
Abstract
Complex hereditary spastic paraplegia (HSP) is a clinically heterogeneous group of disorders usually inherited in an autosomal recessive manner. In the past, complex recessive spastic paraplegias have been frequently associated with SPG11 mutations but also with defects in SPG15, SPG7 and a handful of other rare genes. Pleiotropy exists in HSP genes, exemplified in the recent association of SPG11 mutations with CMT2. In this study, we performed whole exome sequence analysis and identified two siblings with novel compound heterozygous frameshift SPG11 mutations. The mutations segregated with disease were not present in control databases and analysis of skin fibroblast derived mRNA indicated that the SPG11 truncated mRNA species were not degraded significantly by non-sense mediated mRNA decay. These siblings had severe early-onset spastic paraplegia but later in their disease developed severe axonal neuropathy, neuropathic pain and blue/black foot discolouration likely caused by a combination of the severe neuropathy with autonomic dysfunction and peripheral oedema. We also identified a similar late-onset axonal neuropathy in a Cypriot SPG11 family. Although neuropathy is occasionally present in SPG11, in our SPG11 patients reported here it was particularly severe, highlighting the association of axonal neuropathy with SPG11 and the late manifestation of axonal peripheral nerve damage.
Similar content being viewed by others


Introduction
The hereditary spastic paraplegias (HSPs) are heterogeneous inherited neurological disorders, resulting in progressive spasticity of the limbs, bladder dysfunction and walking difficulties. Other features such as weakness, ataxia, cognitive decline and peripheral neuropathy are present in complex HSP. They are classified genetically as autosomal dominant, autosomal recessive and X-linked HSP; and clinically as pure or complicated HSP, based upon the absence (pure) or presence (complex) of additional clinical features. To date, more than 70 different disease-loci and more than 50 spastic paraplegia genes (SPGs) have been identified [1, 2]. Autosomal recessive HSP with a thin corpus callosum is a common subtype of complex HSP. Mutations in SPG11 (encoding for Spastacsin), comprise about 70 % of these cases [2–4] and the onset is usually in the teenage years [5–12].
In the past, axonal neuropathy has been infrequently associated with complex HSP and SPG11 mutations, although this is usually mild [4, 13]. Recently mutations in SPG11 have been shown to cause Charcot–Marie–Tooth disease type 2 (CMT2) as a distinct clinical phenotype [14]. Spatacsin is expressed ubiquitously in the nervous system [1] and the sural nerve biopsy pathology in typical SPG11 cases showed a loss of unmyelinated nerve fibers and accumulation of intra-axonal pleomorphic membranous material. In CMT2 cases sural nerve biopsies showed similar intra-axonal inclusions but also predominantly loss of large myelinated fibers, mainly in fibers of large caliber, in line with the diagnosis of CMT2 [14]. Although the role of the protein is unknown, it seems to be important to the survival of neurons [15].
In this paper we describe novel frameshift mutations in SPG11, identified by whole exome sequencing, segregating with the disease and investigated in mRNA. The causative mutations were found in two sisters with complex HSP consistent with SPG11, associated with the late manifestation of severe axonal neuropathy. A similar presentation was found in a second, unrelated Cypriot family.
Methods
In family F1, two sisters with complex HSP, with clinical features as described below were investigated. Informed consent (UCLH: N99/103) was obtained from all individuals and the institutional review boards at the participating medical centers approved the study. Acquired spastic paraplegia was excluded and neurophysiological studies, MRI scans and skin biopsies were performed using standard methods. Genomic DNA samples from the two affected individuals and two unaffected relatives were used for molecular genetic analyses (supplementary material). The mutation in the Cypriot family was previously reported [4].
Exome sequencing was performed as previously described [16–18] using the Agilent SureSelect kit and run on the Illumina HiSeq2500. Sequences were aligned with the Burrows-Wheeler Aligner, duplicates were removed with Picard, indels aligned and base quality scores recalibrated with the Genome Analysis Toolkit (GATK). The average sequencing depth was 55-fold with variants being filtered according to pathogenicity, inheritance pattern, and segregation in the family. We confirmed causative variant candidates by the Sanger sequencing method using an automatic genetic analyzer. SPG11 PCR primers were designed using Primer3 so that the PCR products would span whole exons and about 35 bp of flanking introns (http://primer3.ut.ee/). Primer sequences are listed in the supplementary file. Touchdown PCR was done using the PCR Master Mix (Roche) and is described in more detail in the supplementary table. PCR amplification products were cleaned with ExoAp. The purified PCR products was split into two and sequenced bidirectionally with the original primers that were used to amplify the region of interest and Big Dye Terminator Kit v.3.1 (Applied Biosystems) [19, 20]. Conditions were as follows: 25 cycles of denaturation at 95 °C for 10 s, annealing at 50 °C for 5 s and extension at 60 °C for 4 min. Sequencing reactions were cleaned using CleanSEQ SPRI beads according to the manufacturer’s protocol (Agencourt). Sequencing was performed using a 3730 DNA Analyzer (Applied Biosystems). SPG11 mutation positions are based on NCBI reference sequences: NM_001160227, NP_001153699 (www.ncbi.nlm.nih.gov).
RNA isolation and reverse transcriptase
Total RNA was extracted from cultured skin fibroblasts using Direct-zol™ RNA Miniprep (Zymo Research, USA) according to the manufacturer’s instructions and reverse transcribed to first strand cDNA by using random primers and Moloney murine leukemia virus (M-MLV) reverse transcriptase (Promega Corporation, Madison, WI, USA). The concentration and purity of RNA was determined spectrophotometrically. PCR amplification and sequencing of cDNA: three primer pairs were designed in flanking exons of those of interest to amplify the coding sequence of exons 22 and 30 of SPG11 mRNA (Supplementary material). Sanger sequencing was performed as above.
Results
Clinical features
The proband and her sister from family F1 (II:1 and II:2; Fig. 1) were the first and second children of healthy, non-consanguineous parents of British descent. At ages 19 and 16 years, respectively, they presented with progressive walking difficulties with additional features of bulbar dysarthria, cognitive problems, limb weakness (proximal power MRC 2/5 and distal 1/5 on examination at the age of 39 and 36, respectively), bladder dysfunction and optic atrophy. At first in their early 20s, limb tone was increased and reflexes were brisk with extensor plantars and no sensory abnormalities but at the ages 39 and 36 reflexes were absent and there was distal sensory loss to pin prick, light touch and temperature. Their fingers were long and tapering, palms were discolored and there was significant limb pain. In addition to having leg oedema the feet of both sisters had strikingly black coloration which improved with massage (Fig. 1). Cognitive tests showed IQs of 54 for the proband, and although only a very limited assessment was possible in the sister due to severe motor and language impairment in the context of complex HSP. The overall picture was one of the global and severe cognitive impairments, occurring at an early age with relative sparing of visual perceptual functions and normal hearing. Pulses were difficult to palpate with the peripheral oedema but capillary refill was normal as was blood pressure testing.

Photographs of the feet in SPG11 patients F1II:1 and F1II:2 (A and B) and F1AII:2 (C and D) showing peripheral oedema and marked discolouration of the feet that reverses with massage. Figure E are feet from a typical SPG11 patient in her mid 30s (Family 4, Tables 1, 2). Figure F to I shows photographs of the hands in the proband F1II:1 and F1II:2 showing generalized tapering of the fingers and swelling of the proximal phalanx, red discolouration of the hands, more marked in the palms. Figure J are hands from a typical SPG11 patient, Family 4. Figure K shows an x-ray of the hands (patient F1II:1) with mild narrowing of the proximal interphalangeal joints bilaterally (arrows) compatible with mild osteoarthritis
The proband in the Cypriot SPG11 (F1AII:2) family had similar clinical features and initially presented at the age of 21 years with a complex HSP phenotype. When this patient was re-examined and investigated at the age of 42 years she had low tone and absent reflexes along with likely distal sensory abnormalities that were difficult to define with the severe cognitive features but these occurred early in the disease. Her feet showed similar discolouration but she did not suffer from any pain (Figs. 1, 2; Tables 1, 2) and strikingly different to typical SPG11 patients.

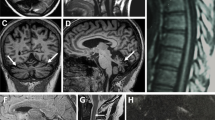
Sagittal MRI sequence; a control MRI with corpus callosum labeling. b Proband II:1 and c case II:2 showing thinning of the corpus callosum and cerebral atrophy. Coronal MRI sequence; d control. e Proband II:1 and f II:2 showing generalized atrophy with periventicular white matter abnormalities. Axial MRI sequence; h control. i Proband II:1 and j II:2 showing periventicular white matter abnormalities
Brain MRIs in both cases showed that the corpus callosum was thin with periventricular white matter changes and possible cerebellum atrophy (Fig. 3). Nerve conduction studies show a severe length-dependent axonal sensory-motor polyneuropathy affecting both lower and upper limbs. The neuropathy was compared with other typical SPG11 cases with complex HSP and was significantly worse (Table 2).

Pedigree of the F1 SPG11 family. Open symbols represent unaffected individuals and filled symbols represent affected individuals. The proband is indicated by an arrow. Stars indicate individuals whose DNA was used for whole exome sequencing and whose skin biopsies were used for fibroblast studies. Sanger sequencing chromatograms surround the symbols and show segregation of mutations. Black bar encompass the sites of interest (mutation over control site)
Genetic studies
To identify the underlying genetic cause, we applied whole-exome sequencing (WES) on the proband (II:1), her affected sister (II:2) and her mother (I:1). Analysis focused on nonsynonymous, splice-site and coding indel variants with a minor allele frequency (MAF) of <0.5 % in the Exome Aggregation Consortium (EXaC; www.exac.broadinstitute.org), Exome Variant Server (EVS; http://evs.gs.washington.edu) and 1000 Genomes databases (1000G; http://www.1000genomes.org). From the variants that met these filtering criteria in the proband, a few co-segregated under an autosomal recessive model and at least one was present in the mother. Of these variants, only two involved a gene associated with complex HSP, i.e., the SPG11 gene. These variants were validated by Sanger sequencing. Segregation analysis confirmed that both affected siblings and their unaffected father were heterozygotes for the SPG11 variant in exon 22, c.3729delC. Moreover, both affected sisters and their mother were heterozygotes for the variant in exon 30 c.5148dupA, also indicating that these two variants were located on the different alleles (Fig. 4). The variant in exon 22 is a one bp deletion which creates a frameshift disrupting the sequence from codon S1243. The new reading frame ends in a stop 6 codon positions downstream. Similarly the variant in exon 30 is a one bp duplication which creates a frameshift disrupting the sequence from codon H1717. The new reading frame ends in a stop 3 position downstream. To analyze the consequence of these mutations, we isolated RNA from the fibroblasts of the two affected sisters and their mother, extracted RNA and carried out RT-PCR. From our data the mutant allele is clearly present in the cDNA (Figs. 5, 6) and likely to exist as a truncated protein, indicating that very little RNA has been targeted for non-sense mediated mRNA decay (Fig. 6). The Cypriot family were found to have a homozygous c.6658_6659delAT, p.M2220Dfs*27 mutation. This is consistent with a typical SPG11 loss of function mutation and the family originated from a small village in Cyprus.

Pedigree of F1A Cypriot SPG11 family. Open symbols represent unaffected individuals and filled symbols represent affected individuals. SPG11 mutation has been previously reported

Schematic representation of the SPG11 protein and position of known (black) and novel (red) mutations. Mutation 1 is predicted to truncate the full length protein from 2443 amino acids (AA) to 1248 AA, mutation 2 truncates to 1718 AA and mutation 3 to 2247 AA. Putative functional domains are depicted as rectangles, and their positions within the amino acid sequence are indicated: the transmembrane domain (yellow box; positions, 163–194, 200–240, 1239–1267, and 1471–1493), glycosyl hydroxylase F1 signature (pink box; position, 482–490), leucine zipper (gray box; position, 611–632), coil–coil domain (brown box; position, 1556–1590), and Myb domain (green box; position 1766–1774). Arrows indicate truncating mutations; dotted arrows indicate missense mutations. Aa = amino acids

A. cDNA sequencing chromatograms of the heterozygous exon 22 SPG11 mutation in the mother (I:1 same as control sequence) and two affected siblings (II:1 and II:2) with deletion of a C (arrows). B. cDNA sequencing chromatograms of the heterozygous exon 30 SPG11 mutation in the mother (I:1 same as affected sequence) and two affected siblings (II:1 and II:2) with the insertion of an A (arrow). The mutation is clearly visible in both chromatograms and the peak heights are the same as genomic DNA (Fig. 3)
Discussion
In this study, we identified novel compound heterozygous mutations in SPG11 in a complex HSP family with thin corpus callosum and severe axonal sensory-motor polyneuropathy as a late manifestation of the disease. Moreover they had feet which were oedematous and strikingly blue–black in color as a likely result of fluid dependency and peripheral neuropathy. A recent study by Montecchiani et al. [14] showed that SPG11 is the causative gene of a wide spectrum of clinical features, including autosomal recessive CMT2. The severe neuropathy in our case is consistent with neuropathy being a major feature in some, but not all patients with SPG11 mutations and a complex HSP phenotype as is confirmed in the Cypriot family. An interesting aspect of the two SPG11 families discussed here is the initial clinical presentation as complex HSP with a later development of an axonal neuropathy clinically and electrophysiologically. As in SPG11 associated with complex HSP, the variants described by Montecchiani et al. [14] were scattered throughout the entire DNA sequence, without evidence of ‘hot spots’, and 93 % were truncating mutations. Our findings are consistent with this and other previous studies describing mostly mutations leading to the truncation of the SPG11 protein and consequent loss of function mechanism [21].
In Family 1 based on our cDNA sequencing data, we would expect the protein to be truncated and only little of the mRNA to be targeted for non-sense mediated decay. In our case, one of the truncated forms of the protein is missing the Myb domain, and the other one is missing both the Myb and the coil–coil domains whose presence may suggest a role of Spatacsin in regulation of gene expression [22]. In terms of pathology, sural nerve biopsy of SPG11 has previously shown loss of unmyelinated nerve fibers and accumulation of intra-axonal pleomorphic membranous material [23]. Also, axonal trafficking of vesicles was demonstrated to be impaired in neurons derived from induced pluripotent stem cells of SPG11 patients [24]. Another alteration is in the secretory pathway. The SPG11 protein Spatacsin has been shown to account for proteins involved in the formation of lysosomes and moreover to interact with components of the AP5 complex involved in membrane sorting of late endosomes [25, 26].
As our understanding of the hereditary spastic paraplegias increases it is clear that the clinical features are very heterogeneous and the spectrum of signs in disease genes such as SPG11 can vary substantially. It is noteworthy for clinicians to consider SPG11 testing in early onset complex HSP or where there is a combination of severe neuropathy and spasticity.
In conclusion, we provide support for the use of whole exome sequencing as a diagnostic tool for identification of mutations in conditions with complex and wide presentations such as HSP. Furthermore, we extend the findings that mutations in SPG11 are the cause of a spectrum of clinical features including the late manifestation of severe axonal neuropathy.
References
Lo Giudice T, Lombardi F, Santorelli FM, Kawarai T, Orlacchio A (2014) Hereditary spastic paraplegia: clinical genetic characteristics and evolving molecular mechanisms. Exp Neurol 261:518–539
Finsterer J, Loscher W, Quasthoff S, Wanschitz J, Auer-Grumbach M, Stevanin G (2012) Hereditary spastic paraplegias with autosomal dominant, recessive, X-linked, or maternal trait of inheritance. J Neurol Sci. 318:1–18
Stevanin G, Azzedine H, Denora P, Boukhris A, Tazie M, Lossos A et al (2008) Mutations in SPG11 are frequent in autosomal recessive spastic paraplegia with thin corpus callosum, cognitive decline and lower motor neuron degeneration. Brain 131:772–784
Kara E, Tucci A, Manzoni C et al (2016) Genetic and phenotypic characterization of complex hereditary spastic paraplegia. Brain 139(Pt 7):1904–1918. doi:10.1093/brain/aww111
Puech B, Lacour A, Stevanin G, Sautiere BG, Devos D, Depienne C, Denis E, Mundwiller E, Ferriby D, Vermersch P, Defoort-Dhellemmes S (2011) Kjellin syndrome: long-term neuro-ophthalmologic follow-up and novel mutations in the SPG11 gene. Ophthalmology 118(3):564–573
Pippucci T, Panza E, Pompilii E et al (2009) Autosomal recessive hereditary spastic paraplegia with thin corpus callosum: a novel mutation in the SPG11 gene and further evidence for genetic heterogeneity. Eur J Neurol 16(1):121–126. doi:10.1111/j.1468-1331.2008.02367.x
Orlen H, Melberg A, Raininko R et al (2009) SPG11 mutations cause Kjellin syndrome, a hereditary spastic paraplegia with thin corpus callosum and central retinal degeneration. Am J Med Genet B Neuropsychiatr Genet 150B(7):984–992. doi:10.1002/ajmg.b.30928
Bauer P, Winner B, Schule R et al (2009) Identification of a heterozygous genomic deletion in the spatacsin gene in SPG11 patients using high-resolution comparative genomic hybridization. Neurogenetics 10(1):43–48. doi:10.1007/s10048-008-0144-2
Samaranch L, Riverol M, Masdeu JC et al (2008) SPG11 compound mutations in spastic paraparesis with thin corpus callosum. Neurology 71(5):332–336. doi:10.1212/01.wnl.0000319646.23052.d1
Erichsen AK, Stevanin G, Denora P, Brice A, Tallaksen CM (2008) SPG11–the most common type of recessive spastic paraplegia in Norway? Acta Neurol Scand Suppl 188:46–50. doi:10.1111/j.1600-0404.2008.01031.x
Paisan-Ruiz C, Nath P, Wood NW, Singleton A, Houlden H (2008) Clinical heterogeneity and genotype–phenotype correlations in hereditary spastic paraplegia because of Spatacsin mutations (SPG11). Eur J Neurol 15(10):1065–1070. doi:10.1111/j.1468-1331.2008.02247.x
Paisan-Ruiz C, Dogu O, Yilmaz A, Houlden H, Singleton A (2008) SPG11 mutations are common in familial cases of complicated hereditary spastic paraplegia. Neurology 70(16 Pt 2):1384–1389. doi:10.1212/01.wnl.0000294327.66106.3d
Hehr U, Bauer P, Winner B et al (2007) Long-term course and mutational spectrum of spatacsin-linked spastic paraplegia. Ann Neurol 62(6):656–665. doi:10.1002/ana.21310
Montecchiani C, Pedace L, Lo Giudice T, Casella A, Mearini M, Gaudiello F et al (2016) ALS5/SPG11/KIAA1840 mutations cause autosomal recessive axonal Charcot-Marie-Tooth disease. Brain 139:73–85
Southgate L, Dafou D, Hoyle J, Li N et al (2010) Novel SPG11 mutations in Asian kindreds and disruption of spatacsin function in the zebrafish. Neurogenetics 11(4):379–389
Sumner CJ, d’Ydewalle C, Wooley J, Fawcett KA, Hernandez D, Gardiner AR, Kalmar B, Baloh RH, Gonzalez M, Zuchner S et al (2013) A dominant mutation in FBXO38 causes distal spinal muscular atrophy with calf predominance. Am J Hum Genet 93:976–983
Sailer A, Scholz SW, Gibbs JR et al (2012) Exome sequencing in an SCA14 family demonstrates its utility in diagnosing heterogeneous diseases. Neurology 79(2):127–131. doi:10.1212/WNL.0b013e31825f048e
Singleton AB, Hardy J, Traynor BJ, Houlden H (2010) Towards a complete resolution of the genetic architecture of disease. Trends Genet 26(10):438–442. doi:10.1016/j.tig.2010.07.004
Houlden H, King RH, Wood NW, Thomas PK, Reilly MM (2001) Mutations in the 5’ region of the myotubularin-related protein 2 (MTMR2) gene in autosomal recessive hereditary neuropathy with focally folded myelin. Brain 124(Pt 5):907–915
Houlden H, King RH, Hashemi-Nejad A et al (2001) A novel TRK A (NTRK1) mutation associated with hereditary sensory and autonomic neuropathy type V. Ann Neurol 49(4):521–525
Pensato V, Castellotti B, Gellera C, Pareyson D, Ciano C, Nanetti L et al (2014) Overlapping phenotypes in complex spastic paraplegias SPG11, SPG15, SPG35 and SPG48. Brain 137:1907–1920
Liu J, Zheng Q, Deng Y, Cheng CS, Kallenbach NR, Lu M (2006) A seven-helix coiled coil. Proc Natl Acad Sci USA 103(42):15457–15462
Hehr U, Bauer P, Winner B, Schule R, Olmez A, Koehler W et al (2007) Long-term course and mutational spectrum of spatacsin-linked spastic paraplegia. Ann Neurol 62:656–665
Perez-Branguli F, Mishra HK, Prots I et al (2014) Dysfunction of spatacsin leads to axonal pathology in SPG11-linked hereditary spastic paraplegia. Hum Mol Genet 23:4859–4874
Chang J, Lee S, Blackstone C (2014) Spastic paraplegia proteins spastizin and spatacsin mediate autophagic lysosome reformation. J Clin Invest 124:5249–5262
Hirst J, Borner GH, Edgar J, Hein MY, Mann M et al (2013) Interaction Between AP-5 and the Hereditary Spastic Paraplegia Proteins SPG11 and SPG15. Mol Biol Cell 24:2558–2569
Acknowledgments
The authors would like to thank the participants of the study for their essential help with this work. This study was supported by the Medical Research Council (MRC UK), The Wellcome Trust [equipment and the Synaptopathies strategic award (104033)] and The UK HSP Society and the EU FP7/2007-2013 under Grant Agreement Number 2012-305121 (NEUROMICS). MMR is grateful to the Medical Research Council (MRC), MRC Centre Grant (G0601943), and the National Institutes of Neurological Diseases and Stroke and office of Rare Diseases (U54NS065712) for their support. The INC (U54NS065712) is a part of the NCATS Rare Diseases Clinical Research Network (RDCRN). We are also supported by the National Institute for Health Research (NIHR) University College London Hospitals (UCLH) Biomedical Research Centre (BRC).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Ethical standard
The work was approved by UCLH ethics committee.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Manole, A., Chelban, V., Haridy, N.A. et al. Severe axonal neuropathy is a late manifestation of SPG11. J Neurol 263, 2278–2286 (2016). https://doi.org/10.1007/s00415-016-8254-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-016-8254-5