respectively (Tselepis et al. 1998). Peptides derived from these sequences were able to block homophilic adhesion mediated by Dsg 1 and Dsc 1 and to inhibit desmosomal adhesion in epithelial cells when the peptides for Dsg 1 and Dsc 1 were applied together, indicating that the CAR site in the EC 1 domain is critical for maintenance of desmosmal adhesion (Runswick et al. 2001; Tselepis et al. 1998). This hypothesis is supported by studies in which mechanisms underlying the loss of keratinocyte cohesion in pemphigus were investigated. AK23, a monoclonal Dsg 3 antibody from a PV mouse model directed against the predicted binding motif of the EC 1, has been shown to be pathogenic in vivo whereas antibodies targeting other parts of the Dsg 3 extracellular domain were not (Tsunoda et al. 2003). Together with the recent finding that AK 23 similar to Dsg 3 antibodies from PV patients, which are known to be also primarily directed against the N-terminal part of EC 1 (Sekiguchi et al. 2001), is able to directly interfere with Dsg 3 binding (Heupel et al. 2007), these data demonstrate that interaction of EC 1 is crucial for Dsg 3 binding. In addition to these functional studies, morphologic studies sought to address the mode of desmosomal cadherin interaction within desmosomes by using electron tomography imaging of epidermal tissue (Al-Amoudi et al. 2007; He et al. 2003). Based on predictions from the C-cadherin crystal structure, He and colleagues reported that in mouse epidermis several desmosomal cadherins form knots in which cadherins display stochastic arrangement. In these knots, desmosomal cadherins seemed to interact via their EC 1 domains with both molecules on the same cell in cis as well as with molecules from opposing cells in trans (He et al. 2003). Al-Amoudi and co-workers refined the technique by employing cryo-electron microscopy in human epidermis. They confirmed cis- and trans-interactions of the EC 1 domains, possibly via insertion of the tryptophane 2 into the hydrophobic pocket of the CAR site (Al-Amoudi et al. 2007). However, they found that cis-interactions of the EC 4–5 regions may also occur and that desmosomal cadherins rather show periodically zipper-like arrangements similar to classical cadherins (Boggon et al. 2002).
Homophilic and heterophilic binding of desmosomal cadherins
In contrast to classical cadherins from adherens junctions which primarily bind in homophilic manner, data indicate that desmosomal cadherins undergo both homophilic and heterophilic transinteraction. Using EC 1–2 fragments of Dsg 2 and Dsc 2, it was shown that homophilic binding occurs in vitro (Syed et al. 2002). Similarly, homophilic binding of Dsg 3 was found (Amagai et al. 1994b). When recombinant proteins consisting of the complete extracellular domain were used for atomic force microscopy (AFM) measurements, it was demonstrated that the unbinding force of single homophilically transinteracting molecules was about 37–68 pN (depending on the retrace velocity 300–6,000 nm/s) for Dsg 1 with a lifetime of 0.17 s and about 50 pN for Dsg 3 (Heupel et al. 2007; Waschke et al. 2005, 2007), which is in the same range as observed for other types of cadherins characterized by the same method such as VE-cadherin, N-cadherin or LI-cadherin (Baumgartner et al. 2003, 2000; Wendeler et al. 2007). These data indicate that the molecular binding properties of homophilic adhesion of desmosomal cadherins may be comparable to other cadherins.
Heterophilic binding of Dsg 2 to Dsc 1 or Dsc 2 was also found on the molecular level (Chitaev and Troyanovsky 1997; Syed et al. 2002) but no interaction of Dsg 1 with Dsg 3 (Heupel et al. 2007). Aggregation assays of transfected cells indicated that in cells, heterophilic binding of Dsgs and Dscs might be of even greater importance than homophilic binding to induce strong intercellular adhesion (Kowalczyk et al. 1996; Marcozzi et al. 1998; Runswick et al. 2001) and that adhesion is strictly dependent on the ratio of the respective Dsgs and Dscs (Getsios et al. 2004a). This view is supported by the recent finding that a conditional Dsc 3-deficiency in mice induced a severe pemphigus-like phenotype with epidermal blistering (Chen et al. 2007). Because antibodies in typical pemphigus are usually not directed against Dsc 3 but against Dsg 1 and Dsg 3, it has to be considered that heterophilic binding of these three molecules is important for epidermal cohesion in vivo.
Armadillo family proteins
From the Armadillo family, plakoglobin and plakophilins 1–3 are important components of desmosomes.
Plakoglobin
Plakoglobin (82 kDa), also termed γ-catenin, is the only essential desmosomal component which is also found in typical adherens junctions (Cowin et al. 1986; Franke et al. 1989, 1983). The gene encoding for plakoglobin was mapped to chromosome 17 (Aberle et al. 1995). Plakoglobin binds to the cytoplasmic cadherin-typical sequence of Dsgs and Dscs via its first three armadillo repeat domains (Chitaev et al. 1998). Because the same binding site is required for interaction of plakoglobin to α-catenin, the latter is excluded from desmosomes. Similarly, although the armadillo repeat domain of β-catenin can also bind to Dsg 2, its flanking domains inhibit this interaction, which may explain the absence of β-catenin from desmosomes (Troyanovsky et al. 1996; Wahl et al. 1996). Plakoglobin has been demonstrated to interact with other desmosomal plaque components such as desmoplakin, plakophilins and also with cytokeratin filaments (Bonne et al. 2003; Chen et al. 2002; Kowalczyk et al. 1997; Smith and Fuchs 1998). The importance of this linker function can be concluded from studies in which inactivation of plakoglobin led to embryonic lethality due to mechanical fragility of the myocardium and, when mice are viable, to subcorneal skin blistering indicating that plakoglobin is essential for desmosomal stability (Table 1) (Bierkamp et al. 1996; Ruiz et al. 1996).
Besides its function as a desmosomal adaptor protein, plakoglobin seems to be involved in nuclear signalling. It has been shown that plakoglobin, comparable to β-catenin in the canonical wnt signalling pathway, confers transcriptional activity together with TCF-4/LEF transcription factors (Maeda et al. 2004). This mechanism seems to interfere with β-catenin-mediated transcription (Hu et al. 2003). Because plakoglobin like β-catenin is a target of glycogen synthase kinase-3 β, which drives proteosomal degradation of both proteins (Kodama et al. 1999; Muller et al. 2008a; Williamson et al. 2006), a complex pattern of direct and indirect transcriptional regulation seems likely. A target gene of Lef-1/plakoglobin signalling is c-Myc, the expression of which was inhibited in keratinocytes but enhanced in transformed rat kidney epithelial cells (Kolligs et al. 2000; Kolly et al. 2007; Williamson et al. 2006). This indicates that the role of plakoglobin transcriptional regulation is strictly cell type-dependent. In keratinoytes, c-Myc repression by plakoglobin is required to stop proliferation and to allow terminal differentiation (Williamson et al. 2006). Another potential target gene is the anti-apoptotic molecule Bcl–XL, which was found to be upregulated in plakoglobin-deficient cells leading to reduced apoptosis and thus might also be repressed by plakoglobin (Dusek et al. 2007a). Taken together, plakoglobin serves as functional linker between intercellular adhesion and regulation of the cell cycle. This might also be important for cancer progression because many tumors are characterized by loss of plakoglobin expression.
Plakophilins
Plakophilin 1 (80 kDa) was first identified as an “accessory” plaque protein because, in contrast to plakoglobin and desmoplakin, it was found in cells from certain stratified and complex epithelia only (Franke et al. 1983; Hatzfeld et al. 1994; Heid et al. 1994). Afterwards, it became clear that plakophilin 2 (100 kDa) is ubiquitously present in all desmosomes and also plakophilin 3 (87 kDa) is present in most simple and stratified epithelia (Mertens et al. 1999; Schmidt et al. 1999). The genes encoding plakophilin 1, 2 and 3 are located on chromosomes 1, 12, and 11, respectively (Bonne et al. 1998). Plakophilin 1 and 2 exist in two splice variants with a shorter “a” and a longer “b” form (Mertens et al. 1996; Schmidt et al. 1997). In addition to their localization in desmosomes, plakophilins 1 and 2 are also found in the karyoplasm in a variety of cells and plakophilin 1 b is exclusively located in the nucleus. Plakophilin 2 deficiency leads to embryonic death due to heart defects indicating that the presence of at least one member of the plakophilin family is required (Table 1) (Grossmann et al. 2004). Under these conditions, cytokeratin filaments were retracted from cell borders and desmoplakin was localized in the cytoplasm rather than at desmosomes in cardiomyocytes, demonstrating the relevance of plakophilin 2 to desmosplakin recruitment. Because cardiomyocytes in contrast to epithelial cells express only plakophilin 2 but not plakophilin 1 and 3, defects were present in the heart only, whereas epithelia were not affected.
Plakophilins can directly interact with all other desmosomal components including cytokeratins via the aminoterminal head domain (Bonne et al. 2003; Hatzfeld 2007; Hatzfeld et al. 2000). Plakophilin 1 recruits desmosomal components to the cell membrane, increases size and number of desmosomes and therefore seems to be a scaffolding protein, which induces desmosome assembly (Hatzfeld et al. 2000; Kowalczyk et al. 1999; Wahl 2005). On the other hand, because plakophilin 1 is located in the dense inner desmosomal plaque whereas cytokeratin filaments only loop into the outer plaque, it is believed that plakophilin 1 enhances desmoplakin lateral interactions but does not directly associate with cytokeratin filaments in vivo (Hatzfeld 2007; North et al. 1999). It has been demonstrated that desmosome formation is mediated by the aminoterminal domain, whereas recruitment of plakophilin 1 itself to the plasma membrane is dependent on the carboxyterminal region (Sobolik-Delmaire et al. 2006).
In addition to its function in the regulation of desmosome assembly, plakophilins may also regulate signalling mechanisms, both at cell borders as well as in the nucleus. Plakophilin 1 associates with actin filaments at cell borders and has been reported to interact with a GTP exchange factor (GEF) for Rho and thereby could regulate activity of Rho GTPases similar to the closely related p120-catenin, which is known to inhibit Rho A and to activate Rac 1 and Cdc42 (Anastasiadis and Reynolds 2001; Hatzfeld 2007). In addition to their localization within the desmosomal plaque, plakophilin 1 and 2 are also present inside nucleus and plakophilin 2 has been found to be part of the polymerase III complex which is responsible for generation of tRNA and rRNA (Mertens et al. 2001). By these two mechanisms, it is possible that plakophilins may regulate cell adhesion and cell growth (Hatzfeld 2007).
Plakin family proteins
Plakin family proteins are linkers between the cytoskeleton and cell–cell or cell–matrix contacts (Jefferson et al. 2007). Desmoplakin, which exists in two spice variants of a protein encoded by a single gene on chromosome 6 (desmoplakin I: 322 kDa; desmoplakin II: 259 kDa), is an essential component of the desmosomal plaque and therefore is regarded as the prototype of this family (Armstrong et al. 1999; Hatsell and Cowin 2001; Mueller and Franke 1983; Sonnenberg and Liem 2007). Other members such as plectin, envoplakin and periplakin were also found in desmosomes, but their significance for the structure and function of desmosomes is less clear. Especially, plectin is primarily important in hemidesmosmes, which anchor epithelia to the extracellular matrix.
Desmoplakin consists of an aminoterminal plakin domain, which can interact with all other desmosomal plaque proteins such as plakoglobin and plakophilins but also with Dsc 1a (Kowalczyk et al. 1997; Smith and Fuchs 1998; Troyanovsky et al. 1994b). The central coiled-coil rod domain, which is important for dimerization, is followed by the carboxyterminal tail consisting of three globular subdomains with several plakin-repeats, which serve as linkers for different intermediate filament types (Choi et al. 2002; Green et al. 1990; Stappenbeck and Green 1992). It is well established that desmoplakin is the main linker protein between the desmosomal cadherin–plakoglobin complex and the intermediate filament cytoskeleton. This has been shown in vitro and was ultimately demonstrated in desmoplakin-deficient mice, which had a reduced number of desmosomes and died at embryonic stage just after implantation (Table 1) (Bornslaeger et al. 1996; Gallicano et al. 1998). Similar to the findings in epidermal-specific desmoplakin-deficient mice, which suffered from skin blistering, desmosomes were not anchored to intermediate filaments (Vasioukhin et al. 2000). Moreover, when desmoplakin was rescued in extraembryonic tissues so that embryos further developed, defects were present not only in the myocardium and epidermis but also in the vasculature and in the neuroepithelium, underlining the importance of desmoplakin for tissue differentiation (Gallicano et al. 2001).
Diversity of desmosomes in different tissues and specific epithelial layers
Although it was discovered about 25 years ago that the structure of desmosomes is not identical in all types of cells and tissues (Giudice et al. 1984; Jones et al. 1986b), the knowledge on the diversity of desmosomes is still uncomplete and matter of discussion (Garrod et al. 2002; Getsios et al. 2004b; Green and Simpson 2007; Hatzfeld 2007; Holthofer et al. 2007; Kottke et al. 2006; Yin and Green 2004). The diversity of desmosomes has implications for tissue differentiation and also is of high-medical relevance because diseases caused by genetic alteration of or by an autoimmune reponse against a specific desmosomal component may affect only certain but not all desmosome-containing tissues.
Some desmosomal components such as Dsg 2, Dsc 2 and the plaque proteins desmoplakin, plakoglobin and plakophilin 2 are ubiquitously expressed in all cells and tissues in which desmosomes are found. Plakophilin 3 is present in most simple epithelia except hepatocytes as well as in stratified epithelia, whereas plakophilin 1 is restricted to stratified and complex epithelia. In epithelia, the desmosomal cadherins show typical expression patterns. Simple epithelia and urothelium usually express Dsg 2 and Dsc 2 only. Apparently, exceptions are the additional presence of Dsg 1 in the mucosa of uterus, stomach, intestine and in epithelia of liver and pancreas as well as the expression of Dsc 1 in intestine and liver or Dsc 3 in stomach, prostate, salivary gland and urothelium. Dsg 4 has a unique tissue distribution in skin and several simple epithelia such as those present in pancreas, salivary glands, testis, prostate and hepatic epithelium.
The Dsg 1/Dsc 1 and Dsg 3/Dsc 3 pairs are largely confined to stratified epithelia where the expression patterns of the Dsg and Dsc isoforms usually conform. Interestingly, in the stratified corneal epithelium, only Dsg 1 and Dsc1 are present indicating that these desmosomal cadherins in the absence of Dsg 1 and Dsg 3 are sufficient to maintain cohesion in stratified epithelia also. In the epidermis, the plaque proteins plakoglobin, desmoplakin and plakophilin 3 are expressed in all layers (Fig. 4). In contrast, plakophilins 1 and 2 display inverse distribution with plakophilin 1 being more abundant in the superficial epidermis. These inverse expression patterns are also typical for the Dsg 1/Dsc 1 and Dsg 3/Dsc 3 pairs. Dsg 1/Dsc 1 are the predominant desmosomal cadherins in the superficial epidermis, whereas Dsg 3/Dsc 3 are primarily expressed in the lower epidermis. In contrast to Dsg 1, which can be detected in some desmosomes in keratinocytes of the basal layer also, Dsc 1 and Dsg 4 are absent in the basal layer (Dusek et al. 2007b; Mahoney et al. 2006; Spindler et al. 2007). Because Dsg 3 is expressed throughout the spinous layers (Fig. 5), the expression patterns of Dsg 1 and Dsg 3 largely overlap in human adult epidermis (Mahoney et al. 2006; Spindler et al. 2007). Dsc 2 is enriched in the deep epidermis with lower levels in the superficial epidermis, whereas Dsg 2 is restricted to basal and suprabasal cells but is present in very faint amounts only (Mahoney et al. 2006) indicating that this pair of proteins may be primarily important for cell cohesion in simple epithelia and myocardium rather than in complex epithelia. However, it is unclear at present which desmosomal cadherin isoforms are capable to heretophilically bind to each other and thus interpretation of these distribution patterns with respect to their relevance for mechanical adhesion is preliminary. In multilayered squamous epithelium of mucous membranes, for instance of the oral cavity, Dsg 1 and Dsg 3 are strongly expressed throughout all layers, whereas Dsg 4 shows strong expression in superficial layers but is missing in the basal layer (Mahoney et al. 2006).

Expression patterns of desmosomal components in the epidermis. The schematic drawing of the epidermis (left) indicates the basal (BL), spinous (SL), granular (GL) and corneal (CL) layer of the epidermis. On the right, the expression patterns of desmosomal components in the specific epidermal layers are illustrated. For instance, Dsg 1 and Pkp 1 are most prominent in the superficial layers, whereas expression of Dsg 3 and Dsc 3 is strongest in the deep epidermis. Dsg desmoglein, Dsc desmocollin, Pkp plakophilin, PG plakoglobin, DP desmoplakin

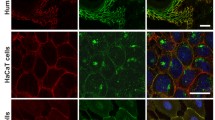
Immunostaining of Dsg 1 and Dsg 3 in human epidermis. Intact human epidermis was immunostained using monoclonal antibodies against Dsg 1 (a) and Dsg 3 (b). A merge of both panels is shown in c. Dsg 1 is most abundant in the superficial epidermis but is also present in the basal layer. Dsg 3 is expressed in the basal layer as well as throughout the spinous layer indicating that in human epidermis the expression patterns of these two proteins broadly overlap. Scale bar is 20 μm
It is important to note that the specific distribution patterns of desmosomal components in stratified epithelia are important for epithelial differentiation and function (Green and Simpson 2007). It was shown that forced overexpression of Dsg 3 in the suprabasal epidermis led to abnormal differentiation and hyperproliferation as well as perinatal lethality due to transepidermal water loss (Elias et al. 2001; Merritt et al. 2002). Similarly, forced suprabasal Dsg 2 and Dsc 3 overexpression resulted in hyperproliferation and formation of papillomas, possibly via altered β-catenin/wnt signalling (Brennan et al. 2007; Hardman et al. 2005).
Desmosome assembly and disassembly
The mechanisms participating in desmosome assembly and disassembly have been reviewed in detail elsewhere (Getsios et al. 2004b; Green and Simpson 2007; Kitajima 2002; Yin and Green 2004). For instance, extracellular Ca2+ and protein kinase C (PKC) signalling are well known to be involved in desmosome assembly. Ca2+ concentrations >0.1 mM allow formation of adherens junctions and desmosomes (Hennings and Holbrook 1983). Desmosomal plaques with inserted cytokeratin filaments became visible as early as after 5 min after the Ca2+ switch followed by appearance of assymetrical desmosomes after 10 min and of symmetric desmosomes after 1 h. Increased extracellular Ca2+ induced incorporation of desmosomal components such as Dsgs, plakoglobin and desmoplakin into the desmosomal plaque (Hennings and Holbrook 1983; Pasdar et al. 1995; Pasdar and Nelson 1988, 1989). Activation of PKC is required for translocation of desmosomal components to the cell membrane and for desmosome assembly (Sheu et al. 1989), but also was found to reduce desmosomal adhesion and to increase Ca2+-dependence of desmosomes (Kimura et al. 2007) indicating that regulation of desmosomal adhesion by PKC is complex.
Before desmosome assembly, adhesion zippers of E-cadherin-containing puncta form on filopodial processes of neighbouring cells, an event that requires both α-catenin and VASP-driven actin reorganization (Vasioukhin et al. 2000). Afterwards, these intermediate junctions mature to adherens junctions and desmosomes are assembled at regions where membranes are brought together. It appears that Dscs initiate the formation of desmosomes. This is based on the observations that Dsc 2 is the first desmosomal component at the cell surface followed by Dsg 2 in MDCK cells (Burdett and Sullivan 2002) and that, in keratinocytes, N-terminally deleted Dsc 3, which compromised desmosome formation was still able to bind to β-catenin. Therefore, it can be speculated that Dsc 3 could localize to pre-existing adherens junctions to induce desmosome formation (Hanakawa et al. 2000). Desmosomal cadherins seem to be transported in vesicles from the Golgi along microtubules whereas non-membranous cytoplasmic particles containing desmoplakin and plakophilin are associated with intermediate filaments and move towards cell-junctions by actin-based motility (Godsel et al. 2005; Green and Simpson 2007). Desmoplakin trafficking seems to be dependent on intracellular Ca2+ levels because patients with Darier’s disease, which results from mutations in a sarcoplasmic reticulum Ca2+ pump show desmoplakin retention in the cytoplasm and altered desmosome structure (Dhitavat et al. 2003; Dhitavat et al. 2004; Sakuntabhai et al. 1999).
On the cell surface, the desmosomal cadherins together with plakoglobin and desmoplakin are sufficient to nucleate a desmosomal plaque (Kowalczyk et al. 1997). Further plaque enlargement and desmoglein clustering are dependent on plakoglobin together with plakophilin (Bornslaeger et al. 2001; Koeser et al. 2003). Therefore, keratinocytes deficient for either plakoglobin or desmoplakin display reduced numbers of desmosomes, disturbed plaque formation and reduced anchorage of cytokeratin filaments (Bierkamp et al. 1999; Vasioukhin et al. 2001). It appears that during desmosome assembly, Dsg3-containing clusters are formed in the beginning, which, upon attachment to cytokeratin filaments, become integrated in desmosomes (Sato et al. 2000). Once they are formed, desmosomes are stable throughout the cell cycle and are not disrupted during mitosis, although the desmosomal components are subjected to a significant turnover with a half-life of about 30 min like it was shown for Dsc 2a (Windoffer et al. 2002). Finally, it has to be emphasized that a reciprocal dependence of desmosomes and adherens junctions seems to exist. This can be concluded from experiments in which expression of N-terminally deleted Dsc 3 or desmoplakin deficiency resulted in impaired formation of both desmosomes and adherens junctions (Hanakawa et al. 2000; Vasioukhin et al. 2001).
Regulation of keratinocyte proliferation by desmosomal cadherins
Evidence is accumulating that desmosomal cadherins such as Dsg 3 regulate keratinocyte proliferation (Muller et al. 2008a). It has been shown that autoantibodies from pemphigus vulgaris patients induce continuing keratinocyte proliferation by impaired Dsg 3/plakoglobin signalling, which finally leads to c-Myc overexpression (Muller et al. 2008a; Williamson et al. 2007, 2006). According to this concept, in healthy epidermis Dsg 3 binding results in inhibition of glycogen synthase kinase 3 (GSK3) via activation of phosphatidylinositol trisphosphate kinase (PI3K) and Akt. In consequence, GSK3 phosphorylation-dependent degradation of plakoglobin is abolished which allows plakoglobin to translocate into the nucleus and to induce growth arrest via suppression of c-Myc (Muller et al. 2008a; Williamson et al. 2006).
Desmosome-associated diseases
Several diseases have been found in which impaired desmosomal adhesion contributes to pathogenesis. Inactivation of desmosomal function may be reduced by completely different mechanisms including genetic defects of desmosomal components, cleavage of desmosomal cadherins by bacterial toxins and binding of autoantibodies to desmogleins 1, the latter of which is the cause of the autoimmune disease pemphigus. Although altered expression of desmosomal cadherins such as Dsg 2/Dsc 2 and Dsg 3/Dsc 3 have been observed in human carcinomas such as squamous cell carcinoma as well as gastric, colorectal and breast carcinomas, mutations are usually absent (Bazzi and Christiano 2007). Therefore, the role of desmosomal cadherins in cancer is unclear at present.
Genetic diseases
Mutations in desmosomal plaque components in humans affect the myocardium as well as the epidermis with its appendages (Table 1). Mutations in genes for the essential desmosomal plaque components desmoplakin and plakoglobin result in heart, skin and hair defects (Bazzi and Christiano 2007; Green and Simpson 2007; McGrath 2005). In contrast, genetic alterations of Dsg 2, Dsc 2 and plakophilin 2 selectively lead to heart defects because these are the only isoforms of their protein families in the myocardium. On the other hand, mutations in Dsg 1 and plakophilin 1, which are primarily expressed in the epidermis, cause skin defects whereas loss of Dsg 4 in hair follicles results in hair loss.
Genetic heart defects
Interestingly, all defects of desmosomal components causing heart defects such as desmoplakin, plakoglobin, plakophilin 2, Dsg 2 and Dsc 2 lead to the phenotype of arrhythmogenic right ventricular cardiomyopathy (ARVC) which is clinically characterized by right bundle block and arrhythmia and histologically by fibrofatty replacement of cardiomyocytes, possibly due to impaired cell adhesion caused by loss and alterations of desmosomes (Asimaki et al. 2007; Gerull et al. 2004; McKoy et al. 2000; Pilichou et al. 2006; Rampazzo et al. 2002; Syrris et al. 2006; Heuser et al. 2006). This is in line with embryonic lethality due to myocardial rupture in mice models deficient in these proteins. Therefore, the thinnest parts of the right ventricle are the most severely affected, but left ventricle involvement also occurs (van Tintelen et al. 2007). However, fibrofatty transdifferentiation of cardiomyocytes cannot be simply explained by impaired desmosomal adhesion, but rather seems to be caused by altered wnt/ β-catenin signalling in response to nuclear translocation of plakoglobin (Garcia-Gras et al. 2006).
Genetic defects of skin and its appendages
Haploinsufficiency of the gene encoding Dsg 1 results in the autosomal dominant skin disease striate palmoplantar keratoderma (SPPK), which is characterized by linearly arranged thickening of the stratum corneum on the palms, soles, knees, ankles and finger knuckles (Milingou et al. 2006; Rickman et al. 1999). However, blisters are absent indicating that disturbed differentiation is the primary mechanism underyling this entity rather than a loss of keratinocyte adhesive function. Similarly, mutated Dsg 4 leads to autosomal recessive inherited hypotrichosis due to defective hair follicle differentiation, a phenotype related to the lanceolate hair mouse (Kljuic et al. 2003). In contrast, ablation of plakophilin 1 results in the recessive skin-fragility ectodermal dysplasia syndrome, which in 1997 was the first genetic desmosome-associated disease to be described (McGrath 2005). Here, both loss and alterations of desmosomes and lacking insertion of cytokeratin filaments due to inability to recruit desmoplakin cause skin blistering around the mouth as well as on palms and soles accompanied by dystrophic hair and nails (McGrath et al. 1997; McMillan and Shimizu 2001).
Mutations in plaque proteins with involvement of various tissues
Mutations in plakoglobin are the cause of Naxos disease in which ARVC and palmoplantar keratoderma are associated with woolly hair (McKoy et al. 2000). Interestingly, in contrast to plakoglobin-deficient mice (Bierkamp et al. 1996), acantholysis is absent indicating that some mechanical functions of plakoglobin are maintained in these patients.
The most variable phenotypes are the consequence of desmoplakin alterations. An autosomal recessive disorder with dilated cardiomyopathy, keratoderma and woolly hair called Carvajal syndrome is comparable to Naxos disease (Norgett et al. 2000). Haploinsufficiency leads to SPPK, whereas non-sense mutations are accompanied by skin fragility leading to blisters in the face as well as on extremities and trunk and also with wolly hair (Armstrong et al. 1999; Whittock et al. 1999, 2002). However, the most severe disorder is lethal acantholytic epidermolyis bullosa, which is caused by C-terminally truncated desmoplakin and was fatal in a 10-day-old hair and nailless newborn due to extensive blistering leading to transcutaneous fluid loss (Jonkman et al. 2005). On the ultra-structural level, desmosomes were reduced in desmoplakin-related SPPK similar to SPPK caused by mutations in Dsg 1 (Wan et al. 2004), but not in lethal acantholytic epidermolysis bullosa. However, desmosome shedding, alterations of desmosomal plaques and impaired cytokeratin insertion were typically associated with desmoplakin mutations (Jonkman et al. 2005; Norgett et al. 2000; Wan et al. 2004). At present, it is unclear why different mutations affect different tissues.
Infectious diseases
Staphylococcal scalded skin syndrome (SSSS), which was first described by Ritter von Rittershain in 1878, is the systemic variant of epidemic pemphigus neonatorum or sporadic bullous impetigo and is characterized by superficial epidermal splitting accompanied by fever, erythema and skin tenderness (Farrell 1999; Lyell 1983; Stanley and Amagai 2006). Most cases are caused by staphylococcal exfoliative toxin (ET), a serine protease, which has been shown to selectively cleave Dsg 1 between EC 3 and 4 in conformation-dependent manner, but not Dsg 3 or E-cadherin (Table 1) (Amagai et al. 2000a, 2002; Hanakawa et al. 2002; Melish and Glasgow 1970). Assuming that ET does not cleave other superficially expressed desmosomal cadherins such as Dsg 4 or Dsc 1, SSSS is a good example that extensive epidermal blistering can be induced by proteolytic cleavage of a single adhesion molecule. According to its bacterial pathogenesis, SSSS can be effectively treated with antibiotics (Stanley and Amagai 2006).
Pemphigus
Pemphigus is an autoimmune blistering skin disease, which is characterized by intraepidermal blistering (Lever 1953). The two major types of pemphigus are the more severe pemphigus vulgaris (PV), which accounts for 80–90% of cases, and pemphigus foliaceus (PF) (Bystryn and Rudolph 2005; Schmidt et al. 2000; Stanley and Amagai 2006). Pemphigus is a rare disease with a yearly incidence of 0.75–5 cases per million and apart from being present in humans, is also found in horses, dogs and cats. In contrast to other autoimmune diseases, which primarily affect women, pemphigus is equally distributed between both genders, and is diagnosed mostly between the fourth and sixth decades. In PV, there are two main forms, the mucosal-dominant and the mucocutaneous type. In both cases, the disease most commonly begins with painful non-healing ulcerations not only in the mucous membranes of the mouth, but also in the larynx, nose and vagina. Later on, flaccid blisters may occur on the scalp, trunk, groin and axillae, which easily rupture, leaving sharply outlined erosions and heal without scarring (Fig. 6). In contrast to PV, PF only affects the epidermis and because epidermal splitting is restricted to the superficial epidermis, lesions appear as crusted erosions on the upper torso, face and scalp. It has to be mentioned that pemphigus can be induced by drugs such as penicillamine, penicillin, captopril and β-blockers and also can occur as a paraneoplastic entity accompanying or preceding lymphoma and lung carcinoma (Yeh et al. 2003). Moreover, in South America an endemic form of PF, called Fogo selvagem, exists, which is thought to be transmitted by insect vectors (Aoki et al. 2004; Diaz et al. 1989). Currently, the therapy of pemphigus is based on immunosupression and reduction of autoantibody load. Conventional therapy includes high-dose corticosteroids, intravenous immune globulin and cytotoxic agents. Before systemic corticosteroids were available, 75% of PV patients died within a year. Second-line therapies for refractory PV include rituximab, an antibody directed against B cell CD20, which reduces autoantibody-producing B cells as well as plasmapheresis to physically remove autoantibodies (Ahmed et al. 2006; Shimanovich et al. 2008).

Clinical phenotype of Pemphigus vulgaris and Pemphigus foliaceus. Patients suffering from the mucocutaneous form of pemphigus vulgaris (PV) usually have flaccid blisters and erosions on the trunk (a) accompanied by mucosal ulcerations in the mouth (b). In contrast, Pemphigus foliaceus patients are characterized by crusted epidermal erosions (c) whereas involvement of mucous membranes is absent
Histology and autoantibody profile in pemphigus
Besides the clinical phenotype, diagnosis of pemphigus is based on histology and the patients’ autoantibody profile (Bystryn and Rudolph 2005). The histologic hallmark of pemphigus is acantholysis, i.e. loss of cell–cell adhesion between keratinocytes. In PV, the epidermal cleavage plane is located in the deep epidermis, usually right above the basal layer (Fig. 7). In contrast, in PF, epidermal splitting occurs between granular layers. Skin blisters can be induced by rubbing on healthy-appearing epidermis, a phenomenon referred to as Nikolsky sign.

Typical histology of epidermal lesions from pemphigus patients. Hematoxylin eosin-stained paraffin sections from PV (a) and PF (b) patients showed suprabasal epidermal cleavage in the PV and superficial granular blistering in PF. Scale bar is 50 μm
Autoantibodies in pemphigus are sufficient to cause blistering in human skin in vivo and in vitro (Anhalt et al. 1982; Schiltz and Michel 1976). In contrast to other autoimmune blistering skin diseases such as bullous pemphigoid or epidermolysis bullosa acquisita (Sitaru and Zillikens 2005; Yancey 2005), pemphigus antibodies do not require the complement system or leukocytes to induce blisters in vivo (Anhalt et al. 1986). It is generally accepted that PV and PF are characterized by different autoantibody profiles, which generally correlate with disease activity (Bystryn and Rudolph 2005; Harman et al. 2001; Ishii et al. 1997; Stanley and Amagai 2006; Stanley et al. 1984; Yeh et al. 2003). Patients suffering from mucosal-dominant PV usually have antibodies directed against Dsg 3 but not Dsg 1, whereas mucocutaneous PV is characterized by both Dsg 3 and Dsg 1 autoantibodies (Amagai et al. 1999; Ding et al. 1997; Jamora et al. 2003; Miyagawa et al. 1999). In contrast, in PF patients usually antibodies against Dsg 1 but not Dsg 3 are detected (Amagai et al. 1999). However, it is also known that in some cases this correlation between the clinical phenotype and the autoantibody profile was not found (Baykal et al. 2002; Jamora et al. 2003; Yoshida et al. 2005; Zagorodniuk et al. 2005).
Over the last decade, there is a debate whether these autoantibodies against desmosomal cadherins are pathogenic (Amagai et al. 2006). It has been shown by passive transfer of affinity-purified Dsg antibody fractions as well as by depletion of pathogenic activity by absorption against desmoglein extracellular domains that Dsg 1 antibodies in PF and the combination of Dsg 1 and Dsg 3 autoantibodies in PV as well as in paraneoplastic pemphigus are sufficient to induce skin blistering (Amagai et al. 1995, 1994a, 1992, 1991, 1998; Koulu et al. 1984; Mahoney et al. 1999). An active PV mouse model in which Dsg 3-deficient mice were immunized with Dsg 3 before splenocytes from these animals were transferred to lymphopenic Rag-2-deficient mice supported the notion that Dsg 3 antibodies alone can cause mucosal erosions (Amagai et al. 2000b). Similar in keratinocyte cultures, depletion of Dsg 1-specific antibodies from PF-IgG by preincubation with recombinant Dsg 1 but not after preincubation with VE-cadherin completely abolished keratinocyte dissociation (Waschke et al. 2005).
Pemphigus IgG were found to include a plethora of more than 20 different autoantibodies against keratinocyte antigens such as antibodies against Dsg 1, Dsg 4, Dsc 1-3, desmoplakin, plakoglobin and E-cadherin and several other proteins not associated with cell junctions (Amagai et al. 2006; Evangelista et al. 2008; Kljuic et al. 2003; Korman et al. 1989; Nguyen et al. 2000c). For instance, in all PF sera as well as in 79% of mucocutaneous PV sera, autoantibody activities against E-cadherin were detected, most of which were due to Dsg 1 autoantibodies cross-reacting with E-cadherin (Evangelista et al. 2008). Some of the different autoantibodies have clearly been shown not to be pathogenic such as the Dsg 4-cross-reacting Dsg 1 antibodies in PF (Nagasaka et al. 2004). Therefore, similar to other autoimmune diseases, the pathogenetic relevance of autoantibodies against a specific protein in pemphigus has to be challenged until it has been convincingly demonstrated (Amagai et al. 2006). However, it has been reported that antibodies others than those directed to desmogleins also contribute to epidermal blistering because PV-IgG not containing Dsg 1 antibodies were effective to cause blistering in Dsg 3-deficient mice (Nguyen et al. 2000c). It is possible that these antibodies include antibodies to cholinergic receptors and to pemphaxin, which have both been detected in 85% of PV and PF sera (Grando 2006a; Nguyen et al. 2000b). The pathogenic relevance of antibodies against cholinergic receptors was concluded from experiments where preincubation of monkey oesophagus with PV-IgG blocked staining by a rabbit acetylcholine receptor antibody and the fact that this antibody induced keratinocyte dissociation in culture (Nguyen et al. 2000a). However, antibodies against pemphaxin alone were not sufficient to induce skin blistering (Nguyen et al. 2000b). Moreover, it has not been demonstrated so far that autoantibodies from pemphigus patients, which target cholinergic receptors are capable to induce acantholysis. Therefore, at present it is safe to believe that epidermal blistering in pemphigus is primarily caused by antibodies against Dsg 1 and Dsg 3. These pathogenic antibodies in PV and PF mainly belong to the IgG 4 and IgG 1 subclasses (Bhol et al. 1995; Rock et al. 1989; Spaeth et al. 2001).
The knowledge that autoantibodies against desmosomal cadherins are sufficient to induce acantholysis in complement- and leukocyte-independent manner makes pemphigus one of the best-characterized models to study the direct mechanisms underlying autoimmune diseases. Besides the role of autoantibodies, the contribution of Dsg 3 autoreactive T helper (Th) cells has also been characterized for PV and endemic PF (Hertl et al. 2006). Th1 and Th2 cells in PV recognize the extracellular domain of Dsg 3 when presented on the HLA class II alleles HLA-DRβ1*0402 and HLA-DQβ1*0503, whereas in Fogo selvagem patients HLA-DRβ1*0402 and HLA-DRβ1*0101 were most common. In PV patients as well as in healthy carriers of the PV-associated HLA II alleles, Dsg 3 and Dsg 1-autoreactive T cells were found. In healthy individuals, Th1 cells with characteristics of regulatory T (Tr1) cells which inhibit T cell activation were most prevalent. In contrast, in PV patients the levels of Tr1 cells were reduced while Th2 cells were increased (Veldman et al. 2004). Therefore, it is possible that an imbalance of autoreactive Tr1 and Th 2 cells plays a role in the induction of PV by promoting the proliferation of anti-Dsg 3 producing B cells.
The mechanisms underlying pemphigus skin blistering
As a first concept it was proposed that proteolytic cleavage of molecules responsible for intercellular adhesion was the mechanism underlying pemphigus skin blistering. Later on, with the identification of desmosomal cadherins as the target antigens of pemphigus autoantibodies and with more sophisticated cell biologic tools at hand, the ideas of direct antibody-mediated inhibition and of indirect signalling-mediated reduction of desmoglein binding were developed (Fig. 8).

The two principal mechanisms underlying pemphigus skin blistering. Two principal mechanisms have been proposed by which autoantibodies specific for Dsg 1 and Dsg 3 could impair desmosomal adhesion. First, antibodies could directly interfere with desmoglein transinteraction (a). Second, antibody binding has been shown to trigger intracellular signalling pathways, which indirectly results in loss of desmoglein-mediated binding (b)
Proteolytic cleavage of desmosomal cadherins
Proteolytic cleavage of cell adhesion molecules has first been suggested to be involved in pemphigus acantholysis because protease inhibitors blocked pemphigus IgG-induced cell detachment in culture (Farb et al. 1978). Moreover, plasminogen activator activity and expression of the urokinase-type plasminogen activator receptor (uPAR) system were found to be increased following treatment with PV- and PF-IgG/serum in keratinocytes in vitro as well as in PV patients’ skin (Feliciani et al. 2003; Hashimoto et al. 1983; Lo Muzio et al. 2002; Schaefer et al. 1996; Seishima et al. 1997; Yamamoto et al. 2007b), possibly via phospholipase C (PLC)-mediated signalling (Esaki et al. 1995). Anti-uPA antibodies and a PA inhibitor were sufficient to block acantholysis induced by PV- or PF-IgG in several studies (Feliciani et al. 2003; Hashimoto et al. 1983; Morioka et al. 1987) but not in that by Schuh et al. (2003). However, a definitive evaluation of the uPAR system for pemphigus acantholysis became possible since a study using uPA- and tissue PA-deficient mice showed extensive skin blistering in response to PV- and PF-IgG (Mahoney et al. 1999). Thus, the plasminogen activator system does not appear to be essential for pemphigus skin blistering but may aggravate the phenotype, especially when secondary inflammatory mediators such as IL-1α and TNF-α are released (Feliciani et al. 2000, 2003).
The same may hold true for other proteases such as matrix metalloproteinases (MMP) or proteases of the ADAM (a disintegrin and metalloproteinase) family. MMP-9, which was overexpressed but not activated following treatment with PV serum was reported to specifically cleave Dsg 3 during apoptosis (Cirillo et al. 2007a, d). ADAM17 on the other hand, was upregulated by activation of the epidermal growth factor receptor (EGFR) and caused shedding of Dsg 2 (Bech-Serra et al. 2006; Santiago-Josefat et al. 2007). These results may be important for PV because EGFR activation was observed following treatment with PV-IgG (Chernyavsky et al. 2007a; Frusic-Zlotkin et al. 2006). The presence of proteolytic enzymes in PV sera may also explain why IgG-depleted PV sera were found to be pathogenic in culture (Cirillo et al. 2007c). However, because no direct evidence was provided that MMP-9 or ADAM17 or any other proteinase cleaves members of the Dsg family in pemphigus, the significance of these findings for acantholysis in pemphigus is unclear and the specific proteolysis hypothesis proposed for pemphigus requires further experimental substantiation (Cirillo et al. 2008). Nevertheless, the fact that specific cleavage of Dsg 1 by staphylococcal exfoliative toxin in bullous impetigo is sufficient to cause a histologic phenotype comparable to PF (Amagai et al. 2000a; Hanakawa et al. 2002) indicates that, in principle, specific proteolysis could be an effective mechanism in pemphigus.
Direct inhibition of desmoglein binding
Since it was discovered that autoantibodies in pemphigus are directed to desmosomal adhesion molecules, it was believed that these autoantibodies might directly interfere with desmoglein binding (Fig. 8) (Amagai et al. 1991; Jones et al. 1986a; Koulu et al. 1984), a mechanism also refered to as “steric hindrance”(Sharma et al. 2007). This model is attractive because it has been shown that autoantibodies against Dsg 3 and Dsg 1 in PV and PF patients primarily target the aminoterminal part of the EC 1 domain (Futei et al. 2000; Hacker-Foegen et al. 2003; Ishii et al. 2008; Muller et al. 2008b; Sekiguchi et al. 2001). The EC 1 domain, according to morphologic studies on desmoglein transinteraction in desmosomes, is increasingly recognized as the part of the desmosomal cadherin ectodomain, responsible for trans-interaction (Al-Amoudi et al. 2007; He et al. 2003) and may harbour the putative transadhesive interface, based on data from the crystal structure of classical cadherins (Boggon et al. 2002; Overduin et al. 1995; Shapiro et al. 1995). Moreover, it seems that autoantibody reactivity to the aminoterminal parts (EC 1) of the Dsg 3 ectodomain correlates with high disease activity as well as epidermal or mucosal involvement in PV although the titers of these antibodies do not show this correlation (Amagai et al. 1992; Muller et al. 2006, 2008b; Salato et al. 2005).
First functional data that anti-Dsg 3 antibodies in PV may directly interfere with Dsg 3 binding were provided using monoclonal antibodies derived from the active PV mouse model (Amagai et al. 2000b). AK 23, which was directed against the aminoterminal part of EC 1 was found to be pathogenic and capable to induce epidermal blistering in vivo, at least when PF-IgG or exfoliative toxin A was added to inactivate Dsg 1 (Shimizu et al. 2005; Tsunoda et al. 2003). Antibodies to other parts of the Dsg 3 extracellular domain such as AK 9 and AK 18 were ineffective to induce blisters. Recently, by using single-molecule atomic force microscopy (AFM), it was shown that PV-IgG as well as AK 23 directly interfered with homophilic Dsg 3 binding under cell free conditions (Heupel et al. 2007) which supports the hypothesis of direct inhibition of Dsg 3 binding in PV (Stanley and Amagai 2006). However, no direct inhibition of Dsg 1 binding by PV-IgG and PF-IgG was detected by AFM. These autoantibodies induced keratinocyte dissociation and reduced binding of both Dsg 3- and Dsg 1-coated microbeads to the surface of cultured keratinocytes, as revealed by laser trapping (Heupel et al. 2007; Waschke et al. 2005). These data suggest that autoantobodies interfere with Dsg 1 binding rather by indirect, cell-dependent mechanisms.
Finally, it has to be noted that, if direct inhibition occurs, it is not possible to discriminate at present whether interference with Dsg 3 binding in PV was mediated by steric hindrance, i.e. by blocking trans-interaction of desmoglein molecules by the bound autoantibody, or rather by allosteric effects, i.e. autoantibody-induced conformational changes of the Dsg 3 ectodomain, which in turn interfere with Dsg 3 transinteraction. An antibody directed against the putative transadhesive interface may directly induce steric hindrance, whereas antibodies directed against other parts of the desmoglein ectodomain could indirectly inhibit desmoglein binding by allosteric mechanisms. The fact that AK 18 and AK 9, which were directed to the middle and the carboxyterminal parts of the Dsg 3 ectodomain, were not pathogenic and did not directly interfere with Dsg 3 binding suggests that these specific antibodies were not capable of causing allosteric hindrance (Heupel et al. 2007; Tsunoda et al. 2003). On the other hand, an antibody directed against the EC2 domain, although this part of the molecule may not be involved in transinteraction, might be large enough to cause steric hindrance of Dsg 3 transinteraction. Therefore, “direct inhibition”, instead of “steric hindrance” of desmoglein binding should be used until discrimination between steric and allosteric effects is possible.
Desmoglein compensation in pemphigus
The desmoglein compensation hypothesis was proposed to explain the different clinical phenotypes of PV and PF on the basis of their different autoantibody profiles (Amagai 2003; Payne et al. 2004; Sharma et al. 2007; Shirakata et al. 1998; Stanley and Amagai 2006; Udey and Stanley 1999). According to this concept, in the deep epidermis which contains both Dsg 1 and Dsg 3, Dsg 3 compensates for the functional loss of Dsg 1 induced by Dsg 1-specific autoantibodies, resulting in more superficial blistering in PF (Fig. 9). In PV, when only Dsg 3 antibodies are present, no epidermal blistering would occur because Dsg 1 is considered to compensate for autoantibody-induced loss of Dsg 3 binding. However, acantholysis occurs in mucous membranes where Dsg 3 is assumed to be the predominantly expressed Dsg isoform, leading to the phenotype of mucosal-dominant PV. When autoantibodies to Dsg 1 are also produced in PV, epidermal blistering occurs. However, it is unclear why the cleavage plane is restricted to the deep epidermis in PV since in PF Dsg 1 autoantibodies cause superficial blistering (Fig. 10). For this reason and other reasons such as the cases of pemphigus in which the autoantibody profiles do not correlate with the clinical phenotype or the presence of other desmosomal cadherin isoforms in the epidermis, this concept has been challenged (Amagai et al. 2006; Bystryn and Rudolph 2005; Muller et al. 2002; Spindler et al. 2007).

The desmoglein compensation hypothesis. Based on the different autoantibody profiles in PV and PF together with the findings that Dsg 3 is present in the deep epidermis only whereas Dsg 1 is primarily expressed in the superficial epidermis, the desmoglein compensation hypothesis has been proposed to explain the epidermal cleavage planes typical for PV and PF. According to this model, blistering in PF affects the superficial epidermis because Dsg 3 is present in the deep epidermis to compensate for the autoantibody-induced loss of Dsg 1 binding. In PV, epidermal involvement would occur only when autoantibodies against both Dsg 1 and Dsg 3 are present because Dsg 1 is found in all epidermal layers and could compensate for loss of Dsg 3 binding when antibodies to Dsg 3 are solely present

Immunostaining of Dsg 1 and Dsg 3 in PV lesional epidermis. Epidermis from a patient with mucocutaneous PV was stained for Dsg 1 (a) and Dsg 3 (b). A merge of both panels is shown in c. Both Dsg 1 and Dsg 3 are expressed in the basal layer underneath the blister as well as in keratinocytes in the blister roof. However, Dsg 3 staining appears to be fragmented throughout the epidermis whereas Dsg 1 staining is more continuous. Note that in the level of the cleavage plane the apical membrane of basal cells shows strong immunostaining for Dsg 1 and Dsg 3 (arrows). Therefore, based on the desmoglein compensation hypothesis the expression patterns of Dsg 1 and Dsg 3 cannot explain why the cleavage plane is located suprabasally in PV but not in other epidermal layers. Scale bar is 20 μm
Experimental support for the desmoglein compensation hypothesis in vivo was obtained in mice. It was shown that PF-IgG were sufficient to cause skin blistering in Dsg 3-deficient mice but not in normal mice (Mahoney et al. 1999). In skin layers where Dsg 1 and Dsg 3 were found, autoantibodies against both desmogleins were required for blistering. In line with these findings, forced expression of Dsg 3 in the superficial epidermis abolished the ability of PF-IgG to induce acantholysis in mice (Wu et al. 2000). In contrast, in human skin and in cultured human keratinocytes in vitro, PF-IgG were effective to induce acantholysis despite of the presence of both Dsg 1 and Dsg 3 (Spindler et al. 2007). The discrepancy between these conflicting findings may be explained in part by the notion that the desmoglein compensation hypothesis is based on the following two assumptions: (1) the expression pattern of Dsg 3 and Dsg 1 do not substantially overlap in epidermal and mucosal layers where the cleavage plane in PV and PF is located. (2) Dsg 1- and Dsg 3-specific autoantibodies only lead to inactivation of either Dsg 1 or Dsg 3, respectively. Because of the latter, the desmoglein compensation hypothesis has been used to promote the idea that autoantibodies reduce Dsg binding by direct inhibition rather than by unspecific proteolysis (Mahoney et al. 1999).
Regarding the distribution of Dsg 1 and Dsg 3, it is important to note that Dsg 3 expression patterns in specific epidermal layers are different in mice and men. In mice, expression of Dsg 3 is restricted to the basal and immediately suprabasal epidermal layer (Mahoney et al. 2006, 1999). In human skin, when PV and PF were used for staining, a similar staining pattern was revealed (Amagai et al. 1996; Shimizu et al. 1995). In contrast, when specific antibodies or in situ hybridisation were used for Dsg 3 mapping in human epidermis, it was demonstrated that Dsg 3 is present throughout the spinous layers and thus Dsg 3 distribution showed substantial overlap with expression of Dsg 1 (Arnemann et al. 1993; Mahoney et al. 2006; Spindler et al. 2007). However, immunostaining of human epidermis using another monoclonal antibody detected expression of Dsg 3 in the lower epidermis only (Wu et al. 2000). In oral mucosa, equally strong expression of Dsg 1 and Dsg 3 was found throughout the epithelium when specific antibodies were used (Mahoney et al. 2006), whereas Dsg 1 staining intensity was found to be much lower when PV-IgG were used for immunstaining (Shirakata et al. 1998). Taken together, the expression patterns of Dsg 1 and Dsg 3 broadly overlap in human epidermis and appear to be identical in oral mucosa.
With respect to the second assumption the desmoglein compensation is based on, i.e. selective inactivation of Dsg 1 but not of Dsg 3 by Dsg 1-specific antibodies, it was shown recently that both PF-IgG (only containing Dsg 1-specific antibodies) and PV-IgG from patients with only Dsg 3-specific antibodies were equally effective to reduce binding of Dsg 1- and Dsg 3-coated beads to the surface of cultured keratinocytes (Heupel et al. 2007; Spindler et al. 2007). These data indicate that PV-IgG and PF-IgG can reduce binding of Dsg 1 and Dsg 3, at least on the keratinocyte cell surface. Taken together, the relevance of desmoglein compensation for pemphigus pathogenesis in humans cannot be concluded from experiments in mice, especially because distribution patterns of Dsg 1 and Dsg 3 substantially differ in the two species. Therefore, alternative models have to be worked out to explain the different epidermal cleavage planes in PV and PF. These may involve different signalling pathways required for maintenance of desmosomal adhesion in the specific epidermal layers as outlined below.
Signalling pathways in pemphigus and in desmosome disassembly
Since it has been shown that PV-IgG upon binding to keratinocytes induced a rapid transient increase of intracellular Ca2+ (Seishima et al. 1995), several signalling pathways have been shown to be involved in pemphigus pathogenesis (Fig. 8) (Kitajima 2002; Lanza et al. 2006; Sharma et al. 2007; Sitaru and Zillikens 2005). Interestingly, evidence has been provided that transadhering non-desmosomal cadherins, for instance Dsg 3, are involved in “outside-in” signalling and that binding of pemphigus IgG interferes with this function (Muller et al. 2008a). This can be concluded from experiments which showed that autoantibody binding as well as keratinocyte separation started between desmosomes (Sato et al. 2000; Takahashi et al. 1985) and that non-junctional Dsg 3 and plakoglobin were depleted first before changes in the desmosomal fractions were present (Aoyama and Kitajima 1999; Williamson et al. 2006; Yamamoto et al. 2007a). Interestingly, to trigger Dsg-induced signalling, autoantibody-mediated cross-linking of Dsg 3 and Dsg 1 seems not to be required because monovalent Fab fragments and single-chain variants of PV- and PF-IgG were effective to cause skin blistering in vivo and to disrupt the desmosomal plaque in vitro (de Bruin et al. 2007; Ishii et al. 2008; Payne et al. 2005; Rock et al. 1990).
Ca2+, PLC and PKC
It was shown that PV-IgG caused a rapid, transient phospholipase C (PLC)-dependent increase of inositol 1,4,5 trisphosphate and of intracellular Ca2+ leading to activation of both PKC and plasminogen activator (PA) (Esaki et al. 1995; Kitajima et al. 1999; Memar et al. 1996; Osada et al. 1997; Seishima et al. 1995, 1999). Because a chelator of intracellular free Ca2+ blocked keratinocyte dissociation in vitro and inhibitors of calmodulin, PLC and PKC were effective to block PV-IgG-induced acantholysis in vivo (Arredondo et al. 2005; Sanchez-Carpintero et al. 2004), it is possible that this signalling pathway may be involved in PV acantholysis. However, because the PA system is not believed to be crucial in this process, PKC signalling may contribute to PV acantholysis by other pathways such as phosphorylation of β-catenin in adherens junctions (Chernyavsky et al. 2007b). This hypothesis is supported by experiments, which showed that keratinocyte adhesion was negatively regulated by pharmacologic PKC activation (Kimura et al. 2007).
p38MAPK
Activation of p38MAPK at present is the most promising signalling mechanism to be responsible for acantholysis in pemphigus. It has been demonstrated in vivo that p38MAPK and one of its downstream targets, heat shock protein (HSP) 25, were phosphorylated in response to PV-IgG and PF-IgG and that pharmacologic inhibition of p38MAPK abolished blister formation (Berkowitz et al. 2006, 2007b). Similarly, p38MAPK and the human homolog HSP 27 were found to be phosphorylated in skin lesions of PV and PF patients (Berkowitz et al. 2007a). In cultured human keratinocytes, phosphorylation of p38MAPK and HSP27 occured after 30 min of exposure to PV-IgG (Berkowitz et al. 2005; Kawasaki et al. 2006). However, another study found that activity of p38MAPK was not increased before 120 min and that activity peaked after 240 min of PV-IgG treatment (Chernyavsky et al. 2007a). In the latter study it was shown that activation was mediated, at least in part, by Dsg 1- and/or Dsg 3-specific antibodies because Dsg depletion by siRNA reduced p38MAPK activation by 50%. Inhibition of p38MAPK blocked autoantibody-induced keratinocyte dissociation, Rho A inactivation, cytokeratin retraction and reorganization of the actin cytoskeleton (Berkowitz et al. 2005; Chernyavsky et al. 2007a; Waschke et al. 2006). These results demonstrate that p38MAPK is involved in the mechanisms leading to acantholysis and that inhibition of p38MAPK could be a beneficial approach to treat pemphigus patients. Nevertheless, based on the finding that activation of p38MAPK can also be a consequence of cell detachment in rat intestinal epithelium (Rosen et al. 2002), it was proposed that p38MAPK activation is a consequence of cell dissociation (Sharma et al. 2007). However, this is unlikely because significant pemphigus-IgG-induced acantholysis usually takes 12–24 h to occur (Berkowitz et al. 2006; Caldelari et al. 2001; Lanza et al. 2008; Mahoney et al. 1999; Nagasaka et al. 2004; Nguyen et al. 2000c; Shu et al. 2007; Spindler et al. 2007; Takahashi et al. 1985; Tsunoda et al. 2003; Waschke et al. 2005; Waschke et al. 2006; Williamson et al. 2006).
The mechanisms by which p38MAPK and HSP27 lead to keratinocyte dissociation are largely unclear but may involve reorganization of the keratinocyte cytoskeleton. It has been shown recently that serine phosphorylation of cytokeratin 8 by p38MAPK induced cytokeratin network disassembly (Woll et al. 2007). Similarly, HSP27 was found to associate with cytokeratin filaments and to inhibit assembly of glial fibrillary acidic protein (GFAP) (Perng et al. 1999). Similarly, HSP25 was found to interact with actin filaments and to prevent formation of aggregates of thermally denatured actin (Panasenko et al. 2003). However, because p38MAPK-mediated phosphorylation of HSP27 or HSP 25 promoted actin assembly and stabilized F-actin against depolymerization in response to cytochalasin D or heat shock (Benndorf et al. 1994; Geum et al. 2002; Guay et al. 1997), one would expect that PV-IgG-induced HSP27 phosphorylation also would stabilize actin filaments. Therefore, it is more likely that HSP27 is part of a salvage pathway in response to PV-IgG and does not directly contribute to PV-IgG-induced actin reorganization.
Rho A GTPase
Rho GTPases are important regulators of the cytoskeleton and of cell adhesion (Braga and Yap 2005; Bustelo et al. 2007; Fukata et al. 1999; Jaffe and Hall 2005). In the epidermis, Rac 1 was found not to be crucial for maintaining epithelial integrity but for stem cell differentiation, presumably via negative regulation of c-Myc expression (Benitah et al. 2005; Chrostek et al. 2006). Recently, it has been shown that PV- and PF-IgG-induced epidermal splitting, keratinocyte dissociation, as well as loss of Dsg 1 and Dsg 3 binding in vitro were accompanied by p38MAPK-dependent inactivation of Rho A and that specific activation of Rho A by the bacterial toxin cytotoxic necrotizing factor y (CNFy) abolished these effects (Spindler et al. 2007; Waschke et al. 2006). Moreover, toxin-mediated inactivation of Rho A and Rac 1 resulted in epidermal splitting, keratinocyte dissociation and actin reorganization similar to treatment with pemphigus IgG. Especially, inactivation of Rho GTPases by toxin B caused deep epidermal acantholysis comparable to the effects of PV-IgG. Based on this observation, it is tempting to speculate that the cleavage plane in pemphigus may in part be explained by different, layer-specific signalling pathways important for keratinocyte cohesion.
Because cytokeratin retraction and actin reorganization in response to pemphigus IgG were also abrogated by activation of Rho A, these results suggested that Rho A could be involved in the regulation of desmoglein cytoskeletal anchorage (Waschke et al. 2006). This idea was supported by Triton extraction experiments which showed that Rho A activation reduced Dsg 3 in the non-cytoskeleton bound fraction. Alternatively, Rho GTPases may regulate endocytosis of desmosomal cadherins similar to their role in E-cadherin turnover (Akhtar and Hotchin 2001; Izumi et al. 2004; Kamei et al. 1999). Interestingly, endocytosis and depletion of Dsg3 are increasingly recognized to contribute to pemphigus acantholysis (Calkins et al. 2006; Yamamoto et al. 2007a). Taken together, Rho GTPases seem to be important for maintenance of desmosomes and activation of Rho A could be used to develop new strategies in pemphigus treatment.
These data are in contrast to previous findings, which suggested that Rho GTPases are not involved in the regulation of desmosomal adhesion. This was concluded from experiments in keratinocytes which showed that Rac 1 inactivation by transfection with a dominant-inactive mutant as well as inactivation of Rho A by C3 toxin for 25 min was sufficient to displace E-cadherin but not desmoplakin from cell junctions (Braga et al. 1997). These studies led to the hypothesis that Rho GTPases are important regulators of keratinocyte adherens junctions but not desmosomes (Braga and Yap 2005). However, because inactivation of Rho A by a cell-permeable C3 fusion toxin was sufficient to displace Dsg 3 from cell borders after 180 min, the negative results from the early study might be caused by the shorter incubation period (Waschke et al. 2006).
Plakoglobin
Besides its involvement in PV pathogenesis, plakoglobin is important for several aspects of desmosomal adhesion. Plakoglobin deficiency resulted in subcorneal acantholysis, loss of desmosomes, and impaired cytoskeletal anchorage of desmoplakin and desmogleins in vivo and in vitro, supporting the notion that plakoglobin is an important cytoskeletal linker and is crucial for desmosome assembly (Bierkamp et al. 1999; Yin et al. 2005). However, at least in part, plakoglobin functions seem to be compensated by other desmosomal plaque proteins such as plakophilin 1 because a plakoglobin-deficient keratinocyte cell line displayed Dsg 3 cytoskeletal anchorage and keratinocyte aggregation similar to wild-type cells (Caldelari et al. 2001). The linker function of plakoglobin seems to be regulated by tyrosine phosphorylation of plakoglobin because following EGFR activation, phosphorylated plakoglobin remained associated with Dsg 2 but not with desmoplakin (Gaudry et al. 2001). Moreover, plakoglobin phosphorylation was shown to be required for EGFR-induced loss of cell adhesion and to regulate binding of plakoglobin to desmoplakin (Miravet et al. 2003; Yin et al. 2005). These data indicate that plakoglobin is required to integrate the effects from extracellular cues and from different signalling pathways in order to allow coordinated modulation of desmosomal adhesion. In addition, plakoglobin regulates the turn over of desmosomal components because the protein levels of desmogleins as well as of plakoglobin and desmoplakin were decreased in plakoglobin-deficient cells (Yin et al. 2005).
With respect to pemphigus, it has been shown that plakoglobin is critical for PV pathogenesis because keratinocytes from plakoglobin-deficient mice were resistant to PV-IgG-induced keratinocyte dissociation, cytokeratin retraction and disruption of the desmosomal plaque (Caldelari et al. 2001; de Bruin et al. 2007). These studies provided first evidence that direct inhibition of Dsg binding alone cannot account for acantholysis in PV and raised the hypothesis that plakoglobin could be part of a receptor complex required to transfer the signal from autoantibody-bound Dsg 3 into the cell, a phenomenon referred to as “outside-in” signalling (Muller et al. 2008a). As outlined above, it has been shown that plakoglobin is involved in c-Myc repression, which is required for keratinocyte differentiation and that PV-IgG by depleting noncytokeratin-anchored plakoglobin on the cell surface also led to c-Myc overexpression (Williamson et al. 2006). However, as discussed below, the role of c-Myc signalling in acantholysis is unclear at present.
EGFR
The involvement of the EGFR in PV pathogenesis has been reported by showing that EGFR activity increased after 30 min and peaked after 60 min of PV-IgG incubation. This was followed by activation of ERK1/2, c-Jun phosphorylation and finally by keratinocyte apoptosis (Chernyavsky et al. 2007a; Frusic-Zlotkin et al. 2006). Inhibition of EGFR reduced EGFR signalling as well as apoptosis and acantholysis (Frusic-Zlotkin et al. 2006). This finding was surprising because EGFR signalling usually is considered to promote cell survival and proliferation (Bazley and Gullick 2005; Muller et al. 2008a). However, because 60 h were required to induce acantholysis, very high concentrations of IgG (5 mg/ml) were used and activation of p38MAPK was not detected in this study, the significance of these results is unclear.
EGFR could contribute to PV pathogenesis by stimulating desmosome disassembly. It has been shown that EGF-induced loss of keratinocyte aggregation was mediated by plakoglobin phosphorylation, which led to uncoupling of the desmoglein 2-plakoglobin complex from desmoplakin and thus from the cytoskeleton (Gaudry et al. 2001; Yin et al. 2005). Moreover, EGFR seems to negatively regulate the protein levels of desmosomal cadherins, in part by promoting their metalloproteinase-mediated degradation, as well as the incorporation of desmosomal cadherins into the desmosomal plaque (Bech-Serra et al. 2006; Lorch et al. 2004; Santiago-Josefat et al. 2007).
Src
The tyrosine kinase Src is activated by PV-IgG within 30 min and seems to contribute to EGFR and p38MAPK activation because inhibition of Src reduced EGFR and p38MAPK phosphorylation by 45 and 30%, respectively (Chernyavsky et al. 2007a). Because Src inhibition reduced PV-IgG-induced loss of keratinocyte cohesion, cytokeratin retraction and apoptosis in vitro and inhition of tyrosin kinases blocked PV-IgG-induced acantholysis in vivo (Chernyavsky et al. 2007a; Sanchez-Carpintero et al. 2004), these results indicate that Src is significantly involved in PV acantholysis. Besides activation of p38MAPK and EGFR, a recent study provided another possible underlying mechanism: Src was shown to directly phosphorylate p120catenin in response to PV-IgG, which correlated with the degree of acantholysis (Chernyavsky et al. 2007b). Because p120catenin is involved in stabilization of classical cadherins such as E-cadherin on the cell surface as well as in cadherin-dependent regulation of Rho A and Rac 1 activity (Alema and Salvatore 2007; Kowalczyk and Reynolds 2004), Src-mediated phosphorylation of p120catenin may be important for PV pathogenesis.
Cholinergic receptors
The cholinergic system of the human epidermis involves two classes of cholinergig receptors, the nicotinic and the muscarinic acetylcholine receptors (nAChR and mAChR), which in keratinocytes are involved in the regulation of cell–cell and cell–matrix adhesion as well as in cell migration (Grando 2006a). As outlined above, it is unclear whether autoantibodies against cholinergic receptors in pemphigus directly contribute to acantholysis. However, it was shown that cholinergic agonists can ameliorate PV acantholysis in vivo and in vitro (Nguyen et al. 2004b) and that local application of cholinergic agonists such as pilocarpine had therapeutic effect on oral and skin lesions in PV patients (Iraji and Yoosefi 2006; Namazi 2004). Cholinergic agonists increased protein levels of Dsg 1, Dsg 3 and E-cadherin and antagonists to cholinergic receptors resulted in keratinocyte dissociation, which was paralleled by phosphorylation of these molecules (Grando 2006a). Recently, a mechanism by which signalling from cholinergic receptors could directly interfere with PV-IgG-induced effects was demonstrated. The M1 mAChR agonist pilocarpine inhibited PV-IgG-induced acantholysis by reducing serine phosphorylation of β-catenin and Src-mediated tyrosine phosphorylation of p120catenin by activation of the specific protein phosphatases (Chernyavsky et al. 2007b). Similarly, an agonist of the α 7 nAChR reduced p120catenin phosphorylation both on the level of Src activation as well as by activation of the tyrosine phosphatase. Moreover, it was shown that M3 and M4 receptors can lead to activation of Rho A and that carbachol, which activates α 3 and α 7 nAChR, activates Rho A, Rac 1 and Cdc42 (Chernyavsky et al. 2004a, b; Ruiz-Velasco et al. 2002), which also might be involved in the inhibition of PV-IgG-induced acantholysis. Taken together, these data indicate that activation of cholinergic receptors could provide a promising strategy to treat PV patients.
Regulation of cell cycle and gene expression
Recent data support the hypothesis that PV-IgG change expression patterns of molecules involved in cell cycle regulation and that these molecules might be involved in acantholysis. As outlined above, PV-IgG-induced plakoglobin depletion resulted in c-Myc overexpression in cultured keratinocytes (Muller et al. 2008a; Williamson et al. 2006). Accordingly, c-Myc overexpression was found in the epidermis of PV patients, but interestingly not in PF skin, indicating that this mechanism may only be important for PV (Williamson et al. 2007, 2006). Because pharmacologic inhibition of c-Myc as well as of plakoglobin degradation by blocking GSK 3 abrogated pemphigus-IgG-induced skin blistering, these results indicate that this mechanism could also be important to induce acantholysis. However, the findings that c-Myc overexpression was found after 24 h, whereas PV-IgG-induced loss of cell aggregation was observed as early as after 12 h and that c-Myc overexpression was absent in PF skin indicate that c-Myc overexpression likely contributes to ongoing proliferation of keratinocytes in PV, whereas it seems not to be essential for acantholysis. Therefore, other mechanisms must exist in addition to induce keratinocyte dissociation. These mechanisms may, amongst others, involve expression of genes different from c-Myc, which is suggested by the fact that plakoglobin deficiency resulted in protection of keratinocytes from apoptosis, possibly via overexpression of the anti-apoptotic molecule Bcl-XL (Dusek et al. 2007a).
Similar to c-Myc, cyclin-dependent kinase 2 (cdk2), another kinase involved in the regulation of cell cycle progression, seems to be involved in keratinocyte proliferation and acantholysis in pemphigus (Lanza et al. 2008). PV serum increased protein levels of cdk2 and PV serum-induced acantholysis in vivo was abolished by pharmacologic inhibition of cdk2. Moreover, siRNA-mediated down-regulation of cdk2 blocked cell dissociation in cultured keratinocytes indicating that cdk2 may be important for PV acantholysis in vivo and in vitro. However, at present, it is unclear how continuing keratinocyte proliferation in consequence to increased c-Myc and cdk2 signalling contributes to acantholysis in PV.
One explanation is that reduced expression of desmosomal components in hyperproliferating cells may foster the loss of keratinocyte cohesion in pemphigus. PV-IgG do not only induce direct signalling effects on cell junctions and the cytoskeleton but also change gene expression patterns, which may also contribute to pemphigus pathogenesis. It has been shown by DNA microarray that PV-IgG within 8 h down-regulated transcription of 198 genes including genes encoding for adhesion molecules including Dsg 3 and desmoplakin, cytoskeletal proteins such as different cytokeratins as well as molecules involved in cell cycle regulation such as p53 and cyclin D2 in cultured keratinocytes and that expression of some of these genes was antagonistically regulated by methylprednisolone (Nguyen et al. 2004a). When PV serum was used for 24 h, the expression of even more genes was altered. In cultured human keratinocytes, transcription of 231 genes was decreased including Dsc 2 and plakophilin 3 whereas transcription of 329 genes was increased. In vivo, expression of 1114 genes was reduced whereas transcription of 349 other genes was inceased (Lanza et al. 2008). Pharmacologic inhibition of cdk2 blunted the effect of PV-IgG on the expression of most of the genes including desmoplakin and also blocked PV-IgG-induced acantholysis. Thus, it is conceivable that altered desmosome formation may be involved in the mechanisms underlying keratinocyte dissociation in pemphigus.
Apoptosis
Apoptosis has been detected in skin lesions and in perilesional skin of PV and PF patients (Gniadecki et al. 1998; Puviani et al. 2003; Wang et al. 2004a) and hallmarks of apoptosis such as DNA fragmentation, increased expression of pro-apoptotic molecules Fas, FasL, Bax, p53, depetion of anti-apoptotic Bcl-2 and FLIPL as well as activation of caspases 1, 3 and 8 have been observed following treatment of cultured keratinocytes with PV-IgG or PV serum (Arredondo et al. 2005; Baroni et al. 2004; Chernyavsky et al. 2007a; Frusic-Zlotkin et al. 2005, 2006; Pelacho et al. 2004; Puviani et al. 2003; Wang et al. 2004a, b). Hence, compelling evidence has been provided for the presence of programmed cell death in PV although the phenotype of acantholytic cells is different from cells undergoing apoptosis (Arredondo et al. 2005). However, for several experiments prolonged incubation times of 48–72 h (Arredondo et al. 2005; Baroni et al. 2004; Frusic-Zlotkin et al. 2005, 2006; Wang et al. 2004a, b) were required whereas in most studies from the literature, acantholysis was clearly present after 18–24 h (Berkowitz et al. 2006; Caldelari et al. 2001; Lanza et al. 2008; Mahoney et al. 1999; Nagasaka et al. 2004; Nguyen et al. 2000c; Shu et al. 2007; Spindler et al. 2007; Takahashi et al. 1985; Tsunoda et al. 2003; Waschke et al. 2005; Waschke et al. 2006; Williamson et al. 2006). In another study, apoptosis was detected by TUNEL reactivity starting after 6 h of PV-IgG treatment (Chernyavsky et al. 2007a). However, in this study, loss of intercellular adhesion was present after 120 min and thus before onset of apoptosis. Therefore, it is conceivable that apoptosis may be an event parallel to or in consequence of acantholysis. On the other hand, it was reported that activated caspase 3 cleaves Dsg 3 (Weiske et al. 2001) and that caspase and calpain inhibitors can block PV-IgG-induced acantholysis in keratinocyte monolayers and in skin organ culture (Arredondo et al. 2005; Wang et al. 2004a, b; Weiske et al. 2001). Taken together, it is unclear at present to which extent PV-IgG-induced acantholysis is caused by apoptosis.
Targets of signalling pathways in pemphigus
Pemphigus is a desmosomal disease because pathogenic autoantibodies are directed against Dsg 1 and Dsg 3. Moreover, these antibodies result in the depletion of desmosomal components from the cell surface, alterations of desmosomal plaques and a loss of desmosomes (Aoyama and Kitajima 1999; Aoyama et al. 1999; Calkins et al. 2006; de Bruin et al. 2007; Sato et al. 2000; Shu et al. 2007; Waschke et al. 2006; Williamson et al. 2006; Yamamoto et al. 2007a). However, it is unclear at present whether desmosomes are the primary targets of autoantibodies or whether components outside of desmosomes cause disassembly of desmosomes via mechanisms involving adherens junctions or the cytoskeleton, which finally results in acantholysis. The latter hypothesis is supported by the observation that intercellular spaces between desmosomes widen before desmosomes separate (Takahashi et al. 1985).
Effects on desmosomes and adherens junctions
It is well established that pemphigus autoantibodies lead to depletion of desmosomal components from desmosomes as well as from the cell surface. Within 30–60 min, PV-IgG induced internalization and lysosomal degradation of cytoskeleton-unbound Dsg 3 together with plakoglobin (Aoyama and Kitajima 1999; Aoyama et al. 1999; Calkins et al. 2006; Sato et al. 2000; Williamson et al. 2006; Yamamoto et al. 2007a). After 24 h, Dsg 3 but not Dsg 2, plakoglobin or desmoplakin was also depleted from the cytoskeletal fractions leading to reduced total cellular Dsg 3 levels (Calkins et al. 2006; Yamamoto et al. 2007a). This effect was mediated by Dsg 3-specific antibodies because monoclonal Dsg 3 antibodies such as AK23 were effective to induce Dsg 3 depletion and this effect correlated with the pathogenic activity of these autoantibodies in vivo and in vitro (Shu et al. 2007; Yamamoto et al. 2007a). However, no strong correlation between Dsg 3 depletion and keratinocyte dissociation was observed. Dsg 3 depletion was observed in DJM-1 cells at 40% confluence as well as in normal human keratinocytes. In contrast, in confluent HaCaT monolayers, a reduction of Dsg 3 half-life but no depletion of total cellular Dsg 3 was detected (Cirillo et al. 2006; Waschke et al. 2006). Because depletion of Dsg 3 was first found in parallel with serine phosphorylation of Dsg 3, it was suggested that phosphorylation might be required for Dsg 3 degradation (Aoyama and Kitajima 1999; Aoyama et al. 1999; Bystryn and Rodriguez 1978; Rodriguez and Bystryn 1977). However, phosphorylation of Dsg 3 was observed in some studies but not in others, which might be due to different cell culture models indicating that Dsg phosphorylation is not necessary for acantholysis (Chernyavsky et al. 2007b; Nguyen et al. 2004a).
Some evidence exists that depletion of Dsg 1 may also occur in pemphigus. However, studies to systematically investigate Dsg 1 turnover in pemphigus are lacking. Dsg 1 internalization and decreased immunostaining of Dsg 1 have been observed following treatment with PF-IgG/-serum and PV serum (Cirillo et al. 2007b; Waschke et al. 2005). This is in line with previous findings from the literature, which demonstrated that PF-IgG did not bind to keratinocytes in the lower epidermis of PF patients but strongly labelled basal keratinocytes in the epidermis of healthy subjects suggesting that in PF the lower epidermis was depleted from Dsg 1 (Bystryn and Rodriguez 1978; Rodriguez and Bystryn 1977). Taken together, it is unclear at present to which extent Dsg depletion from desmosomes causes antibody-mediated acantholysis. However, even if depletion of desmosomal components occured secondary to keratinocyte dissociation, it likely would further aggravate the loss of keratinocyte cohesion.
The involvement of adherens junctions in pemphigus pathogenesis is unclear at present. It has been shown that formation of adherens junctions is a prerequisite for desmosome assembly and α-catenin-deficient keratinocytes had reduced numbers of desmosmes (Vasioukhin et al. 2000). Because Rho GTPases are known to primarily regulate adhesion mediated by classical cadherins in adherens junctions whereas the requirement of Rho A and Rac 1 activity for maintenance of desmosomal adhesion was reported just recently (Braga and Yap 2005; Fukata et al. 1999; Spindler et al. 2007; Waschke et al. 2006), it was suggested that pemphigus IgG-induced inactivation of Rho A causes disassembly of desmosomes via destabilization of adherens junctions (Sharma et al. 2007). Although alterations of adherens junctions in response to PV-IgG were negligible compared to the effects on desmosomes (Calkins et al. 2006; de Bruin et al. 2007; Muller et al. 2008a; Waschke et al. 2006), this scenario cannot be completely ruled out at present. In fact it was reported recently that PV-IgG lead to phosphorylation of β-catenin and p120catenin by PKC and Src, respectively (Chernyavsky et al. 2007b). However, more studies are required to address the question how catenin phosphorylation in adherens junctions is associated with keratinocyte dissociation in pemphigus.
Effects on the cytoskeleton
Alterations of the cytoskeleton have been described both in pemphigus skin lesions as well as in keratinocytes exposed to pemphigus autoantibodies,. These effects include dysorganization of cytokeratin filaments, the actin cytoskeleton as well as of microtubules.
First, it has been described that cytokeratin filaments become detached from cell junctions and accumulate in the perinuclear region, a phenomenon commonly referred to as “keratin retraction” (Berkowitz et al. 2005; Caldelari et al. 2001; Calkins et al. 2006; Chernyavsky et al. 2007a; Jinbu et al. 1992; Kitajima et al. 1987; Waschke et al. 2006; Wilgram et al. 1961, 1964; Williamson et al. 2006). It was shown that cytokeratin retraction could be detected as early as 4–6 h after addition of PV-IgG, i.e. when first alterations of desmosomes became visible, and was fully established after 24 h (Berkowitz et al. 2005; Calkins et al. 2006; Chernyavsky et al. 2007a). However, because no cytokeratin filament reorganization was observed before cell detachment and the desmosomes of acantholytic cells remained anchored to cytokeratin filament bundles (Kitajima et al. 1986; Shimizu et al. 2004; Spindler et al. 2007), it appears that cytokeratin retraction is not required for acantholysis but may be a secondary event.
Recent studies demonstrated that acantholysis in pemphigus was paralleled by alterations of actin filaments including increased stress fiber formation as well as fragmentation of actin filament bundles. Moreover, pharmacological inhibition of pemphigus IgG-induced acantholysis also was effective to reverse the effects on the cytoskeleton (Berkowitz et al. 2005; Spindler et al. 2007; Waschke et al. 2005; Waschke et al. 2006). Similarly, microtubule distribution was found to be altered by pemphigus IgG (Kitajima et al. 1986). Taken together, pemphigus IgG cause profound cytoskeletal reorganization but it is unclear at present whether these effects contribute to acantholysis or are triggered consequent to keratinocyte dissociation. Nevertheless, it is likely that alterations in the cytoskeleton may account for the changes in cell shape during keratinocyte dissociation, which seem to start between existing desmosomes (Bystryn and Grando 2006; Takahashi et al. 1985).
The role of the different mechanisms in blister formation
As outlined above, both direct and indirect mechanisms finally leading to a loss of desmoglein-mediated adhesion have been found to be involved in pemphigus acantholysis. At present, compelling evidence indicates that acantholysis in PV and in PF is initiated by cellular signalling pathways rather than by direct inhibition of Dsg binding (Fig. 11). This was first shown using plakoglobin-deficient keratinocytes, which were resistant against PV-IgG-induced acantholysis and was further supported by the findings that inhibition of p38MAPK and activation of Rho A is sufficient to abolish PV-IgG and PF-IgG-induced skin blistering (Berkowitz et al. 2005, 2006, 2007b; Caldelari et al. 2001; Waschke et al. 2006). Moreover, low temperature-abrogated keratinocyte dissociation but not antibody binding to the keratinocyte cell surface indicating that direct inhibition of cadherin transinteraction by autoantibody binding is not sufficient to cause acantholysis (Calkins et al. 2006). If extracellular autoantibody-mediated direct inhibition of Dsg binding would be the primary cause of acantholysis, it is hard to imagine how this process should be blocked by modified intracellular signalling or low temperature. It is possible that antibody-mediated direct inhibition of Dsg binding may contribute to trigger cellular signalling events (Muller et al. 2008a; Sharma et al. 2007; Tsunoda et al. 2003). However, because PF-IgG were shown to induce cellular signalling events but not to directly reduce Dsg 1 binding, it seems that direct inhibition of Dsg transinteraction is not essential to alter Dsg-mediated signalling (Heupel et al. 2007; Waschke et al. 2005).

The mechanisms involved in pemphigus acantholysis. Accumulating evidence indicates that PV-IgG and PF-IgG initiate keratinocyte dissociation via intracellular signalling pathways including p38 MAPK, Rho A and plakoglobin (PV only). In addition, other mechanisms such as direct inhibition of Dsg 3 binding and Dsg 3 depletion from desmosomes as well as other signalling events seem to contribute to PV pathogenesis, whereas their role for acantholysis in PF is unclear. These mechanisms may account for the more severe clinical phenotype of PV compared to PF. PLC phospholipase C, PKC protein kinase C, cdk 2 cyclin-dependent kinase 2, EGFR epidermal growth factor receptor
It is tempting to speculate that the clinical phenotype of PV may be more severe compared to PF because additional mechanisms could be involved in PV (Fig. 11). Indeed, some mechanisms have only been found to contribute to acantholysis in PV but not in PF. For instance, direct inhibition of Dsg 3 transinteraction was found in PV but not in PF under conditions where antibodies caused acantholysis (Heupel et al. 2007). Therefore, it is possible that direct inhibition as a second pathway to reduce Dsg 3 binding in PV may explain the more severe phenotype of PV. Similarly, depletion of Dsg 3 has been convincingly shown in PV whereas depletion of Dsg 1 in both PV and PF is less clear (Yamamoto et al. 2007a). Moreover, altered plakoglobin signalling leading to c-Myc overexpression was found in PV patients’s skin but not in PF (Williamson et al. 2007, 2006). Other pathways have been investigated for PV only whereas data for PF are lacking. As outlined above, inhibitors of calmodulin, PKC, EGFR, cdk2 as well as of tyrosin kinases have been shown to inhibit PV-IgG-induced acantholysis.
Finally, it has to be emphasized that desmoglein proteolysis, mechanical stress as well as secondary changes for example in the extracellular Ca2+ concentration might contribute to skin blistering. For instance, acantholysis may foster skin blistering by derangement of the epidermal Ca2+ gradient, which in turn would result in loss of desmosomal cadherin binding, especially when the tight junctions of the granular layer are affected.
Concluding remarks
The major goal for the future is to elucidate the primary signalling pathways responsible for the diverse effects of pemphigus IgG such as inhibition of desmoglein binding, depletion of desmosomal components, loss of desmosomes, reorganization of the cytoskeleton and finally the induction of acantholysis. Other pathways will be identified to represent salvage mechanisms to rescue keratinocyte cohesion (Grando 2006b). Moreover, future experiments may reveal how the different signalling pathways are involved in the regulation of desmosomal adhesion in the specific epidermal layers. This knowledge may elucidate why the cleavage planes in PV and PF usually are different. The second goal is to characterize whether acantholysis in pemphigus is caused by mechanisms directly targeting desmosomes or whether cellular signalling pathways regulate desmosomal adhesion via reorganisation of the cytoskeleton or via other types of cell contacts such as adherens junctions.
References
Aberle H, Bierkamp C, Torchard D, Serova O, Wagner T, Natt E, Wirsching J, Heidkamper C, Montagna M, Lynch HT et al (1995) The human plakoglobin gene localizes on chromosome 17q21 and is subjected to loss of heterozygosity in breast and ovarian cancers. Proc Natl Acad Sci USA 92:6384–6388
Ahmed AR, Spigelman Z, Cavacini LA, Posner MR (2006) Treatment of pemphigus vulgaris with rituximab and intravenous immune globulin. New Engl J Med 355:1772–1779
Akat K, Bleck CK, Lee YM, Haselmann-Weiss U, Kartenbeck J (2008) Characterization of a novel type of adherens junction in meningiomas and the derived cell line HBL-52. Cell Tissue Res 331:401–412
Akhtar N, Hotchin NA (2001) RAC1 regulates adherens junctions through endocytosis of E-cadherin. Molec Biol Cell 12:847–862
Al-Amoudi A, Diez DC, Betts MJ, Frangakis AS (2007) The molecular architecture of cadherins in native epidermal desmosomes. Nature 450:832–837
Alema S, Salvatore AM (2007) p120 catenin and phosphorylation: mechanisms and traits of an unresolved issue. Biochim Biophys Acta 1773:47–58
Amagai M (2003) Desmoglein as a target in autoimmunity and infection. J Am Acad Dermatol 48:244–252
Amagai M, Klaus-Kovtun V, Stanley JR (1991) Autoantibodies against a novel epithelial cadherin in pemphigus vulgaris, a disease of cell adhesion. Cell 67:869–877
Amagai M, Karpati S, Prussick R, Klaus-Kovtun V, Stanley JR (1992) Autoantibodies against the amino-terminal cadherin-like binding domain of pemphigus vulgaris antigen are pathogenic. J Clin Invest 90:919–926
Amagai M, Hashimoto T, Shimizu N, Nishikawa T (1994a) Absorption of pathogenic autoantibodies by the extracellular domain of pemphigus vulgaris antigen (Dsg3) produced by baculovirus. J Clin Invest 94:59–67
Amagai M, Karpati S, Klaus-Kovtun V, Udey MC, Stanley JR (1994b) Extracellular domain of pemphigus vulgaris antigen (desmoglein 3) mediates weak homophilic adhesion. J Invest Dermatol 103:609–615
Amagai M, Hashimoto T, Green KJ, Shimizu N, Nishikawa T (1995) Antigen-specific immunoadsorption of pathogenic autoantibodies in pemphigus foliaceus. J Invest Dermatol 104:895–901
Amagai M, Koch PJ, Nishikawa T, Stanley JR (1996) Pemphigus vulgaris antigen (desmoglein 3) is localized in the lower epidermis, the site of blister formation in patients. J Invest Dermatol 106:351–355
Amagai M, Nishikawa T, Nousari HC, Anhalt GJ, Hashimoto T (1998) Antibodies against desmoglein 3 (pemphigus vulgaris antigen) are present in sera from patients with paraneoplastic pemphigus and cause acantholysis in vivo in neonatal mice. J Clin Invest 102:775–782
Amagai M, Tsunoda K, Zillikens D, Nagai T, Nishikawa T (1999) The clinical phenotype of pemphigus is defined by the anti-desmoglein autoantibody profile. J Am Acad Dermatol 40:167–170
Amagai M, Matsuyoshi N, Wang ZH, Andl C, Stanley JR (2000a) Toxin in bullous impetigo and staphylococcal scalded-skin syndrome targets desmoglein 1. Nat Med 6:1275–1277
Amagai M, Tsunoda K, Suzuki H, Nishifuji K, Koyasu S, Nishikawa T (2000b) Use of autoantigen-knockout mice in developing an active autoimmune disease model for pemphigus. J Clin Invest 105:625–631
Amagai M, Yamaguchi T, Hanakawa Y, Nishifuji K, Sugai M, Stanley JR (2002) Staphylococcal exfoliative toxin B specifically cleaves desmoglein 1. J Invest Dermatol 118:845–850
Amagai M, Ahmed AR, Kitajima Y, Bystryn JC, Milner Y, Gniadecki R, Hertl M, Pincelli C, Fridkis-Hareli M, Aoyama Y, Frusic-Zlotkin M, Muller E, David M, Mimouni D, Vind-Kezunovic D, Michel B, Mahoney M, Grando S (2006) Are desmoglein autoantibodies essential for the immunopathogenesis of pemphigus vulgaris, or just ‘witnesses of disease’? Exp Dermatol 15:815
Anastasiadis PZ, Reynolds AB (2001) Regulation of Rho GTPases by p120-catenin. Curr Opin Cell Biol 13:604–610
Angst BD, Khan LU, Severs NJ, Whitely K, Rothery S, Thompson RP, Magee AI, Gourdie RG (1997) Dissociated spatial patterning of gap junctions and cell adhesion junctions during postnatal differentiation of ventricular myocardium. Circ Res 80:88–94
Anhalt GJ, Labib RS, Voorhees JJ, Beals TF, Diaz LA (1982) Induction of pemphigus in neonatal mice by passive transfer of IgG from patients with the disease. New Engl J Med 306:1189–1196
Anhalt GJ, Patel HP, Labib RS, Diaz LA, Proud D (1986) Dexamethasone inhibits plasminogen activator activity in experimental pemphigus in vivo but does not block acantholysis. J Immunol 136:113–117
Aoki V, Millikan RC, Rivitti EA, Hans-Filho G, Eaton DP, Warren SJ, Li N, Hilario-Vargas J, Hoffmann RG, Diaz LA (2004) Environmental risk factors in endemic pemphigus foliaceus (fogo selvagem). J Invest Dermatol Symp Proc/Soc Invest Dermatol Inc 9:34–40
Aoyama Y, Kitajima Y (1999) Pemphigus vulgaris-IgG causes a rapid depletion of desmoglein 3 (Dsg3) from the Triton X-100 soluble pools, leading to the formation of Dsg3-depleted desmosomes in a human squamous carcinoma cell line, DJM-1 cells. J Invest Dermatol 112:67–71
Aoyama Y, Owada MK, Kitajima Y (1999) A pathogenic autoantibody, pemphigus vulgaris-IgG, induces phosphorylation of desmoglein 3, and its dissociation from plakoglobin in cultured keratinocytes. Eur J Immunol 29:2233–2240
Armstrong DK, McKenna KE, Purkis PE, Green KJ, Eady RA, Leigh IM, Hughes AE (1999) Haploinsufficiency of desmoplakin causes a striate subtype of palmoplantar keratoderma. Hum Mol Genet 8:143–148
Arnemann J, Spurr NK, Wheeler GN, Parker AE, Buxton RS (1991) Chromosomal assignment of the human genes coding for the major proteins of the desmosome junction, desmoglein DGI (DSG), desmocollins DGII/III (DSC), desmoplakins DPI/II (DSP), and plakoglobin DPIII (JUP). Genomics 10:640–645
Arnemann J, Sullivan KH, Magee AI, King IA, Buxton RS (1993) Stratification-related expression of isoforms of the desmosomal cadherins in human epidermis. J Cell Sci 104(Pt 3):741–750
Arredondo J, Chernyavsky AI, Karaouni A, Grando SA (2005) Novel mechanisms of target cell death and survival and of therapeutic action of IVIg in Pemphigus. Am J Pathol 167:1531–1544
Asimaki A, Syrris P, Wichter T, Matthias P, Saffitz JE, McKenna WJ (2007) A novel dominant mutation in plakoglobin causes arrhythmogenic right ventricular cardiomyopathy. Am J Hum Genet 81:964–973
Baroni A, Buommino E, Paoletti I, Orlando M, Ruocco E, Ruocco V (2004) Pemphigus serum and captopril induce heat shock protein 70 and inducible nitric oxide synthase overexpression, triggering apoptosis in human keratinocytes. Br J Dermatol 150:1070–1080
Baumgartner W, Hinterdorfer P, Ness W, Raab A, Vestweber D, Schindler H, Drenckhahn D (2000) Cadherin interaction probed by atomic force microscopy. Proc Natl Acad Sci USA 97:4005–4010
Baumgartner W, Golenhofen N, Grundhofer N, Wiegand J, Drenckhahn D (2003) Ca2+ dependency of N-cadherin function probed by laser tweezer and atomic force microscopy. J Neurosci 23:11008–11014
Baykal C, Azizlerli G, Thoma-Uszynski S, Hertl M (2002) Pemphigus vulgaris localized to the nose and cheeks. J Am Acad Dermatol 47:875–880
Bazley LA, Gullick WJ (2005) The epidermal growth factor receptor family. Endocr Relat Cancer 12(Suppl 1):S17–S27
Bazzi H, Christiano AM (2007) Broken hearts, woolly hair, and tattered skin: when desmosomal adhesion goes awry. Curr Opin Cell Biol 19:515–520
Bech-Serra JJ, Santiago-Josefat B, Esselens C, Saftig P, Baselga J, Arribas J, Canals F (2006) Proteomic identification of desmoglein 2 and activated leukocyte cell adhesion molecule as substrates of ADAM17 and ADAM10 by difference gel electrophoresis. Mol Cell Biol 26:5086–5095
Benitah SA, Frye M, Glogauer M, Watt FM (2005) Stem cell depletion through epidermal deletion of Rac1. Science (New York, NY) 309:933–935
Benndorf R, Hayess K, Ryazantsev S, Wieske M, Behlke J, Lutsch G (1994) Phosphorylation and supramolecular organization of murine small heat shock protein HSP25 abolish its actin polymerization-inhibiting activity. J Biol Chem 269:20780–20784
Berkowitz P, Hu P, Liu Z, Diaz LA, Enghild JJ, Chua MP, Rubenstein DS (2005) Desmosome signaling. Inhibition of p38MAPK prevents pemphigus vulgaris IgG-induced cytoskeleton reorganization. J Biol Chem 280:23778–23784
Berkowitz P, Hu P, Warren S, Liu Z, Diaz LA, Rubenstein DS (2006) p38MAPK inhibition prevents disease in pemphigus vulgaris mice. Proc Natl Acad Sci USA 103:12855–12860
Berkowitz P, Diaz LA, Hall RP, Rubenstein DS (2007a) Induction of p38MAPK and HSP27 phosphorylation in pemphigus patient skin. J Invest Dermatol 128(3):738–740
Berkowitz P, Liu Z, Diaz LA, Rubenstein DS (2007b) Autoantibodies in the autoimmune disease pemphigus foliaceus induce blistering in vivo via p38MAPK-dependent signalling. J Invest Dermatol 127(Suppl):34 (abstract 201)
Beutner EH, Jordon RE (1964) Demonstration of skin antibodies in sera of pemphigus vulgaris patients by indirect immunofluorescent staining. Proc Soc Exp Biol Med 117:505–510
Bhol K, Natarajan K, Nagarwalla N, Mohimen A, Aoki V, Ahmed AR (1995) Correlation of peptide specificity and IgG subclass with pathogenic and nonpathogenic autoantibodies in pemphigus vulgaris: a model for autoimmunity. Proc Natl Acad Sci USA 92:5239–5243
Bierkamp C, McLaughlin KJ, Schwarz H, Huber O, Kemler R (1996) Embryonic heart and skin defects in mice lacking plakoglobin. Dev Biol 180:780–785
Bierkamp C, Schwarz H, Huber O, Kemler R (1999) Desmosomal localization of beta-catenin in the skin of plakoglobin null-mutant mice. Development (Cambridge, England) 126:371–381
Bizzozero G (1864) Delle cellule cigliate, del reticulo Malpighiani d’ell epiderme. Ann Univ Med 190
Blaschuk OW, Sullivan R, David S, Pouliot Y (1990) Identification of a cadherin cell adhesion recognition sequence. Dev Biol 139:227–229
Boggon TJ, Murray J, Chappuis-Flament S, Wong E, Gumbiner BM, Shapiro L (2002) C-cadherin ectodomain structure and implications for cell adhesion mechanisms. Science (New York, NY 296:1308–1313
Bonne S, van Hengel J, van Roy F (1998) Chromosomal mapping of human armadillo genes belonging to the p120(ctn)/plakophilin subfamily. Genomics 51:452–454
Bonne S, Gilbert B, Hatzfeld M, Chen X, Green KJ, van Roy F (2003) Defining desmosomal plakophilin-3 interactions. J Cell Biol 161:403–416
Bornslaeger EA, Corcoran CM, Stappenbeck TS, Green KJ (1996) Breaking the connection: displacement of the desmosomal plaque protein desmoplakin from cell–cell interfaces disrupts anchorage of intermediate filament bundles and alters intercellular junction assembly. J Cell Biol 134:985–1001
Bornslaeger EA, Godsel LM, Corcoran CM, Park JK, Hatzfeld M, Kowalczyk AP, Green KJ (2001) Plakophilin 1 interferes with plakoglobin binding to desmoplakin, yet together with plakoglobin promotes clustering of desmosomal plaque complexes at cell–cell borders. J Cell Sci 114:727–738
Borrmann CM, Grund C, Kuhn C, Hofmann I, Pieperhoff S, Franke WW (2006) The area composita of adhering junctions connecting heart muscle cells of vertebrates. II. Colocalizations of desmosomal and fascia adhaerens molecules in the intercalated disk. Euro J Cell Biol 85:469–485
Braga VM, Yap AS (2005) The challenges of abundance: epithelial junctions and small GTPase signalling. Curr Opin Cell Biol 17:466–474
Braga VM, Machesky LM, Hall A, Hotchin NA (1997) The small GTPases Rho and Rac are required for the establishment of cadherin-dependent cell–cell contacts. J Cell Biol 137:1421–1431
Brennan D, Hu Y, Joubeh S, Choi YW, Whitaker-Menezes D, O’Brien T, Uitto J, Rodeck U, Mahoney MG (2007) Suprabasal Dsg2 expression in transgenic mouse skin confers a hyperproliferative and apoptosis-resistant phenotype to keratinocytes. J Cell Sci 120:758–771
Burdett ID, Sullivan KH (2002) Desmosome assembly in MDCK cells: transport of precursors to the cell surface occurs by two phases of vesicular traffic and involves major changes in centrosome and Golgi location during a Ca(2+) shift. Exp Cell Res 276:296–309
Bustelo XR, Sauzeau V, Berenjeno IM (2007) GTP-binding proteins of the Rho/Rac family: regulation, effectors and functions in vivo. Bioessays 29:356–370
Buxton RS, Cowin P, Franke WW, Garrod DR, Green KJ, King IA, Koch PJ, Magee AI, Rees DA, Stanley JR et al (1993) Nomenclature of the desmosomal cadherins. J Cell Biol 121:481–483
Bystryn JC, Grando SA (2006) A novel explanation for acantholysis in pemphigus vulgaris: the basal cell shrinkage hypothesis. J Am Acad Dermatol 54:513–516
Bystryn JC, Rodriguez J (1978) Absence of intercellular antigens in the deep layers of the epidermis in pemphigus foliaceus. J Clin Invest 61:339–348
Bystryn JC, Rudolph JL (2005) Pemphigus. Lancet 366:61–73
Caldelari R, de Bruin A, Baumann D, Suter MM, Bierkamp C, Balmer V, Muller E (2001) A central role for the armadillo protein plakoglobin in the autoimmune disease pemphigus vulgaris. J Cell Biol 153:823–834
Calkins CC, Setzer SV (2007) Spotting desmosomes:the first 100 years. J Invest Dermatol 127:E2–E3
Calkins CC, Setzer SV, Jennings JM, Summers S, Tsunoda K, Amagai M, Kowalczyk AP (2006) Desmoglein endocytosis and desmosome disassembly are coordinated responses to pemphigus autoantibodies. J Biol Chem 281:7623–7634
Chen X, Bonne S, Hatzfeld M, van Roy F, Green KJ (2002) Protein binding and functional characterization of plakophilin 2. Evidence for its diverse roles in desmosomes and beta-catenin signaling. J Biol Chem 277:10512–10522
Chen J, Den Z, Merched-Sauvage M, Koch PJ (2007) Loss of desmocollin 3 in the epidermis of mice causes epidermal blistering and telogen hair loss. J Invest Dermatol 127 (Suppl 1)
Chernyavsky AI, Arredondo J, Marubio LM, Grando SA (2004a) Differential regulation of keratinocyte chemokinesis and chemotaxis through distinct nicotinic receptor subtypes. J Cell Sci 117:5665–5679
Chernyavsky AI, Arredondo J, Wess J, Karlsson E, Grando SA (2004b) Novel signaling pathways mediating reciprocal control of keratinocyte migration and wound epithelialization through M3 and M4 muscarinic receptors. J Cell Biol 166:261–272
Chernyavsky AI, Arredondo J, Kitajima Y, Sato-Nagai M, Grando SA (2007a) Desmoglein versus non-desmoglein signaling in pemphigus acantholysis: characterization of novel signaling pathways downstream of pemphigus vulgaris antigens. J Biol Chem 282:13804–13812
Chernyavsky AI, Arredondo J, Piser T, Karlsson E, Grando SA (2007b) Differential coupling of M1 muscarinic and alpha 7 nicotinic receptors to inhibition of pemphigus acantholysis. J Biol Chem 283(6):3401–3408
Chidgey M, Brakebusch C, Gustafsson E, Cruchley A, Hail C, Kirk S, Merritt A, North A, Tselepis C, Hewitt J, Byrne C, Fassler R, Garrod D (2001) Mice lacking desmocollin 1 show epidermal fragility accompanied by barrier defects and abnormal differentiation. J Cell Biol 155:821–832
Chitaev NA, Troyanovsky SM (1997) Direct Ca2+-dependent heterophilic interaction between desmosomal cadherins, desmoglein and desmocollin, contributes to cell–cell adhesion. J Cell Biol 138:193–201
Chitaev NA, Averbakh AZ, Troyanovsky RB, Troyanovsky SM (1998) Molecular organization of the desmoglein-plakoglobin complex. J Cell Sci 111(Pt 14):1941–1949
Choi HJ, Park-Snyder S, Pascoe LT, Green KJ, Weis WI (2002) Structures of two intermediate filament-binding fragments of desmoplakin reveal a unique repeat motif structure. Nat Struct Biol 9:612–620
Chrostek A, Wu X, Quondamatteo F, Hu R, Sanecka A, Niemann C, Langbein L, Haase I, Brakebusch C (2006) Rac1 is crucial for hair follicle integrity but is not essential for maintenance of the epidermis. Mol Cell Biol 26:6957–6970
Cirillo N, Femiano F, Gombos F, Lanza A (2006) Serum from pemphigus vulgaris reduces desmoglein 3 half-life and perturbs its de novo assembly to desmosomal sites in cultured keratinocytes. FEBS Lett 580:3276–3281
Cirillo N, Femiano F, Gombos F, Lanza A (2007a) Metalloproteinase 9 is the outer executioner of desmoglein 3 in apoptotic keratinocytes. Oral Diseases 13:341–345
Cirillo N, Gombos F, Lanza A (2007b) Changes in desmoglein 1 expression and subcellular localization in cultured keratinocytes subjected to anti-desmoglein 1 pemphigus autoimmunity. J Cell Physiol 210:411–416
Cirillo N, Lanza M, Femiano F, Gaeta GM, De Rosa A, Gombos F, Lanza A (2007c) If pemphigus vulgaris IgG are the cause of acantholysis, new IgG-independent mechanisms are the concause. J Cell Physiol 212:563–567
Cirillo N, Lanza M, Rossiello L, Gombos F, Lanza A (2007d) Defining the involvement of proteinases in pemphigus vulgaris: evidence of matrix metalloproteinase-9 overexpression in experimental models of disease. J Cell Physiol 212:36–41
Cirillo N, Dell’ Ermo A, Gombos F, Lanza A (2008) The specific proteolysis hypothesis of pemphigus: does the song remain the same? Med Hypotheses 70:333–337
Collins JE, Legan PK, Kenny TP, MacGarvie J, Holton JL, Garrod DR (1991) Cloning and sequence analysis of desmosomal glycoproteins 2 and 3 (desmocollins): cadherin-like desmosomal adhesion molecules with heterogeneous cytoplasmic domains. J Cell Biol 113:381–391
Cowin P, Mattey D, Garrod D (1984) Identification of desmosomal surface components (desmocollins) and inhibition of desmosome formation by specific Fab’. J Cell Sci 70:41–60
Cowin P, Kapprell HP, Franke WW, Tamkun J, Hynes RO (1986) Plakoglobin: a protein common to different kinds of intercellular adhering junctions. Cell 46:1063–1073
Cowley CM, Simrak D, Marsden MD, King IA, Arnemann J, Buxton RS (1997) A YAC contig joining the desmocollin and desmoglein loci on human chromosome 18 and ordering of the desmocollin genes. Genomics 42:208–216
de Bruin A, Caldelari R, Williamson L, Suter MM, Hunziker T, Wyder M, Muller EJ (2007) Plakoglobin-dependent disruption of the desmosomal plaque in pemphigus vulgaris. Exp Dermatol 16:468–475
Den Z, Cheng X, Merched-Sauvage M, Koch PJ (2006) Desmocollin 3 is required for pre-implantation development of the mouse embryo. J Cell Sci 119:482–489
Dhitavat J, Dode L, Leslie N, Sakuntabhai A, Lorette G, Hovnanian A (2003) Mutations in the sarcoplasmic/endoplasmic reticulum Ca2+ ATPase isoform cause Darier’s disease. J Invest Dermatol 121:486–489
Dhitavat J, Fairclough RJ, Hovnanian A, Burge SM (2004) Calcium pumps and keratinocytes: lessons from Darier’s disease and Hailey–Hailey disease. Br J Dermatol 150:821–828
Diaz LA, Sampaio SA, Rivitti EA, Martins CR, Cunha PR, Lombardi C, Almeida FA, Castro RM, Macca ML, Lavrado C et al (1989) Endemic pemphigus foliaceus (Fogo Selvagem): II. Current and historic epidemiologic studies. J Invest Dermatol 92:4–12
Ding X, Aoki V, Mascaro JM Jr, Lopez-Swiderski A, Diaz LA, Fairley JA (1997) Mucosal and mucocutaneous (generalized) pemphigus vulgaris show distinct autoantibody profiles. J Invest Dermatol 109:592–596
Drochmans P, Freudenstein C, Wanson JC, Laurent L, Keenan TW, Stadler J, Leloup R, Franke WW (1978) Structure and biochemical composition of desmosomes and tonofilaments isolated from calf muzzle epidermis. J Cell Biol 79:427–443
Dusek RL, Godsel LM, Chen F, Strohecker AM, Getsios S, Harmon R, Muller EJ, Caldelari R, Cryns VL, Green KJ (2007a) Plakoglobin deficiency protects keratinocytes from apoptosis. J Invest Dermatol 127:792–801
Dusek RL, Godsel LM, Green KJ (2007b) Discriminating roles of desmosomal cadherins: beyond desmosomal adhesion. J Dermatol Sci 45:7–21
Elias PM, Matsuyoshi N, Wu H, Lin C, Wang ZH, Brown BE, Stanley JR (2001) Desmoglein isoform distribution affects stratum corneum structure and function. J Cell Biol 153:243–249
Elias P, Ahn S, Brown B, Crumrine D, Feingold KR (2002) Origin of the epidermal calcium gradient: regulation by barrier status and role of active vs. passive mechanisms. J Invest Dermatol 119:1269–1274
Esaki C, Seishima M, Yamada T, Osada K, Kitajima Y (1995) Pharmacologic evidence for involvement of phospholipase C in pemphigus IgG-induced inositol 1,4,5-trisphosphate generation, intracellular calcium increase, and plasminogen activator secretion in DJM-1 cells, a squamous cell carcinoma line. J Invest Dermatol 105:329–333
Eshkind L, Tian Q, Schmidt A, Franke WW, Windoffer R, Leube RE (2002) Loss of desmoglein 2 suggests essential functions for early embryonic development and proliferation of embryonal stem cells. Euro J Cell Biol 81:592–598
Evangelista F, Dasher DA, Diaz LA, Prisayanh PS, Li N (2008) E-cadherin is an additional immunological target for pemphigus autoantibodies. J Invest Dermatol (in press)
Eyre RW, Stanley JR (1987) Human autoantibodies against a desmosomal protein complex with a calcium-sensitive epitope are characteristic of pemphigus foliaceus patients. J Exp Med 165:1719–1724
Eyre RW, Stanley JR (1988) Identification of pemphigus vulgaris antigen extracted from normal human epidermis and comparison with pemphigus foliaceus antigen. J Clin Invest 81:807–812
Farb RM, Dykes R, Lazarus GS (1978) Anti-epidermal-cell-surface pemphigus antibody detaches viable epidermal cells from culture plates by activation of proteinase. Proc Natl Acad Sci USA 75:459–463
Farquhar MG, Palade GE (1963) Junctional complexes in various epithelia. J Cell Biol 17:375–412
Farrell AM (1999) Staphylococcal scalded-skin syndrome. Lancet 354:880–881
Fawcett DW, McNutt NS (1969) The ultrastructure of the cat myocardium. I. Ventricular papillary muscle. J Cell Biol 42:1–45
Fawcett DW, Selby CC (1958) Observations on the fine structure of the turtle atrium. J Biophys Biochem Cytol 4:63–72
Feliciani C, Toto P, Amerio P, Pour SM, Coscione G, Shivji G, Wang B, Sauder DN (2000) In vitro and in vivo expression of interleukin-1alpha and tumor necrosis factor-alpha mRNA in pemphigus vulgaris: interleukin-1alpha and tumor necrosis factor-alpha are involved in acantholysis. J Invest Dermatol 114:71–77
Feliciani C, Toto P, Wang B, Sauder DN, Amerio P, Tulli A (2003) Urokinase plasminogen activator mRNA is induced by IL-1alpha and TNF-alpha in in vitro acantholysis. Exp Dermatol 12:466–471
Franke WW, Mueller H, Mittnacht S, Kapprell HP, Jorcano JL (1983) Significance of two desmosome plaque-associated polypeptides of molecular weights 75000 and 83000. EMBO J 2:2211–2215
Franke WW, Goldschmidt MD, Zimbelmann R, Mueller HM, Schiller DL, Cowin P (1989) Molecular cloning and amino acid sequence of human plakoglobin, the common junctional plaque protein. Proc Natl Acad Sci USA 86:4027–4031
Franke WW, Borrmann CM, Grund C, Pieperhoff S (2006) The area composita of adhering junctions connecting heart muscle cells of vertebrates. I. Molecular definition in intercalated disks of cardiomyocytes by immunoelectron microscopy of desmosomal proteins. Euro J Cell Biol 85:69–82
Frusic-Zlotkin M, Pergamentz R, Michel B, David M, Mimouni D, Bregegere F, Milner Y (2005) The interaction of pemphigus autoimmunoglobulins with epidermal cells: activation of the fas apoptotic pathway and the use of caspase activity for pathogenicity tests of pemphigus patients. Ann N Y Acad Sci 1050:371–379
Frusic-Zlotkin M, Raichenberg D, Wang X, David M, Michel B, Milner Y (2006) Apoptotic mechanism in pemphigus autoimmunoglobulins-induced acantholysis–possible involvement of the EGF receptor. Autoimmunity 39:563–575
Fukata M, Nakagawa M, Kuroda S, Kaibuchi K (1999) Cell adhesion and Rho small GTPases. J Cell Sci 112(Pt 24):4491–4500
Futei Y, Amagai M, Sekiguchi M, Nishifuji K, Fujii Y, Nishikawa T (2000) Use of domain-swapped molecules for conformational epitope mapping of desmoglein 3 in pemphigus vulgaris. J Invest Dermatol 115:829–834
Gallicano GI, Kouklis P, Bauer C, Yin M, Vasioukhin V, Degenstein L, Fuchs E (1998) Desmoplakin is required early in development for assembly of desmosomes and cytoskeletal linkage. J Cell Biol 143:2009–2022
Gallicano GI, Bauer C, Fuchs E (2001) Rescuing desmoplakin function in extra-embryonic ectoderm reveals the importance of this protein in embryonic heart, neuroepithelium, skin and vasculature. Development (Cambridge, England) 128:929–941
Garcia-Gras E, Lombardi R, Giocondo MJ, Willerson JT, Schneider MD, Khoury DS, Marian AJ (2006) Suppression of canonical Wnt/beta-catenin signaling by nuclear plakoglobin recapitulates phenotype of arrhythmogenic right ventricular cardiomyopathy. J Clin Invest 116:2012–2021
Garrod DR, Merritt AJ, Nie Z (2002) Desmosomal cadherins. Curr Opin Cell Biol 14:537–545
Gaudry CA, Palka HL, Dusek RL, Huen AC, Khandekar MJ, Hudson LG, Green KJ (2001) Tyrosine-phosphorylated plakoglobin is associated with desmogleins but not desmoplakin after epidermal growth factor receptor activation. J Biol Chem 276:24871–24880
Gerull B, Heuser A, Wichter T, Paul M, Basson CT, McDermott DA, Lerman BB, Markowitz SM, Ellinor PT, MacRae CA, Peters S, Grossmann KS, Drenckhahn J, Michely B, Sasse-Klaassen S, Birchmeier W, Dietz R, Breithardt G, Schulze-Bahr E, Thierfelder L (2004) Mutations in the desmosomal protein plakophilin-2 are common in arrhythmogenic right ventricular cardiomyopathy. Nat Genet 36:1162–1164
Getsios S, Amargo EV, Dusek RL, Ishii K, Sheu L, Godsel LM, Green KJ (2004a) Coordinated expression of desmoglein 1 and desmocollin 1 regulates intercellular adhesion. Differ Res Biol Divers 72:419–433
Getsios S, Huen AC, Green KJ (2004b) Working out the strength and flexibility of desmosomes. Nat Rev Mol Cell Biol 5:271–281
Geum D, Son GH, Kim K (2002) Phosphorylation-dependent cellular localization and thermoprotective role of heat shock protein 25 in hippocampal progenitor cells. J Biol Chem 277:19913–19921
Giudice GJ, Cohen SM, Patel NH, Steinberg MS (1984) Immunological comparison of desmosomal components from several bovine tissues. J Cell Biochem 26:35–45
Gniadecki R, Jemec GB, Thomsen BM, Hansen M (1998) Relationship between keratinocyte adhesion and death: anoikis in acantholytic diseases. Arch Dermatol Res 290:528–532
Godsel LM, Hsieh SN, Amargo EV, Bass AE, Pascoe-McGillicuddy LT, Huen AC, Thorne ME, Gaudry CA, Park JK, Myung K, Goldman RD, Chew TL, Green KJ (2005) Desmoplakin assembly dynamics in four dimensions: multiple phases differentially regulated by intermediate filaments and actin. J Cell Biol 171:1045–1059
Gorbsky G, Steinberg MS (1981) Isolation of the intercellular glycoproteins of desmosomes. J Cell Biol 90:243–248
Grando SA (2006a) Cholinergic control of epidermal cohesion. Exp Dermatol 15:265–282
Grando SA (2006b) Pemphigus in the XXI century: new life to an old story. Autoimmunity 39:521–530
Green KJ, Parry DA, Steinert PM, Virata ML, Wagner RM, Angst BD, Nilles LA (1990) Structure of the human desmoplakins. Implications for function in the desmosomal plaque. J Biol Chem 265:2603–2612
Green KJ, Simpson CL (2007) Desmosomes: new perspectives on a classic. J Invest Dermatol 127:2499–2515
Grossmann KS, Grund C, Huelsken J, Behrend M, Erdmann B, Franke WW, Birchmeier W (2004) Requirement of plakophilin 2 for heart morphogenesis and cardiac junction formation. J Cell Biol 167:149–160
Guay J, Lambert H, Gingras-Breton G, Lavoie JN, Huot J, Landry J (1997) Regulation of actin filament dynamics by p38 map kinase-mediated phosphorylation of heat shock protein 27. J Cell Sci 110(Pt 3):357–368
Gusek W (1962) Submicroscopic studies as a contribution to the structure and oncology of meningeoma. Beitr Pathol Anat 127:274–326
Gutstein DE, Liu FY, Meyers MB, Choo A, Fishman GI (2003) The organization of adherens junctions and desmosomes at the cardiac intercalated disc is independent of gap junctions. J Cell Sci 116:875–885
Hacker-Foegen MK, Janson M, Amagai M, Fairley JA, Lin MS (2003) Pathogenicity and epitope characteristics of anti-desmoglein-1 from pemphigus foliaceus patients expressing only IgG1 autoantibodies. J Invest Dermatol 121:1373–1378
Hammerling B, Grund C, Boda-Heggemann J, Moll R, Franke WW (2006) The complexus adhaerens of mammalian lymphatic endothelia revisited: a junction even more complex than hitherto thought. Cell Tiss Res 324:55–67
Hanakawa Y, Amagai M, Shirakata Y, Sayama K, Hashimoto K (2000) Different effects of dominant negative mutants of desmocollin and desmoglein on the cell–cell adhesion of keratinocytes. J Cell Sci 113(Pt 10):1803–1811
Hanakawa Y, Schechter NM, Lin C, Garza L, Li H, Yamaguchi T, Fudaba Y, Nishifuji K, Sugai M, Amagai M, Stanley JR (2002) Molecular mechanisms of blister formation in bullous impetigo and staphylococcal scalded skin syndrome. J Clin Invest 110:53–60
Hardman MJ, Liu K, Avilion AA, Merritt A, Brennan K, Garrod DR, Byrne C (2005) Desmosomal cadherin misexpression alters beta-catenin stability and epidermal differentiation. Molec Cell Biol 25:969–978
Harman KE, Seed PT, Gratian MJ, Bhogal BS, Challacombe SJ, Black MM (2001) The severity of cutaneous and oral pemphigus is related to desmoglein 1 and 3 antibody levels. Br J Dermatol 144:775–780
Hashimoto K, Shafran KM, Webber PS, Lazarus GS, Singer KH (1983) Anti-cell surface pemphigus autoantibody stimulates plasminogen activator activity of human epidermal cells. A mechanism for the loss of epidermal cohesion and blister formation. J Exp Med 157:259–272
Hashimoto T (2003) Recent advances in the study of the pathophysiology of pemphigus. Arch Dermatol Res 295(Suppl 1):S2–S11
Hatsell S, Cowin P (2001) Deconstructing desmoplakin. Nat Cell Biol 3:E270–272
Hatzfeld M (2007) Plakophilins: multifunctional proteins or just regulators of desmosomal adhesion? Biochim Biophys Acta 1773:69–77
Hatzfeld M, Kristjansson GI, Plessmann U, Weber K (1994) Band 6 protein, a major constituent of desmosomes from stratified epithelia, is a novel member of the armadillo multigene family. J Cell Sci 107(Pt 8):2259–2270
Hatzfeld M, Haffner C, Schulze K, Vinzens U (2000) The function of plakophilin 1 in desmosome assembly and actin filament organization. J Cell Biol 149:209–222
He W, Cowin P, Stokes DL (2003) Untangling desmosomal knots with electron tomography. Science (New York, NY) 302:109–113
Heid HW, Schmidt A, Zimbelmann R, Schafer S, Winter-Simanowski S, Stumpp S, Keith M, Figge U, Schnolzer M, Franke WW (1994) Cell type-specific desmosomal plaque proteins of the plakoglobin family: plakophilin 1 (band 6 protein). Differ Res Biol Divers 58:113–131
Hennings H, Holbrook KA (1983) Calcium regulation of cell–cell contact and differentiation of epidermal cells in culture. An ultrastructural study. Exp Cell Res 143:127–142
Hertl M, Eming R, Veldman C (2006) T cell control in autoimmune bullous skin disorders. J Clin Invest 116:1159–1166
Heupel W, Drenckhahn D, Zillikens D, Waschke J (2007) Steric hindrance of desmoglein 3 transinteraction by pemphigus vulgaris IgG. J Invest Dermatol 127(Suppl 1):5 (abstract 29)
Heuser A, Plovie ER, Ellinor PT, Grossmann KS, Shin JT, Wichter T, Basson CT, Lerman BB, Sasse-Klaassen S, Thierfelder L, MacRae CA, Gerull B (2006) Mutant desmocollin-2 causes arrhythmogenic right ventricular cardiomyopathy. Am J Hum Genet 79(6):1081–1088
Holthofer B, Windoffer R, Troyanovsky S, Leube RE (2007) Structure and function of desmosomes. Int Rev Cytol 264:65–163
Hu P, Berkowitz P, O’Keefe EJ, Rubenstein DS (2003) Keratinocyte adherens junctions initiate nuclear signaling by translocation of plakoglobin from the membrane to the nucleus. J Invest Dermatol 121:242–251
Iraji F, Yoosefi A (2006) Healing effect of Pilocarpine gel 4% on skin lesions of pemphigus vulgaris. Int J Dermatol 45:743–746
Ishii K, Amagai M, Hall RP, Hashimoto T, Takayanagi A, Gamou S, Shimizu N, Nishikawa T (1997) Characterization of autoantibodies in pemphigus using antigen-specific enzyme-linked immunosorbent assays with baculovirus-expressed recombinant desmogleins. J Immunol 159:2010–2017
Ishii K, Lin C, Siegel DL, Stanley JR (2008) Isolation of pathogenic monoclonal anti-desmoglein 1 human antibodies by phage display of pemphigus foliaceus autoantibodies. J Invest Dermatol 128(4):939–948
Izumi G, Sakisaka T, Baba T, Tanaka S, Morimoto K, Takai Y (2004) Endocytosis of E-cadherin regulated by Rac and Cdc42 small G proteins through IQGAP1 and actin filaments. J Cell Biol 166:237–248
Jaffe AB, Hall A (2005) Rho GTPases: biochemistry and biology. Annu Rev Cell Dev Biol 21:247–269
Jamora MJ, Jiao D, Bystryn JC (2003) Antibodies to desmoglein 1 and 3, and the clinical phenotype of pemphigus vulgaris. J Am Acad Dermatol 48:976–977
Jefferson JJ, Ciatto C, Shapiro L, Liem RK (2007) Structural analysis of the plakin domain of bullous pemphigoid antigen1 (BPAG1) suggests that plakins are members of the spectrin superfamily. J Molec Biol 366:244–257
Jinbu Y, Kitajima Y, Koto S, Akasaka Y, Yaoita H (1992) Different effects of pemphigus antibody and plasmin on the distribution of keratin intermediate filaments and desmoplakins between cultured oral and epidermal keratinocytes. J Dermatol Sci 3:6–12
Jones JC, Yokoo KM, Goldman RD (1986a) A cell surface desmosome-associated component: identification of tissue-specific cell adhesion molecule. Proc Natl Acad Sci USA 83:7282–7286
Jones JC, Yokoo KM, Goldman RD (1986b) Further analysis of pemphigus autoantibodies and their use in studies on the heterogeneity, structure, and function of desmosomes. J Cell Biol 102:1109–1117
Jonkman MF, Pasmooij AM, Pasmans SG, van den Berg MP, Ter Horst HJ, Timmer A, Pas HH (2005) Loss of desmoplakin tail causes lethal acantholytic epidermolysis bullosa. Am J Hum Genet 77:653–660
Kamei T, Matozaki T, Sakisaka T, Kodama A, Yokoyama S, Peng YF, Nakano K, Takaishi K, Takai Y (1999) Coendocytosis of cadherin and c-Met coupled to disruption of cell–cell adhesion in MDCK cells-regulation by Rho, Rac and Rab small G proteins. Oncogene 18:6776–6784
Karpati S, Amagai M, Prussick R, Cehrs K, Stanley JR (1993) Pemphigus vulgaris antigen, a desmoglein type of cadherin, is localized within keratinocyte desmosomes. J Cell Biol 122:409–415
Kawasaki Y, Aoyama Y, Tsunoda K, Amagai M, Kitajima Y (2006) Pathogenic monoclonal antibody against desmoglein 3 augments desmoglein 3 and p38 MAPK phosphorylation in human squamous carcinoma cell line. Autoimmunity 39:587–590
Kelly DE (1966) Fine structure of desmosomes, hemidesmosomes, and an adepidermal globular layer in developing newt epidermis. J Cell Biol 28:51–72
Kimura TE, Merritt AJ, Garrod DR (2007) Calcium-independent desmosomes of keratinocytes are hyper-adhesive. J Invest Dermatol 127:775–781
King IA, Sullivan KH, Bennett R Jr, Buxton RS (1995) The desmocollins of human foreskin epidermis: identification and chromosomal assignment of a third gene and expression patterns of the three isoforms. J Invest Dermatol 105:314–321
Kitajima Y (2002) Mechanisms of desmosome assembly and disassembly. Clin Exp Dermatol 27:684–690
Kitajima Y, Inoue S, Yaoita H (1986) Effects of pemphigus antibody on the organization of microtubules and keratin-intermediate filaments in cultured human keratinocytes. Br J Dermatol 114:171–179
Kitajima Y, Inoue S, Yaoita H (1987) Effects of pemphigus antibody on the regeneration of cell–cell contact in keratinocyte cultures grown in low to normal Ca++ concentration. J Invest Dermatol 89:167–171
Kitajima Y, Aoyama Y, Seishima M (1999) Transmembrane signaling for adhesive regulation of desmosomes and hemidesmosomes, and for cell–cell datachment induced by pemphigus IgG in cultured keratinocytes: involvement of protein kinase C. J Invest Dermatol Symp Proc/Soc Invest Dermatol Inc 4:137–144
Kljuic A, Bazzi H, Sundberg JP, Martinez-Mir A, O’Shaughnessy R, Mahoney MG, Levy M, Montagutelli X, Ahmad W, Aita VM, Gordon D, Uitto J, Whiting D, Ott J, Fischer S, Gilliam TC, Jahoda CA, Morris RJ, Panteleyev AA, Nguyen VT, Christiano AM (2003) Desmoglein 4 in hair follicle differentiation and epidermal adhesion: evidence from inherited hypotrichosis and acquired pemphigus vulgaris. Cell 113:249–260
Koch PJ, Walsh MJ, Schmelz M, Goldschmidt MD, Zimbelmann R, Franke WW (1990) Identification of desmoglein, a constitutive desmosomal glycoprotein, as a member of the cadherin family of cell adhesion molecules. Euro J Cell Biol 53:1–12
Koch PJ, Goldschmidt MD, Walsh MJ, Zimbelmann R, Franke WW (1991a) Complete amino acid sequence of the epidermal desmoglein precursor polypeptide and identification of a second type of desmoglein gene. Euro J Cell Biol 55:200–208
Koch PJ, Goldschmidt MD, Walsh MJ, Zimbelmann R, Schmelz M, Franke WW (1991b) Amino acid sequence of bovine muzzle epithelial desmocollin derived from cloned cDNA: a novel subtype of desmosomal cadherins. Differ Res Biol Divers 47:29–36
Koch PJ, Mahoney MG, Cotsarelis G, Rothenberger K, Lavker RM, Stanley JR (1998) Desmoglein 3 anchors telogen hair in the follicle. J Cell Sci 111(Pt 17):2529–2537
Kodama S, Ikeda S, Asahara T, Kishida M, Kikuchi A (1999) Axin directly interacts with plakoglobin and regulates its stability. J Biol Chem 274:27682–27688
Koeser J, Troyanovsky SM, Grund C, Franke WW (2003) De novo formation of desmosomes in cultured cells upon transfection of genes encoding specific desmosomal components. Exp Cell Res 285:114–130
Kolligs FT, Kolligs B, Hajra KM, Hu G, Tani M, Cho KR, Fearon ER (2000) Gamma-catenin is regulated by the APC tumor suppressor and its oncogenic activity is distinct from that of beta-catenin. Genes Dev 14:1319–1331
Kolly C, Zakher A, Strauss C, Suter MM, Muller EJ (2007) Keratinocyte transcriptional regulation of the human c-Myc promoter occurs via a novel Lef/Tcf binding element distinct from neoplastic cells. FEBS Lett 581:1969–1976
Korman NJ, Eyre RW, Klaus-Kovtun V, Stanley JR (1989) Demonstration of an adhering-junction molecule (plakoglobin) in the autoantigens of pemphigus foliaceus and pemphigus vulgaris. New Engl J Med 321:631–635
Kottke MD, Delva E, Kowalczyk AP (2006) The desmosome: cell science lessons from human diseases. J Cell Sci 119:797–806
Koulu L, Kusumi A, Steinberg MS, Klaus-Kovtun V, Stanley JR (1984) Human autoantibodies against a desmosomal core protein in pemphigus foliaceus. J Exp Med 160:1509–1518
Kowalczyk AP, Reynolds AB (2004) Protecting your tail: regulation of cadherin degradation by p120-catenin. Curr Opin Cell Biol 16:522–527
Kowalczyk AP, Borgwardt JE, Green KJ (1996) Analysis of desmosomal cadherin-adhesive function and stoichiometry of desmosomal cadherin-plakoglobin complexes. J Invest Dermatol 107:293–300
Kowalczyk AP, Bornslaeger EA, Borgwardt JE, Palka HL, Dhaliwal AS, Corcoran CM, Denning MF, Green KJ (1997) The amino-terminal domain of desmoplakin binds to plakoglobin and clusters desmosomal cadherin-plakoglobin complexes. J Cell Biol 139:773–784
Kowalczyk AP, Hatzfeld M, Bornslaeger EA, Kopp DS, Borgwardt JE, Corcoran CM, Settler A, Green KJ (1999) The head domain of plakophilin-1 binds to desmoplakin and enhances its recruitment to desmosomes. Implications for cutaneous disease. J Biol Chem 274:18145–18148
Kowalczyk AP, Navarro P, Dejana E, Bornslaeger EA, Green KJ, Kopp DS, Borgwardt JE (1998) VE-cadherin and desmoplakin are assembled into dermal microvascular endothelial intercellular junctions: a pivotal role for plakoglobin in the recruitment of desmoplakin to intercellular junctions. J Cell Sci 111(Pt 20):3045–3057
Lanza A, Cirillo N, Femiano F, Gombos F (2006) How does acantholysis occur in pemphigus vulgaris: a critical review. J Cutan Pathol 33:401–412
Lanza A, Cirillo N, Rossiello R, Rienzo M, Cutillo L, Casamassimi A, de Nigris F, Schiano C, Rossiello L, Femiano F, Gombos F, Napoli C (2008) Evidence of key role of CDK-2 overexpression in pemphigus vulgaris. J Biol Chem. doi:10.1074/jbc.M702186200
Lever WF (1953) Pemphigus. Medicine 32:2–132
Lo Muzio L, Pannone G, Staibano S, Mignogna MD, Rubini C, Farronato G, Ferrari F, Nocini PF, De Rosa G (2002) Strict correlation between uPAR and plakoglobin expression in pemphigus vulgaris. J Cutan Pathol 29:540–548
Lorch JH, Klessner J, Park JK, Getsios S, Wu YL, Stack MS, Green KJ (2004) Epidermal growth factor receptor inhibition promotes desmosome assembly and strengthens intercellular adhesion in squamous cell carcinoma cells. J Biol Chem 279:37191–37200
Lyell A (1983) The staphylococcal scalded skin syndrome in historical perspective: emergence of dermopathic strains of Staphylococcus aureus and discovery of the epidermolytic toxin. A review of events up to 1970. J Am Acad Dermatol 9:285–294
Maeda O, Usami N, Kondo M, Takahashi M, Goto H, Shimokata K, Kusugami K, Sekido Y (2004) Plakoglobin (gamma-catenin) has TCF/LEF family-dependent transcriptional activity in beta-catenin-deficient cell line. Oncogene 23:964–972
Mahoney MG, Wang Z, Rothenberger K, Koch PJ, Amagai M, Stanley JR (1999) Explanations for the clinical and microscopic localization of lesions in pemphigus foliaceus and vulgaris. J Clin Invest 103:461–468
Mahoney MG, Hu Y, Brennan D, Bazzi H, Christiano AM, Wahl JK III (2006) Delineation of diversified desmoglein distribution in stratified squamous epithelia: implications in diseases. Exp Dermatol 15:101–109
Marcozzi C, Burdett ID, Buxton RS, Magee AI (1998) Coexpression of both types of desmosomal cadherin and plakoglobin confers strong intercellular adhesion. J Cell Sci 111(Pt 4):495–509
Mathur M, Goodwin L, Cowin P (1994) Interactions of the cytoplasmic domain of the desmosomal cadherin Dsg1 with plakoglobin. J Biol Chem 269:14075–14080
McGrath JA (2005) Inherited disorders of desmosomes. Austr J Dermatol 46:221–229
McGrath JA, McMillan JR, Shemanko CS, Runswick SK, Leigh IM, Lane EB, Garrod DR, Eady RA (1997) Mutations in the plakophilin 1 gene result in ectodermal dysplasia/skin fragility syndrome. Nat Genet 17:240–244
McKoy G, Protonotarios N, Crosby A, Tsatsopoulou A, Anastasakis A, Coonar A, Norman M, Baboonian C, Jeffery S, McKenna WJ (2000) Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease). Lancet 355:2119–2124
McMillan JR, Shimizu H (2001) Desmosomes: structure and function in normal and diseased epidermis. J Dermatol 28:291–298
McNutt NS, Fawcett DW (1969) The ultrastructure of the cat myocardium. II. Atrial muscle. J Cell Biol 42:46–67
Melish ME, Glasgow LA (1970) The staphylococcal scalded-skin syndrome. New Engl J Med 282:1114–1119
Memar O, Christensen B, Rajaraman S, Goldblum R, Tyring SK, Brysk MM, McCormick DJ, el-Zaim H, Fan JL, Prabhakar BS (1996) Induction of blister-causing antibodies by a recombinant full-length, but not the extracellular, domain of the pemphigus vulgaris antigen (desmoglein 3). J Immunol 157:3171–3177
Menon GK, Elias PM (1991) Ultrastructural localization of calcium in psoriatic and normal human epidermis. Arch Dermatol 127:57–63
Merritt AJ, Berika MY, Zhai W, Kirk SE, Ji B, Hardman MJ, Garrod DR (2002) Suprabasal desmoglein 3 expression in the epidermis of transgenic mice results in hyperproliferation and abnormal differentiation. Molec Cell Biol 22:5846–5858
Mertens C, Kuhn C, Franke WW (1996) Plakophilins 2a and 2b: constitutive proteins of dual location in the karyoplasm and the desmosomal plaque. J Cell Biol 135:1009–1025
Mertens C, Kuhn C, Moll R, Schwetlick I, Franke WW (1999) Desmosomal plakophilin 2 as a differentiation marker in normal and malignant tissues. Differ Res Biol Divers 64:277–290
Mertens C, Hofmann I, Wang Z, Teichmann M, Sepehri Chong S, Schnolzer M, Franke WW (2001) Nuclear particles containing RNA polymerase III complexes associated with the junctional plaque protein plakophilin 2. Proc Natl Acad Sci USA 98:7795–7800
Milingou M, Wood P, Masouye I, McLean WH, Borradori L (2006) Focal palmoplantar keratoderma caused by an autosomal dominant inherited mutation in the desmoglein 1 gene. Dermatology (Basel, Switzerland) 212:117–122
Miranda KC, Joseph SR, Yap AS, Teasdale RD, Stow JL (2003) Contextual binding of p120ctn to E-cadherin at the basolateral plasma membrane in polarized epithelia. J Biol Chem 278:43480–43488
Miravet S, Piedra J, Castano J, Raurell I, Franci C, Dunach M, Garcia de Herreros A (2003) Tyrosine phosphorylation of plakoglobin causes contrary effects on its association with desmosomes and adherens junction components and modulates beta-catenin-mediated transcription. Molec Cell Biol 23:7391–7402
Miyagawa S, Amagai M, Iida T, Yamamoto Y, Nishikawa T, Shirai T (1999) Late development of antidesmoglein 1 antibodies in pemphigus vulgaris: correlation with disease progression. Br J Dermatol 141:1084–1087
Moll R, Franke WW (1982) Intermediate filaments and their interaction with membranes. The desmosome-cytokeratin filament complex and epithelial differentiation. Pathol Res Pract 175:146–161
Moll R, Cowin P, Kapprell HP, Franke WW (1986) Desmosomal proteins: new markers for identification and classification of tumors. Lab Invest J Tech Methods Pathol 54:4–25
Morioka S, Lazarus GS, Jensen PJ (1987) Involvement of urokinase-type plasminogen activator in acantholysis induced by pemphigus IgG. J Invest Dermatol 89:474–477
Mueller H, Franke WW (1983) Biochemical and immunological characterization of desmoplakins I and II, the major polypeptides of the desmosomal plaque. J Molec Biol 163:647–671
Muller E, Kernland K, Caldelari R, Wyder M, Balmer V, Hunziker T (2002) Unusual pemphigus phenotype in the presence of a Dsg1 and Dsg3 autoantibody profile. J Invest Dermatol 118:551–555
Muller R, Svoboda V, Wenzel E, Gebert S, Hunzelmann N, Muller HH, Hertl M (2006) IgG reactivity against non-conformational NH-terminal epitopes of the desmoglein 3 ectodomain relates to clinical activity and phenotype of pemphigus vulgaris. Exp Dermatol 15:606–614
Muller EJ, Williamson L, Kolly C, Suter MM (2008a) Outside-in signaling through integrins and cadherins: a central mechanism to control epidermal growth and differentiation? J Invest Dermatol 128:501–516
Muller R, Svoboda V, Wenzel E, Muller HH, Hertl M (2008b) IgG against extracellular subdomains of desmoglein 3 relates to clinical phenotype of pemphigus vulgaris. Exp Dermatol 17:35–43
Nagasaka T, Nishifuji K, Ota T, Whittock NV, Amagai M (2004) Defining the pathogenic involvement of desmoglein 4 in pemphigus and staphylococcal scalded skin syndrome. J Clin Invest 114:1484–1492
Namazi MR (2004) Practice pearl: gargling with cholinergic ophthalmic drops for treating the oral lesions of pemphigus vulgaris. J Drugs Dermatol 3:484–485
Nguyen VT, Ndoye A, Grando SA (2000a) Novel human alpha9 acetylcholine receptor regulating keratinocyte adhesion is targeted by Pemphigus vulgaris autoimmunity. Am J Pathol 157:1377–1391
Nguyen VT, Ndoye A, Grando SA (2000b) Pemphigus vulgaris antibody identifies pemphaxin. A novel keratinocyte annexin-like molecule binding acetylcholine. J Biol Chem 275:29466–29476
Nguyen VT, Ndoye A, Shultz LD, Pittelkow MR, Grando SA (2000c) Antibodies against keratinocyte antigens other than desmogleins 1 and 3 can induce pemphigus vulgaris-like lesions. J Clin Invest 106:1467–1479
Nguyen VT, Arredondo J, Chernyavsky AI, Kitajima Y, Pittelkow M, Grando SA (2004a) Pemphigus vulgaris IgG and methylprednisolone exhibit reciprocal effects on keratinocytes. J Biol Chem 279:2135–2146
Nguyen VT, Arredondo J, Chernyavsky AI, Pittelkow MR, Kitajima Y, Grando SA (2004b) Pemphigus vulgaris acantholysis ameliorated by cholinergic agonists. Arch Dermatol 140:327–334
Nollet F, Kools P, van Roy F (2000) Phylogenetic analysis of the cadherin superfamily allows identification of six major subfamilies besides several solitary members. J Molec Biol 299:551–572
Norgett EE, Hatsell SJ, Carvajal-Huerta L, Cabezas JC, Common J, Purkis PE, Whittock N, Leigh IM, Stevens HP, Kelsell DP (2000) Recessive mutation in desmoplakin disrupts desmoplakin-intermediate filament interactions and causes dilated cardiomyopathy, woolly hair and keratoderma. Hum Molec Genet 9:2761–2766
North AJ, Bardsley WG, Hyam J, Bornslaeger EA, Cordingley HC, Trinnaman B, Hatzfeld M, Green KJ, Magee AI, Garrod DR (1999) Molecular map of the desmosomal plaque. J Cell Sci 112(Pt 23):4325–4336
Odland GF (1958) The fine structure of the interrelationship of cells in the human epidermis. J Biophys Biochem Cytol 4:529–538
Osada K, Seishima M, Kitajima Y (1997) Pemphigus IgG activates and translocates protein kinase C from the cytosol to the particulate/cytoskeleton fractions in human keratinocytes. J Invest Dermatol 108:482–487
Overduin M, Harvey TS, Bagby S, Tong KI, Yau P, Takeichi M, Ikura M (1995) Solution structure of the epithelial cadherin domain responsible for selective cell adhesion. Science (New York, NY) 267:386–389
Panasenko OO, Kim MV, Marston SB, Gusev NB (2003) Interaction of the small heat shock protein with molecular mass 25 kDa (hsp25) with actin. Euro J Biochem/FEBS 270:892–901
Pasdar M, Nelson WJ (1988) Kinetics of desmosome assembly in Madin–Darby canine kidney epithelial cells: temporal and spatial regulation of desmoplakin organization and stabilization upon cell–cell contact. II. Morphological analysis. J Cell Biol 106:687–695
Pasdar M, Nelson WJ (1989) Regulation of desmosome assembly in epithelial cells: kinetics of synthesis, transport, and stabilization of desmoglein I, a major protein of the membrane core domain. J Cell Biol 109:163–177
Pasdar M, Li Z, Chlumecky V (1995) Plakoglobin: kinetics of synthesis, phosphorylation, stability, and interactions with desmoglein and E-cadherin. Cell Motility Cytoskeleton 32:258–272
Payne AS, Hanakawa Y, Amagai M, Stanley JR (2004) Desmosomes and disease: pemphigus and bullous impetigo. Curr Opin Cell Biol 16:536–543
Payne AS, Ishii K, Kacir S, Lin C, Li H, Hanakawa Y, Tsunoda K, Amagai M, Stanley JR, Siegel DL (2005) Genetic and functional characterization of human pemphigus vulgaris monoclonal autoantibodies isolated by phage display. J Clin Invest 115:888–899
Pelacho B, Natal C, Espana A, Sanchez-Carpintero I, Iraburu MJ, Lopez-Zabalza MJ (2004) Pemphigus vulgaris autoantibodies induce apoptosis in HaCaT keratinocytes. FEBS Lett 566:6–10
Perng MD, Cairns L, van den Ijssel P, Prescott A, Hutcheson AM, Quinlan RA (1999) Intermediate filament interactions can be altered by HSP27 and alphaB-crystallin. J Cell Sci 112(Pt 13):2099–2112
Pertz O, Bozic D, Koch AW, Fauser C, Brancaccio A, Engel J (1999) A new crystal structure, Ca2+ dependence and mutational analysis reveal molecular details of E-cadherin homoassociation. EMBO J 18:1738–1747
Pieperhoff S, Franke WW (2007) The area composita of adhering junctions connecting heart muscle cells of vertebrates—IV: coalescence and amalgamation of desmosomal and adhaerens junction components—late processes in mammalian heart development. Euro J Cell Biol 86:377–391
Pieperhoff S, Schumacher H, Franke WW (2008) The area composita of adhering junctions connecting heart muscle cells of vertebrates. V. The importance of plakophilin-2 demonstrated by small interference RNA-mediated knockdown in cultured rat cardiomyocytes. Euro J Cell Biol (in press)
Pilichou K, Nava A, Basso C, Beffagna G, Bauce B, Lorenzon A, Frigo G, Vettori A, Valente M, Towbin J, Thiene G, Danieli GA, Rampazzo A (2006) Mutations in desmoglein-2 gene are associated with arrhythmogenic right ventricular cardiomyopathy. Circulation 113:1171–1179
Porter KR (1956) Observations on the fine structure of animal epidermis. In: Proceedings of the International Congress on Electron Microscopy. Royal Microscopical Society, London, p 539
Puviani M, Marconi A, Cozzani E, Pincelli C (2003) Fas ligand in pemphigus sera induces keratinocyte apoptosis through the activation of caspase-8. J Invest Dermatol 120:164–167
Rampazzo A, Nava A, Malacrida S, Beffagna G, Bauce B, Rossi V, Zimbello R, Simionati B, Basso C, Thiene G, Towbin JA, Danieli GA (2002) Mutation in human desmoplakin domain binding to plakoglobin causes a dominant form of arrhythmogenic right ventricular cardiomyopathy. Am J Hum Genet 71:1200–1206
Rickman L, Simrak D, Stevens HP, Hunt DM, King IA, Bryant SP, Eady RA, Leigh IM, Arnemann J, Magee AI, Kelsell DP, Buxton RS (1999) N-terminal deletion in a desmosomal cadherin causes the autosomal dominant skin disease striate palmoplantar keratoderma. Hum Molec Genet 8:971–976
Rock B, Labib RS, Diaz LA (1990) Monovalent Fab’ immunoglobulin fragments from endemic pemphigus foliaceus autoantibodies reproduce the human disease in neonatal Balb/c mice. J Clin Invest 85:296–299
Rock B, Martins CR, Theofilopoulos AN, Balderas RS, Anhalt GJ, Labib RS, Futamura S, Rivitti EA, Diaz LA (1989) The pathogenic effect of IgG4 autoantibodies in endemic pemphigus foliaceus (fogo selvagem). New Engl J Med 320:1463–1469
Rodriguez J, Bystryn JC (1977) Pemphigus foliaceus associated with absence of intercellular antigens in lower layers of epidermis. Arch Dermatol 113:1696–1699
Rosen K, Shi W, Calabretta B, Filmus J (2002) Cell detachment triggers p38 mitogen-activated protein kinase-dependent overexpression of Fas ligand. A novel mechanism of Anoikis of intestinal epithelial cells. J Biol Chem 277:46123–46130
Ruiz P, Brinkmann V, Ledermann B, Behrend M, Grund C, Thalhammer C, Vogel F, Birchmeier C, Gunthert U, Franke WW, Birchmeier W (1996) Targeted mutation of plakoglobin in mice reveals essential functions of desmosomes in the embryonic heart. J Cell Biol 135:215–225
Ruiz-Velasco R, Lanning CC, Williams CL (2002) The activation of Rac1 by M3 muscarinic acetylcholine receptors involves the translocation of Rac1 and IQGAP1 to cell junctions and changes in the composition of protein complexes containing Rac1, IQGAP1, and actin. J Biol Chem 277:33081–33091
Runswick SK, O’Hare MJ, Jones L, Streuli CH, Garrod DR (2001) Desmosomal adhesion regulates epithelial morphogenesis and cell positioning. Nat Cell Biol 3:823–830
Sakuntabhai A, Ruiz-Perez V, Carter S, Jacobsen N, Burge S, Monk S, Smith M, Munro CS, O’Donovan M, Craddock N, Kucherlapati R, Rees JL, Owen M, Lathrop GM, Monaco AP, Strachan T, Hovnanian A (1999) Mutations in ATP2A2, encoding a Ca2+ pump, cause Darier disease. Nat Genet 21:271–277
Salato VK, Hacker-Foegen MK, Lazarova Z, Fairley JA, Lin MS (2005) Role of intramolecular epitope spreading in pemphigus vulgaris. Clin Immunol (Orlando, FL) 116:54–64
Sanchez-Carpintero I, Espana A, Pelacho B, Lopez Moratalla N, Rubenstein DS, Diaz LA, Lopez-Zabalza MJ (2004) In vivo blockade of pemphigus vulgaris acantholysis by inhibition of intracellular signal transduction cascades. Br J Dermatol 151:565–570
Santiago-Josefat B, Esselens C, Bech-Serra JJ, Arribas J (2007) Post-transcriptional up-regulation of ADAM17 upon epidermal growth factor receptor activation and in breast tumors. J Biol Chem 282:8325–8331
Sato M, Aoyama Y, Kitajima Y (2000) Assembly pathway of desmoglein 3 to desmosomes and its perturbation by pemphigus vulgaris-IgG in cultured keratinocytes, as revealed by time-lapsed labeling immunoelectron microscopy. Lab Invest J Tech Methods Pathol 80:1583–1592
Schaefer BM, Jaeger CJ, Kramer MD (1996) Plasminogen activator system in pemphigus vulgaris. Br J Dermatol 135:726–732
Schafer S, Koch PJ, Franke WW (1994) Identification of the ubiquitous human desmoglein, Dsg2, and the expression catalogue of the desmoglein subfamily of desmosomal cadherins. Exp Cell Res 211:391–399
Schaffer J (1920) Vorlesungen über Histologie und Histogenese. Wien
Schiltz JR, Michel B (1976) Production of epidermal acantholysis in normal human skin in vitro by the IgG fraction from pemphigus serum. J Invest Dermatol 67:254–260
Schmelz M, Franke WW (1993) Complexus adhaerentes, a new group of desmoplakin-containing junctions in endothelial cells: the syndesmos connecting retothelial cells of lymph nodes. Euro J Cell Biol 61:274–289
Schmelz M, Duden R, Cowin P, Franke WW (1986a) A constitutive transmembrane glycoprotein of Mr 165,000 (desmoglein) in epidermal and non-epidermal desmosomes. I. Biochemical identification of the polypeptide. Euro J Cell Biol 42:177–183
Schmelz M, Duden R, Cowin P, Franke WW (1986b) A constitutive transmembrane glycoprotein of Mr 165,000 (desmoglein) in epidermal and non-epidermal desmosomes. II. Immunolocalization and microinjection studies. Euro J Cell Biol 42:184–199
Schmelz M, Moll R, Kuhn C, Franke WW (1994) Complexus adhaerentes, a new group of desmoplakin-containing junctions in endothelial cells: II. Different types of lymphatic vessels. Differ Res Biol Divers 57:97–117
Schmidt A, Langbein L, Rode M, Pratzel S, Zimbelmann R, Franke WW (1997) Plakophilins 1a and 1b: widespread nuclear proteins recruited in specific epithelial cells as desmosomal plaque components. Cell Tissue Res 290:481–499
Schmidt A, Langbein L, Pratzel S, Rode M, Rackwitz HR, Franke WW (1999) Plakophilin 3—a novel cell-type-specific desmosomal plaque protein. Differ Res Biol Divers 64:291–306
Schmidt E, Brocker EB, Zillikens D (2000) [Pemphigus. Loss of desmosomal cell–cell contact]. Der Hautarzt; Zeitschrift Dermatol Venerologie verwandte Gebiete 51:309–318
Schuh T, Besch R, Braungart E, Flaig MJ, Douwes K, Sander CA, Magdolen V, Probst C, Wosikowski K, Degitz K (2003) Protease inhibitors prevent plasminogen-mediated, but not pemphigus vulgaris-induced, acantholysis in human epidermis. Biol Chem 384:311–315
Schwarz MA, Owaribe K, Kartenbeck J, Franke WW (1990) Desmosomes and hemidesmosomes: constitutive molecular components. Annu Rev Cell Biol 6:461–491
Seishima M, Esaki C, Osada K, Mori S, Hashimoto T, Kitajima Y (1995) Pemphigus IgG, but not bullous pemphigoid IgG, causes a transient increase in intracellular calcium and inositol 1,4,5-triphosphate in DJM-1 cells, a squamous cell carcinoma line. J Invest Dermatol 104:33–37
Seishima M, Satoh S, Nojiri M, Osada K, Kitajima Y (1997) Pemphigus IgG induces expression of urokinase plasminogen activator receptor on the cell surface of cultured keratinocytes. J Invest Dermatol 109:650–655
Seishima M, Iwasaki-Bessho Y, Itoh Y, Nozawa Y, Amagai M, Kitajima Y (1999) Phosphatidylcholine-specific phospholipase C, but not phospholipase D, is involved in pemphigus IgG-induced signal transduction. Arch Dermatol Res 291:606–613
Sekiguchi M, Futei Y, Fujii Y, Iwasaki T, Nishikawa T, Amagai M (2001) Dominant autoimmune epitopes recognized by pemphigus antibodies map to the N-terminal adhesive region of desmogleins. J Immunol 167:5439–5448
Shapiro L, Fannon AM, Kwong PD, Thompson A, Lehmann MS, Grubel G, Legrand JF, Als-Nielsen J, Colman DR, Hendrickson WA (1995) Structural basis of cell–cell adhesion by cadherins. Nature 374:327–337
Sharma P, Mao X, Payne AS (2007) Beyond steric hindrance: the role of adhesion signaling pathways in the pathogenesis of pemphigus. J Dermatol Sci 48:1–14
Sheu HM, Kitajima Y, Yaoita H (1989) Involvement of protein kinase C in translocation of desmoplakins from cytosol to plasma membrane during desmosome formation in human squamous cell carcinoma cells grown in low to normal calcium concentration. Exp Cell Res 185:176–190
Shimada T, Kawazato H, Yasuda A, Ono N, Sueda K (2004) Cytoarchitecture and intercalated disks of the working myocardium and the conduction system in the mammalian heart. Anat Rec 280:940–951
Shimanovich I, Nitschke M, Rose C, Grabbe J, Zillikens D (2008) Treatment of severe pemphigus with protein A immunoadsorption, rituximab and intravenous immunoglobulins. Br J Dermatol 158:382–388
Shimizu H, Masunaga T, Ishiko A, Kikuchi A, Hashimoto T, Nishikawa T (1995) Pemphigus vulgaris and pemphigus foliaceus sera show an inversely graded binding pattern to extracellular regions of desmosomes in different layers of human epidermis. J Invest Dermatol 105:153–159
Shimizu A, Ishiko A, Ota T, Tsunoda K, Amagai M, Nishikawa T (2004) IgG binds to desmoglein 3 in desmosomes and causes a desmosomal split without keratin retraction in a pemphigus mouse model. J Invest Dermatol 122:1145–1153
Shimizu A, Ishiko A, Ota T, Saito H, Oka H, Tsunoda K, Amagai M, Nishikawa T (2005) In vivo ultrastructural localization of the desmoglein 3 adhesive interface to the desmosome mid-line. J Invest Dermatol 124:984–989
Shirakata Y, Amagai M, Hanakawa Y, Nishikawa T, Hashimoto K (1998) Lack of mucosal involvement in pemphigus foliaceus may be due to low expression of desmoglein 1. J Invest Dermatol 110:76–78
Shu E, Yamamoto Y, Aoyama Y, Kitajima Y (2007) Intraperitoneal injection of pemphigus vulgaris-IgG into mouse depletes epidermal keratinocytes of desmoglein 3 associated with generation of acantholysis. Arch Dermatol Res 299:165–167
Sitaru C, Zillikens D (2005) Mechanisms of blister induction by autoantibodies. Exp Dermatol 14:861–875
Skerrow CJ, Matoltsy AG (1974) Chemical characterization of isolated epidermal desmosomes. J Cell Biol 63:524–530
Smith EA, Fuchs E (1998) Defining the interactions between intermediate filaments and desmosomes. J Cell Biol 141:1229–1241
Sobolik-Delmaire T, Katafiasz D, Wahl JK 3rd (2006) Carboxyl terminus of Plakophilin-1 recruits it to plasma membrane, whereas amino terminus recruits desmoplakin and promotes desmosome assembly. J Biol Chem 281:16962–16970
Sonnenberg A, Liem RK (2007) Plakins in development and disease. Exp Cell Res 313:2189–2203
Spaeth S, Riechers R, Borradori L, Zillikens D, Budinger L, Hertl M (2001) IgG, IgA and IgE autoantibodies against the ectodomain of desmoglein 3 in active pemphigus vulgaris. Br J Dermatol 144:1183–1188
Spindler V, Drenckhahn D, Zillikens D, Waschke J (2007) Pemphigus IgG causes skin splitting in the presence of both desmoglein 1 and desmoglein 3. Am J Pathol 171:906–916
Stanley JR, Amagai M (2006) Pemphigus, bullous impetigo, and the staphylococcal scalded-skin syndrome. New Engl J Med 355:1800–1810
Stanley JR, Koulu L, Thivolet C (1984) Distinction between epidermal antigens binding pemphigus vulgaris and pemphigus foliaceus autoantibodies. J Clin Invest 74:313–320
Stappenbeck TS, Green KJ (1992) The desmoplakin carboxyl terminus coaligns with and specifically disrupts intermediate filament networks when expressed in cultured cells. J Cell Biol 116:1197–1209
Swartzendruber DC (1965) Desmosomes in germinal centers of mouse spleen. Exp Cell Res 40:429–432
Syed SE, Trinnaman B, Martin S, Major S, Hutchinson J, Magee AI (2002) Molecular interactions between desmosomal cadherins. Biochem J 362:317–327
Syrris P, Ward D, Asimaki A, Sen-Chowdhry S, Ebrahim HY, Evans A, Hitomi N, Norman M, Pantazis A, Shaw AL, Elliott PM, McKenna WJ (2006) Clinical expression of plakophilin-2 mutations in familial arrhythmogenic right ventricular cardiomyopathy. Circulation 113:356–364
Takahashi Y, Patel HP, Labib RS, Diaz LA, Anhalt GJ (1985) Experimentally induced pemphigus vulgaris in neonatal BALB/c mice: a time-course study of clinical, immunologic, ultrastructural, and cytochemical changes. J Invest Dermatol 84:41–46
Troyanovsky SM, Eshkind LG, Troyanovsky RB, Leube RE, Franke WW (1993) Contributions of cytoplasmic domains of desmosomal cadherins to desmosome assembly and intermediate filament anchorage. Cell 72:561–574
Troyanovsky SM, Troyanovsky RB, Eshkind LG, Krutovskikh VA, Leube RE, Franke WW (1994a) Identification of the plakoglobin-binding domain in desmoglein and its role in plaque assembly and intermediate filament anchorage. J Cell Biol 127:151–160
Troyanovsky SM, Troyanovsky RB, Eshkind LG, Leube RE, Franke WW (1994b) Identification of amino acid sequence motifs in desmocollin, a desmosomal glycoprotein, that are required for plakoglobin binding and plaque formation. Proc Natl Acad Sci USA 91:10790–10794
Troyanovsky RB, Chitaev NA, Troyanovsky SM (1996) Cadherin binding sites of plakoglobin: localization, specificity and role in targeting to adhering junctions. J Cell Sci 109(Pt 13):3069–3078
Tselepis C, Chidgey M, North A, Garrod D (1998) Desmosomal adhesion inhibits invasive behavior. Proc Natl Acad Sci USA 95:8064–8069
Tsunoda K, Ota T, Aoki M, Yamada T, Nagai T, Nakagawa T, Koyasu S, Nishikawa T, Amagai M (2003) Induction of pemphigus phenotype by a mouse monoclonal antibody against the amino-terminal adhesive interface of desmoglein 3. J Immunol 170:2170–2178
Udey MC, Stanley JR (1999) Pemphigus–diseases of antidesmosomal autoimmunity. Jama 282:572–576
van Tintelen JP, Hofstra RM, Wiesfeld AC, van den Berg MP, Hauer RN, Jongbloed JD (2007) Molecular genetics of arrhythmogenic right ventricular cardiomyopathy: emerging horizon? Curr Opin Cardiol 22:185–192
Vasioukhin V, Bauer C, Yin M, Fuchs E (2000) Directed actin polymerization is the driving force for epithelial cell–cell adhesion. Cell 100:209–219
Vasioukhin V, Bowers E, Bauer C, Degenstein L, Fuchs E (2001) Desmoplakin is essential in epidermal sheet formation. Nat Cell Biol 3:1076–1085
Veldman CM, Gebhard KL, Uter W, Wassmuth R, Grotzinger J, Schultz E, Hertl M (2004) T cell recognition of desmoglein 3 peptides in patients with pemphigus vulgaris and healthy individuals. J Immunol 172:3883–3892
Wahl JK III (2005) A role for plakophilin-1 in the initiation of desmosome assembly. J Cell Biochem 96:390–403
Wahl JK, Sacco PA, McGranahan-Sadler TM, Sauppe LM, Wheelock MJ, Johnson KR (1996) Plakoglobin domains that define its association with the desmosomal cadherins and the classical cadherins: identification of unique and shared domains. J Cell Sci 109(Pt 5):1143–1154
Wan H, Dopping-Hepenstal PJ, Gratian MJ, Stone MG, Zhu G, Purkis PE, South AP, Keane F, Armstrong DK, Buxton RS, McGrath JA, Eady RA (2004) Striate palmoplantar keratoderma arising from desmoplakin and desmoglein 1 mutations is associated with contrasting perturbations of desmosomes and the keratin filament network. Br J Dermatol 150:878–891
Wang X, Bregegere F, Frusic-Zlotkin M, Feinmesser M, Michel B, Milner Y (2004a) Possible apoptotic mechanism in epidermal cell acantholysis induced by pemphigus vulgaris autoimmunoglobulins. Apoptosis 9:131–143
Wang X, Bregegere F, Soroka Y, Frusic-Zlotkin M, Milner Y (2004b) Replicative senescence enhances apoptosis induced by pemphigus autoimmune antibodies in human keratinocytes. FEBS Lett 567:281–286
Waschke J, Bruggeman P, Baumgartner W, Zillikens D, Drenckhahn D (2005) Pemphigus foliaceus IgG causes dissociation of desmoglein 1-containing junctions without blocking desmoglein 1 transinteraction. J Clin Invest 115:3157–3165
Waschke J, Spindler V, Bruggeman P, Zillikens D, Schmidt G, Drenckhahn D (2006) Inhibition of Rho A activity causes pemphigus skin blistering. J Cell Biol 175:721–727
Waschke J, Menendez-Castro C, Bruggeman P, Koob R, Amagai M, Gruber HJ, Drenckhahn D, Baumgartner W (2007) Imaging and force spectroscopy on desmoglein 1 using atomic force microscopy reveal multivalent Ca(2+)-dependent, low-affinity trans-interaction. J Membrane Biol 216:83–92
Weiske J, Schoneberg T, Schroder W, Hatzfeld M, Tauber R, Huber O (2001) The fate of desmosomal proteins in apoptotic cells. J Biol Chem 276:41175–41181
Wendeler MW, Drenckhahn D, Gessner R, Baumgartner W (2007) Intestinal LI-cadherin acts as a Ca2+-dependent adhesion switch. J Molec Biol 370:220–230
Whittock NV, Bower C (2003) Genetic evidence for a novel human desmosomal cadherin, desmoglein 4. J Invest Dermatol 120:523–530
Whittock NV, Ashton GH, Dopping-Hepenstal PJ, Gratian MJ, Keane FM, Eady RA, McGrath JA (1999) Striate palmoplantar keratoderma resulting from desmoplakin haploinsufficiency. J Invest Dermatol 113:940–946
Whittock NV, Wan H, Morley SM, Garzon MC, Kristal L, Hyde P, McLean WH, Pulkkinen L, Uitto J, Christiano AM, Eady RA, McGrath JA (2002) Compound heterozygosity for non-sense and mis-sense mutations in desmoplakin underlies skin fragility/woolly hair syndrome. J Invest Dermatol 118:232–238
Wilgram GF, Caulfield JB, Lever WF (1961) An electron microscopic study of acantholysis in pemphigus vulgaris. J Invest Dermatol 36:373–382
Wilgram GF, Caulfield JB, Madgic EB (1964) An Electron Microscopic Study of Acantholysis and Dyskeratosis in Pemphigus Foliaceus: with a Special Note on Peculiar Intracytoplasmic Bodies. J Invest Dermatol 43:287–299
Williamson L, Raess NA, Caldelari R, Zakher A, de Bruin A, Posthaus H, Bolli R, Hunziker T, Suter MM, Muller EJ (2006) Pemphigus vulgaris identifies plakoglobin as key suppressor of c-Myc in the skin. EMBO J 25:3298–3309
Williamson L, Hunziker T, Suter MM, Muller EJ (2007) Nuclear c-Myc: a molecular marker for early stage pemphigus vulgaris. J Invest Dermatol 127:1549–1555
Windoffer R, Borchert-Stuhltrager M, Leube RE (2002) Desmosomes: interconnected calcium-dependent structures of remarkable stability with significant integral membrane protein turnover. J Cell Sci 115:1717–1732
Woll S, Windoffer R, Leube RE (2007) p38 MAPK-dependent shaping of the keratin cytoskeleton in cultured cells. J Cell Biol 177:795–807
Wu H, Wang ZH, Yan A, Lyle S, Fakharzadeh S, Wahl JK, Wheelock MJ, Ishikawa H, Uitto J, Amagai M, Stanley JR (2000) Protection against pemphigus foliaceus by desmoglein 3 in neonates. New Engl J Med 343:31–35
Yamamoto Y, Aoyama Y, Shu E, Tsunoda K, Amagai M, Kitajima Y (2007a) Anti-desmoglein 3 (Dsg3) monoclonal antibodies deplete desmosomes of Dsg3 and differ in their Dsg3-depleting activities related to pathogenicity. J Biol Chem 282:17866–17876
Yamamoto Y, Aoyama Y, Shu E, Tsunoda K, Amagai M, Kitajima Y (2007b) No activation of urokinase plasminogen activator by anti-desmoglein 3 monoclonal IgG antibodies in cultured human keratinocytes. J Dermatol Sci 47:119–125
Yancey KB (2005) The pathophysiology of autoimmune blistering diseases. J Clin Invest 115:825–828
Yeh SW, Ahmed B, Sami N, Razzaque Ahmed A (2003) Blistering disorders: diagnosis and treatment. Dermatol Therapy 16:214–223
Yin T, Green KJ (2004) Regulation of desmosome assembly and adhesion. Semin Cell Dev Biol 15:665–677
Yin T, Getsios S, Caldelari R, Godsel LM, Kowalczyk AP, Muller EJ, Green KJ (2005) Mechanisms of plakoglobin-dependent adhesion: desmosome-specific functions in assembly and regulation by epidermal growth factor receptor. J Biol Chem 280:40355–40363
Yoshida K, Takae Y, Saito H, Oka H, Tanikawa A, Amagai M, Nishikawa T (2005) Cutaneous type pemphigus vulgaris: a rare clinical phenotype of pemphigus. J Am Acad Dermatol 52:839–845
Zagorodniuk I, Weltfriend S, Shtruminger L, Sprecher E, Kogan O, Pollack S, Bergman R (2005) A comparison of anti-desmoglein antibodies and indirect immunofluorescence in the serodiagnosis of pemphigus vulgaris. Int J Dermatol 44:541–544
Acknowledgments
The author is grateful to Detlev Drenckhahn for helpful comments and for critically reading the manuscript as well as to Michael Christof for figure design. Photographs of pemphigus patients were generously contributed by D. Zillikens and E. Schmidt (both Department of Dermatology, University of Lübeck). Special thanks to the members of the PTF (Wolfgang-Moritz Heupel, Volker Spindler, Peter Engerer, Judith Gutberlet, Seraj Khan) for inspiring discussion. The author’s work was supported by grants from the Deutsche Forschungsgemeinschaft (SFB 487, TP B5 to Detlev Drenckhahn) and the IZKF Würzburg (TP A-51).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Waschke, J. The desmosome and pemphigus. Histochem Cell Biol 130, 21–54 (2008). https://doi.org/10.1007/s00418-008-0420-0
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00418-008-0420-0