
Abstract
Genome amplification (DNA synthesis) is one of the most demanding cellular processes in all proliferative cells. The DNA replication machinery (also known as the replisome) orchestrates genome amplification during S-phase of the cell cycle. Genetic material is particularly vulnerable to various events that can challenge the replisome during its assembly, activation (firing), progression (elongation) and disassembly from chromatin (termination). Any disturbance of the replisome leads to stalling of the DNA replication fork and firing of dormant replication origins, a process known as DNA replication stress. DNA replication stress is considered to be one of the main causes of sporadic cancers and other pathologies related to tissue degeneration and ageing. The mechanisms of replisome assembly and elongation during DNA synthesis are well understood. However, once DNA synthesis is complete, the process of replisome disassembly, and its removal from chromatin, remains unclear. In recent years, a growing body of evidence has alluded to a central role in replisome regulation for the ubiquitin-dependent protein segregase p97, also known as valosin-containing protein (VCP) in metazoans and Cdc48 in lower eukaryotes. By orchestrating the spatiotemporal turnover of the replisome, p97 plays an essential role in DNA replication. In this review, we will summarise our current knowledge about how p97 controls the replisome from replication initiation, to elongation and finally termination. We will also further examine the more recent findings concerning the role of p97 and how mutations in p97 cofactors, also known as adaptors, cause DNA replication stress induced genomic instability that leads to cancer and accelerated ageing. To our knowledge, this is the first comprehensive review concerning the mechanisms involved in the regulation of DNA replication by p97.
Similar content being viewed by others

Introduction
Duplication of the genomic information (DNA) is a key objective for any dividing cell. All nuclear DNA has to be duplicated in an accurate way during S-phase of the cell cycle to ensure that the genetic information stays preserved for the next generation (Masai et al. 2010; Toueille and Hubscher 2004). In human cells, DNA replication has to accurately copy around 6 × 109 bases over approximately 10 h during S-phase (Goren and Cedar 2003); therefore, it must be spatiotemporally regulated at different levels. Any disturbance of this process might introduce mutations and chromosomal aberrations that could lead to various diseases such as stem cell depletion, neurodegeneration, cancer and accelerated (premature) ageing (Brambati et al. 2015; Fragkos et al. 2015; Macheret and Halazonetis 2015). The initiation of DNA replication starts at multiple origins of replication along chromosomes. Here, a pre-replicative complex (Pre-RC) is formed in a process known as licensing (Heller et al. 2011). Licensing begins at the end of mitosis and early G1-phase of the cell cycle with the loading of the main component of the replicative helicase: the hexameric minichromosome maintenance 2–7 (Mcm) complex. Loading of the Mcm2-7 complex, and therefore origin licensing, requires the six subunit origin recognition complex (ORC1-6), cell division cycle 6 (Cdc6) and chromatin licensing and DNA replication factor 1 (Cdt1) (Mehanna and Diffley 2012; Nishitani and Lygerou 2002; Yardimci and Walter 2014). Licensing is complete when ORC1-6 and Cdc6 convert individual heptamers of Mcm2-7-Cdt1 into head-to-head double hexamers of Mcm2-7 around double-stranded DNA, with the subsequent release of Cdt1 and Cdc6 (Yekezare et al. 2013). Subsequently, origin activation (firing) starts in S-phase of the cell cycle with the activation of Mcm2-7. The main firing factors, cell division cycle 45 (Cdc45) and the GINS (for Go, Ichi, Nii and San: five, one, two and three in Japanese) complex, associate with Mcm2-7 and form a functional CMG (Cdc45, Mcm2-7, GINS) helicase that powers the progression of the replication fork. The association of Cdc45 and GINS with Mcm2-7 depends on additional firing factors such as DNA Topoisomerase 2-binding protein (TopBP1), the TopBP1 binding protein Treslin and the DNA replication regulators Sld2 and Dpb11 (Boos et al. 2012; Heller et al. 2011). Activation of the pre-RC, and subsequent origin firing, is largely regulated by the CDK (cyclin-dependent kinase) and DDK (Dbf4-dependent kinase) kinases, but also an additional factor, Mcm10, whose binding to the Mcm2-7 complex is essential for the recruitment of the single-stranded DNA binding protein replication protein A (RPA), resulting in the initiation of DNA replication (Yeeles et al. 2015). Origin firing occurs at random locations and different timing throughout S-phase, classified as early and late origin firing, and proceeds in a bi-directional manner along the DNA. Once an origin has fired, the CMG helicase complex starts to unwind the double-stranded DNA ahead of the replication fork. This is then followed by the core DNA replication machinery [PCNA (proliferating cell nuclear antigen), the DNA polymerases α, δ and ε, RPA and Fen1 (Flap endonuclease 1), amongst others] which carries out synthesis of the new ‘daughter’ DNA strand (Barlow and Nussenzweig 2014; Fragkos et al. 2015; Yeeles et al. 2015). High topological stress ahead of the DNA replication fork, produced during DNA unwinding by the CMG helicase complex, is released by a specialised group of enzymes called topoisomerases. Topoisomerases cleave, untwist and re-ligate single- or double-stranded DNA ahead of the DNA replication fork, thus releasing topological stress to ensure undisturbed DNA synthesis (Chen et al. 2013). When an ongoing DNA replication fork from one origin meets an incoming DNA replication fork from an adjacent origin, DNA replication is stopped and disassembled in a process called termination (Bailey et al. 2015; Dewar et al. 2015). It is worth mentioning that licensing occurs only once per cell cycle, and that all the potential origins are licensed at the beginning of S-phase. On the contrary, only a handful of the origins that are licensed are actually utilised to complete replication. Most licensed origins remain dormant (dormant origins) during ongoing replication and are passively replicated by forks arising from neighbouring origins (Alver et al. 2014).
Besides the spatiotemporal orchestration of replication licensing, firing, initiation and termination, various additional obstacles constantly challenge the progression of a replication fork. This can include a variety of DNA lesions, DNA secondary structures, tightly bound DNA–protein complexes, collisions with transcription machinery, an inefficient pool of dNTPs, or unscheduled oncogene-activated firing of DNA replication (Gaillard et al. 2015; Petermann and Helleday 2010). Each of these challenges may affect the stability of the DNA replication fork. The inability of the replisome to cope with any of those challenges causes DNA replication stress and potentially error-prone DNA synthesis (Macheret and Halazonetis 2015). DNA replication stress is characterised by several parameters: (i) the velocity of replication fork is severely reduced, (ii) increase of fork stalling, (iii) firing of dormant origins, (iv) increased amounts of long stretches of single stranded DNA coated by RPA, (v) activation of the ataxia telangiectasia-mutated and Rad3-related (ATR)/Checkpoint kinase 1 (CHK1) signalling pathway and consequently the intra-S-phase checkpoint, (vi) formation of DNA double-strand breaks due to nucelolytic attack of long stretches of unprotected DNA and consequently activation of the ataxia telangiectasia-mutated (ATM)/Checkpoint kinase 2 (CHK2) signalling pathway, (vii) increased DNA end resection, (viii) increased levels of ultrafine chromosomal bridges and finally (ix) elevated levels of p53-binding protein 1 (53BP1) bodies in G1-phase of the cell cycle. Together, these hallmarks of DNA replication stress are considered to be one of the main sources of genomic instability that can ultimately lead to ageing-related phenotypes such as cancer (Da-Re and Halazonetis 2015; Macheret and Halazonetis 2015), stem cell depletion, tissue degeneration and even neurodegeneration (Burhans and Weinberger 2007; Yurov et al. 2011). Phosphorylation events, mostly controlled by CDK and DDK kinases, are known to play a major role in the coordination of the DNA replication fork (Sclafani and Holzen 2007).
However, in the last decade, other posttranslational modifications, such as ubiquitination and SUMOylation, have emerged as being equally as important, but their role and function in the regulation of DNA replication are poorly understood (Bergink and Jentsch 2009; Moreno and Gambus 2015; Ulrich 2014). The molecular segregase p97 also known as valsoin-containing protein (VCP) in metazoan or Cdc48 in lower eukaryotes is a central molecule in the regulation of ubiquitinated substrates and thus plays an essential role in protein homeostasis. There is a growing amount of evidence that ubiquitin-dependent regulation of the DNA replication machinery by p97 is essential for faithful DNA synthesis and genome stability (Vaz et al. 2013). In this review, we will focus on the role of p97 in regulating the DNA replication machinery. The ultimate goal of this review is to inform the scientific community about the essential and necessary functions of the ubiquitin-dependent segregase p97 in coordinating DNA replication.
Ubiquitin-dependent molecular segregase p97 (VCP or Cdc48)
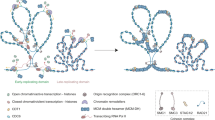
p97, also known as VCP in metazoans, CDC-48 in Caenorhabditis elegans, Cdc48 in yeast and Ter94 in insects, is an ubiquitin-dependent unfoldase/segregase that plays a central role in the ubiquitin-proteasome system (UPS) and in the regulation of protein homeostasis (Meyer et al. 2012; Richly et al. 2005). For the sake of simplicity, we will use the following abbreviations when specifically referring to p97 in metazoans (p97/VCP), in yeast (p97/Cdc48) or in worms (p97/CDC-48); otherwise, p97 will be used for general statements. p97 recognises substrates via their ubiquitin modifications and subsequently transfers them either to the proteasome for degradation or facilitates substrate recycling. Similarly to ubiquitin and the proteasome, the p97 system (p97 together with its cofactors, see below) is involved in a variety of cellular processes (Meyer et al. 2012; Meyer and Weihl 2014) (Fig. 1). The specificity of p97 toward diverse substrates, cellular locations and pathways is coordinated by a plethora of adaptor proteins, also known as p97 cofactors (Buchberger et al. 2015; Yeung et al. 2008). There are about 30 known p97 cofactors that in various combinations form numerous p97-subcomplexes, which are then directed towards specific substrates and pathways (Meyer et al. 2012; Meyer and Weihl 2014). All of the p97 cofactors are characterised by p97 interacting motifs, and the majority of them bind the N-terminal part of p97 (Yeung et al. 2008). However, we still do not know exactly how many different p97-subcomplexes exist or the specificity of different cofactor combinations. Regardless of the cofactor composition, p97, via its cofactors, predominantly binds ubiquitinated substrates (Fig. 2) (Alexandru et al. 2008; Ye 2006). Once bound to a substrate, p97 hydrolyses ATP, via the ATPase cassette in its D2 domain, and utilises the released energy to undergo a conformational change across its homohexamer structure, also referred to as an interprotomer motion transmission mechanism (Huang et al. 2012; Li et al. 2012). This ATP-driven interprotomer motion enables p97 to translocate substrates through its narrow central pore (Tonddast-Navaei and Stan 2013), thus removing them from different cellular locations such as the endoplasmic reticulum, chromatin, ribosomes or protein-macro complexes. Consequently, unfolded and/or segregated substrates are now easily recognised by the proteasome and are degraded or recycled with the help of p97-associated deubiquitinating enzymes (see below). In addition to its segregase and/or unfoldase activity, p97 also plays an important role as a recruitment platform, although not necessarily independently of the proteasome. This may serve as an additional layer of substrate regulation involving deubiquitinating enzymes, such as VCIP135 (valosin-containing interacting protein complex p97/p47 interacting-protein 135), Ataxin-3 and OTU1 (ovarian tumour domain-containing protein 1) and E3-ubiquitin ligases, such as AMFR (Autocrine Motility Factor Receptor)/gp78 and E4-ubiquitin ligase Ufd2 (Ubiquitin fusion degradation protein 2) in yeast (UBE4A and UBE4B in mammals) (Hoppe 2005; Meyer and Weihl 2014). These aforementioned functions are evolutionarily conserved and are essential for cell survival. For instance, knockdown of both p97 alleles (p97−/−) causes early-embryonic lethality in mice (Muller et al. 2007). Supporting this finding, siRNA-depletion of p97 in cells causes apoptosis (Wojcik et al. 2004).

The role of p97 in various cellular processes. The specificity of p97 function towards designated pathways and substrates is coordinated by a plethora of cofactors (depicted by the arrows). ER endoplasmic reticulum

The ubiquitinated substrate is bound to a cofactor that contains a ubiquitin-binding domain and is presented to p97. The substrate is shifted into the central pore of p97 and remodelled by ATPase activity of p97. This allows substrate unfolding and/or extraction from different cellular compartments of protein-protein complexes
Since the discovery of p97 as cold-sensitive cell-division-cycle mutant 48 (Cdc48) in yeast (Moir et al. 1982), VCP in pigs (Koller and Brownstein 1987) and finally biochemically and structurally characterised as the ring-like homohexameric ATPase p97 (Peters et al. 1990), its known functions were solely related to processes in the cytoplasm (Halawani and Latterich 2006; Meyer 2005; Ye 2006) for more than two decades. In 2007, it was discovered that p97/CDC-48 binds and extracts polyubiquitinated protein kinase Aurora B from chromatin during mitosis (Ramadan et al. 2007). Since this discovery, chromatin-associated protein removal and degradation (CAD) has been emerging as an essential process in the maintenance of genome stability (Beli and Jackson 2015; Dantuma et al. 2014; Dantuma and Hoppe 2012; Moreno and Gambus 2015; Ramadan 2012). It seems that p97 plays a central role in CAD, and therefore, it has been recognised as a genome caretaker (Vaz et al. 2013) (Fig. 3). p97, with the help of its cofactors, recognises and remodels a diverse set of chromatin-associated substrates. These are removed from chromatin and, in the majority of cases, degraded by the proteasome, or alternatively some of the substrates can also be recycled (Acs et al. 2011; Bailey et al. 2015; Franz et al. 2011; Meerang et al. 2011; Puumalainen et al. 2014; Ramadan 2012; Ramadan et al. 2007). In relation to CAD, impairment of the segregase or unfoldase activity of p97 causes the hyperaccumulation of ubiquitinated proteins on chromatin. In time, this interferes negatively with chromatin-related processes such as mitosis, DNA replication, DNA repair and transcription (Dantuma et al. 2014; Dantuma and Hoppe 2012; Vaz et al. 2013). Ultimately, the inhibition of CAD leads to protein-induced chromatin stress (PICHROS) and, consequently, severe DNA damage and genomic instability (Vaz et al. 2013). This is best demonstrated in the case where p97 and its cofactors Ufd1 (ubiquitin fusion degradation 1), Npl4 (nuclear protein localisation protein 4) and UBXD7 (UBX domain-containing protein 7), the p97-Ufd1-Npl4-UBXD7 complex were inhibited, resulting in the hyperaccumulation of the DNA damage sensors DDB2 (damage specific DNA binding protein 2) and XPC (Xeroderma pigmentosum, complementation group C) at sites of UV-induced chromatin lesions (Puumalainen et al. 2014).

p97 in chromatin-associated degradation (CAD). p97, together with its cofactors (Ufd1-Npl4 complex, UBXD7, SPRTN and FAF1), binds and processes the ubiquitinated substrate (S) on chromatin. The substrate (S) is then released from chromatin and either degraded by the proteasome or recycled
As the scope of this article concerns the role of p97 in DNA replication, we will only discuss chromatin-associated protein turnover at the site of DNA replication. To gain a deeper understanding of the role of p97 in other CAD processes, and how exactly p97 protects cells from PICHROS and genome instability, we suggest reading the following review articles (Buchberger et al. 2015; Dantuma et al. 2014; Dantuma and Hoppe 2012; Meyer et al. 2012; Meyer and Weihl 2014; Vaz et al. 2013).
Multiple roles of p97 in DNA replication
p97 was shown to physically interact with DNA replication associated proteins, such as the RecQ DNA helicase Werner (Indig et al. 2004; Partridge et al. 2003) in human cells and HIM-6 (Bloom helicases homolog) in C. elegans (Caruso et al. 2008). The first evidence that p97 is required for DNA replication emerged from the Hoppe laboratory (Mouysset et al. 2008).
The Hoppe group has demonstrated in C. elegans the importance of the p97/CDC-48 UFD-1 NPL-4 complex (p97/CDC-48-UFD-1-NPL-4) for DNA replication in dividing cells which are essential for embryonic cell division and germ line development of adult worms. Depletion of any of the components of the p97/CDC-48-UFD-1-NPL-4 complex in C. elegans embryos leads to severe replication-related DNA stress. This was manifested as: (i) increased levels of chromosomal aberrations such as chromosome bridges, (ii) decreased DNA content, (iii) increased amounts of stalled and collapsed DNA replication forks visualised by Rad51 foci formation, (iv) delayed S-phase progression due to activation of the DNA replication-related checkpoint ATR-CHK1 signalling cascade and (v) hypersensitivity of embryos to hydroxyurea-induced replication stress. Whilst co-depletion of ATR or CHK-1 in p97/CDC-48-UFD-1-NPL-4 depleted cells restored S-phase progression, it was not able to rescue the formation of chromosomal bridges. This suggests that the role of p97/CDC-48-UFD-1-NPL-4 is directly linked to DNA replication and not to the DNA replication-related checkpoint signalling cascade (Deichsel et al. 2009; Mouysset et al. 2008).
Interestingly, the authors concluded that worms depleted for p97/CDC-48, UFD-1 and NPL-4 show a strong reduction in DNA content, which resembles the loss of the DNA replication licensing factors Cdt1 and CDC6. However, the follow-up work has further demonstrated that p97/CDC-48 together with its cofactor Fas-associated 1 (FAF1) binds ubiquitinated Cdt1 and facilitates its degradation (Franz et al. 2016). Altogether, these data suggest that p97/CDC-48 regulates and promotes Cdt1 degradation, and therefore, the inactivation of p97/CDC-48 should instead cause re-replication due to high level of Cdt1 rather than a reduction in DNA content. Why p97/CDC-48-depleted worm cells, despite the high level of Cdt1, have less DNA content and whether p97/CDC-48 depleted cells show signs of re-replication still remains unclear and needs further study.
This initial work (Mouysset et al. 2008) highlighted the role of p97 in DNA replication and also provided a platform for further influential research in that direction. Indeed, recent work from several laboratories has highlighted the complexity of the p97 system in DNA replication (Fig. 4) (Davis et al. 2012; Franz et al. 2011, 2016; Ghosal et al. 2012; Maric et al. 2014; Moreno et al. 2014; Mosbech et al. 2012; Raman et al. 2011; Stingele et al. 2014). Taking into consideration the spatiotemporal dynamics of the DNA replication machinery during DNA synthesis and the capability of p97 to remodel a broad range of substrates, it is very likely that p97, together with its cofactors, is involved in the processing and remodelling of other, as of yet undiscovered, substrates associated with the DNA replication machinery.

The multiple roles of p97 in DNA replication. a Licencing and firing of DNA replication origins is controlled by the p97-Ufd1-Npl4 dependent degradation of Cdt1. b A speculative model: The bidirectional progression of the DNA replication fork is controlled by p97/Cdc48-WSS1 and p97/VCP-SPRTN proteolytic removal of DPCs in front of DNA replication fork. c The switch between replicative and TLS DNA polymerases at sites of stalled replication forks is facilitated by p97-Ufd1-Npl4-SPRTN. d The inactivation of FANCI and FANCD2 by p97-SPRTN in the FA pathway. e The disassembly of the MCM complex (a core component of CMG helicase) at the end of DNA replication (termination of the DNA replication fork) by p97-dependent extraction of ubiquitinated MCM7 (in red)
Initiation of DNA replication
A critical step in the initiation of DNA replication is the recruitment of the licensing factor Cdt1 during G1 phase to the origin of replication by the ORC complex (Mehanna and Diffley 2012). Cdt1 licenses origins by recruiting the CMG helicase which unwinds DNA, thus allowing the progression of DNA replication (Maiorano et al. 2000; Nishitani et al. 2000). Cdt1 must be inactivated during S-phase to prevent re-licensing of origins with the CMG helicase and consequently re-replicating an already replicated part of the DNA (Blow and Dutta 2005). Cdt1 inactivation is orchestrated by binding of the inhibitory protein Geminin and by ubiquitination by two cullin-RING-based E3-ubiquitin ligases (CRL): SCF-Skp2 and CRL4-DDB1-Cdt2. This ubiquitination results in Cdt1 proteasomal degradation (Hu et al. 2004; Kondo et al. 2004; Nishitani et al. 2006; Wohlschlegel et al. 2000; Zhong et al. 2003). Cdt1 proteasomal degradation has long been recognised as an essential step in regulating re-replication and in the maintenance of genome stability. This has recently been expanded upon by new findings from two independent laboratories who have demonstrated that Cdt1 degradation is regulated by the p97/VCP/Cdc-48-Ufd1-Npl4 complex, and that this mechanism is conserved from worms to humans (Franz et al. 2011; Raman et al. 2011) (Fig. 4a). The Harper lab has investigated the factors necessary for Cdt1 degradation by performing a genome-wide siRNA screen in HeLa cells (Raman et al. 2011). This approach identified p97 and its cofactor Ufd1 (p97-ufd1), but not Npl4, as essential factors for the proteasomal degradation of Cdt1. Furthermore, they have also identified another PCNA-dependent substrate of the E3-ubiquitin ligase CRL4A-DDB1-Cdt2, the histone methyltransferase SET8, which is also targeted for degradation during replication (Raman et al. 2011). SET8 is essential for cell cycle progression and its depletion leads to slow S-phase progression and extensive DNA damage, most probably due to high levels of histone H4 methylation at lysine 20 (H4K20) and consequently high chromatin condensation (Jorgensen et al. 2007). During S-phase, SET8 is kept low by CRL4A-DDB1-Cdt2 ubiquitination and p97-dependent degradation to maintain a chromatin structure that allows DNA replication progression (Jorgensen et al. 2011; Raman et al. 2011). By using a combination of mammalian and Xenopus cell-free extracts, the authors demonstrated that the p97/VCP-Ufd1-Npl4 complex binds and removes the ubiquitinated form of Cdt1 from chromatin (Raman et al. 2011). Similar results were obtained by independent work performed in the Hoppe lab (Franz et al. 2011). They have systematically analysed the subcellular localisation and dynamics of several conserved replication factors by time lapse microscopy in C. elegans. Whilst in wild-type C. elegans cells the CDC-45/GINS complex dissociates from chromatin after S-phase, inactivation of p97/CDC-48, or its core cofactors UFD-1 and NPL-4, leads to the persistent accumulation of CDC-45 and the GINS subunit SLD-5 throughout the cell cycle. However, inactivation of the p97/CDC-48-UFD-1-NPL-4 complex does not affect other replication factors such as ORC-2, CDC-6 or MCM-2, suggesting a substrate-specific role for p97 in DNA replication. In agreement with the previous results, P1 cell division delay in p97/CDC-48-depleted C. elegans mitotic embryos could be restored by depletion of either the licensing factor CDT-1 or the firing factors CDC-45 and GINS, suggesting that the removal of the licensing and firing factors is controlled by p97.
It was just recently demonstrated that another p97 cofactor, Fas-associated factor 1 (FAF1), plays an essential role in the stability of DNA replication forks in worms and several human cell lines (Franz et al. 2016). The p97-FAF1 complex, and its worm homolog, CDC-48-UBXN-3, directly binds the ubiquitinated licensing factor Cdt1, resulting in its removal from chromatin and finally Cdt1 proteasomal degradation. Inactivation of UBXN-3/FAF1 stabilises Cdt1 and Cdc45/GINS on chromatin, causing severe defects in replication fork dynamics (also see below) accompanied by an increased firing of dormant origins, and activation of the DNA damage checkpoint.
Elongation of DNA replication and DNA replication-associated processes: translesion DNA synthesis (TLS), the Fanconi anaemia (FA) pathway and DNA-protein crosslink (DPC) repair
After firing of the DNA replication factories at various origins, bidirectional and semiconservative DNA replication in eukaryotes allows the progression of the DNA replication fork in both directions from the origin (Burhans et al. 1990; Romero and Lee 2008). This process is called elongation of DNA replication and continues until two opposite replication forks converge. The DNA replication elongation process is governed by the active replicative helicase CMG complex which unwinds double stranded DNA, thus allowing unperturbed DNA synthesis (Moyer et al. 2006). Currently, there is almost no evidence as to whether p97 or its cofactors play a role in the elongation process of DNA replication (Fig. 4b).
However, monogenic and biallelic mutations in SPRTN (Lessel et al. 2014), also known as DVC1, a recently identified p97 cofactor (Davis et al. 2012; Ghosal et al. 2012; Mosbech et al. 2012), or its depletion in various human cell lines, causes severe DNA replication elongation defects, as measured by the DNA fibre assays (Lessel et al. 2014). Patients with mutated SPRTN (Lessel et al. 2014) also present a decrease in replication fork velocity, which could be explained as an indirect consequence of dormant origin firing (Alver et al. 2014).
Also, another cofactor of p97, FAF1, as mentioned earlier in the regulation of DNA initiation, also regulates DNA replication elongation (Franz et al. 2016). Depletion of FAF1 in various human cell lines causes a severe reduction in DNA replication fork velocity and an increased level of stalled forks as measured by the DNA fibre assay. Decreased DNA fork velocity and an increased level of stalled forks in FAF1 depleted cells can be almost completely restored by co-depletion of Cdt1. It is still not yet completely clear why co-depletion of Cdt1 restores the fork progression in FAF1-depleted cells. It could be as an indirect effect of increased loading of the Mcm2-7 complex that is stimulated by elevated levels of Cdt1. An increased level of Mcm2-7 might consequently cause an increased level of dormant origin firing once cells enter S-phase.
Translesion DNA synthesis
Various obstacles such as unrepaired DNA damage, DNA secondary structures, or DNA-protein crosslinks (DPCs) constantly challenge the DNA replication machinery during DNA elongation. Similarly, various metabolic factors such as depletion of the free dNTP pool also strongly threaten the progression of the DNA replication fork (Mirkin and Mirkin 2007). An inability of the DNA replication machinery to properly cope with all of these obstacles causes DNA replication fork stalling, persistence of which causes the stalled forks to collapse, leading to the formation of potentially cytotoxic DNA double-strand breaks. To prevent stalling and collapse of DNA replication forks, cells have evolved specialised DNA polymerases that are able to cope with various obstacles and bulky-DNA lesions in front of the DNA replication fork. This mechanism is called translesion DNA synthesis (TLS) (Daigaku et al. 2010) and is coordinated by ubiquitin and SUMO modifications on PCNA (Chen et al. 2011; Plosky and Woodgate 2004). One of the most common bulky-DNA lesions are thymidine dimers induced by UV-light or other metabolic processes (Spivak and Hanawalt 2015). Replicative DNA polymerases are unable to incorporate nucleotides opposite these lesions, causing the DNA replication machinery to stall and the replicative DNA polymerase to be replaced with a TLS DNA polymerase. After the lesion has been bypassed by the TLS DNA polymerase(s), the replicative DNA polymerases resume DNA synthesis. The process of DNA polymerase exchange at sites of damage is called DNA polymerase switching (Lehmann 2006).
Since the discovery of SPRTN, a body of evidence has accumulated indicating that SPRTN, together with p97, regulates DNA polymerase switching at sites of UV-induced DNA lesions (Davis et al. 2012; Ghosal et al. 2012; Kim et al. 2013; Machida et al. 2012; Mosbech et al. 2012). SPRTN, through its PCNA binding motif (PIP box) and ubiquitin binding domain (UBZ), is recruited to sites of stalled DNA replication forks and regulates DNA polymerase switching (Fig. 4c). It seems that SPRTN plays multiple roles during DNA polymerases switching: (i) removing the replicative DNA polymerase δ (Ghosal et al. 2012), (ii) facilitating DNA polymerase η recruitment (Ghosal et al. 2012), (iii) removing DNA polymerase η beyond the lesion (Davis et al. 2012; Mosbech et al. 2012), (iv) regulating error-free TLS (Kim et al. 2013; Machida et al. 2012) and (v) enhancing the recruitment of the E3 ligase Rad18 which monoubiquitinates PCNA (Centore et al. 2012), an essential process for TLS DNA polymerase recruitment. Furthermore, it was also shown that SPRTN protects polyubiquitinated PCNA from deubiquitination by antagonising USP1 protease activity, thus promoting error-free TLS DNA damage bypass at sites of stalled DNA replication forks and preventing stalled forks from recombination (Juhasz et al. 2012). It is well documented that SPRTN plays multiple roles at sites of DNA replication. It seems that the ubiquitination status of PCNA is monitored by the p97-SPRTN complex, which can then further enhance the ubiquitination of PCNA, governing either error-free TLS or the DNA damage avoidance pathway (template switch) at sites of stalled DNA replication forks (Kim et al. 2013; Machida et al. 2012). It was demonstrated that SPRTN, with its putative metalloprotease active centre [glutamic acid at position 112 (E112) located in the SprT-domain], prevents DNA damage induced mutagenesis at sites of stalled DNA replication forks. SPRTN prevents complex formation between DNA polymerase δ subunit 3 (POLD3), REV1 and DNA polymerase ζ that are involved in mutagenic (error-prone) TLS. In this way, SPRTN stimulates error-free TLS orchestrated by DNA polymerase η and extended by error-free DNA polymerases δ and ε.
SPRTN has many different domains (see Fig. 6 for domains in SPRTN) that could direct its function towards specific substrates, which might explain the diverse functions of SPRTN in DNA replication. Once recruited to sites of stalled replication forks in a PIP-box and UBZ-dependent manner, SPRTN recruits the p97/VCP-Ufd1-Npl4 core complex via its SHP domain to remove either DNA polymerase δ or η from sites of DNA replication blocks (Davis et al. 2012; Ghosal et al. 2012; Mosbech et al. 2012). Regardless of the precise molecular insights into the function of SPRTN in TLS, it is very likely that p97/VCP-Ufd1-Npl4, together with SPRTN (p97/VCP-Ufd1-Npl4-SPRTN), is of major relevance for DNA replication fork progression past replication-blocking damage, the mechanism known as DNA damage tolerance (DDT). Further work is necessary to understand how the p97-Ufd1-Npl4-SPRTN complex controls DDT, as well as the substrates which it processes.
Fanconi anaemia pathway
In addition to TLS synthesis, it was also recently demonstrated that p97, together with SPRTN, plays a role in the Fanconi anaemia (FA) DNA repair pathway (Gibbs-Seymour et al. 2015) (Fig. 4d). FA is a rare autosomal recessive disorder, in which at least 16 different genes have so far been identified, relating to the FA pathway (Longerich et al. 2014). Haematological abnormalities, due to failure of the bone marrow, and cancer predisposition are the main clinical characteristics of FA (Garaycoechea and Patel 2014). FA patient cells have increased levels of chromosomal aberrations and are hypersensitive to DNA-interstrand crosslinking (ICL) agents. ICLs represent a strong physical barrier to moving DNA replication forks, causing fork stalling and collapse. The FA pathway protein complex is recruited to sites of ICLs and, with the action of its endonuclease activity, cleaves DNA around the lesion, which then activates subsequent DNA repair pathways, such as HR (homologous recombination), TLS or NER (nucleotide excision repair) (Crossan and Patel 2012; Niedzwiedz et al. 2004). However, not only ICL-inducing agents activate the FA pathway, but also many other DNA replication stress-inducing agents such as hydroxyurea or aphidicolin. This suggests that the role of the FA pathway is not ICLs specific but rather a DNA replication-associated process (Jo and Kim 2015; Longerich et al. 2014). FA pathway activation is potentially cytotoxic due to its strong nuclease activity and, therefore, must be tightly regulated. Regulation of the FA complex is under the jurisdiction of various PTMs; it was recently demonstrated that SUMOylation and ubiquitination play a role in FA-dependent chromatin associated protein removal and degradation (Gibbs-Seymour et al. 2015; Kim and D’Andrea 2012). Two components of the FA pathway, FANCI (FA, complementation group I) and FANCD2 (FA, complementation group D2), are heavily SUMOylated and consequently ubiquitinated by the SUMO-targeted E3-ubiquitin ligase RNF4 (ring finger protein 4). The p97/VCP-SPRTN complex, but not the three most abundant p97-cofactors Ufd1, Npl4 and p47, recognises and extracts FANCI and FANCD2 from sites of DNA damage (Gibbs-Seymour et al. 2015). Their extraction and removal by the p97/VCP-SPRTN complex strictly depend on the segregase activity of p97, as a p97-segregase defective mutant is not able to remove FANCI and FANCD2 from chromatin (Gibbs-Seymour et al. 2015). Based on the abovementioned work, it is difficult to conclude whether p97/VCP-SPRTN is the sole p97 complex that extracts and inactivates the FA pathway complex, or whether other p97 cofactors are involved in this process as well. Furthermore, although these results suggest the importance of the p97-SPRTN complex in the FA pathway, SPRTN-depleted cell show a very mild sensitivity to MMC when compared to depletion of FANCD2 (Gibbs-Seymour et al. 2015). Also, SPRTN-mutated patients (Lessel et al. 2014) or SPRTN haploinsuficiency mice (Maskey et al. 2014) (for details see below) do not have any haematological abnormalities which would indicate an essential role for SPRTN in the FA pathway. Therefore, the role of SPRTN in the FA pathway might only be minor or represent a backup mechanism when other components of the FA pathway are affected.
DNA-protein crosslink repair
In yeast, the essential role of the p97/Cdc48 system during the progression of DNA synthesis and its function in coping with various obstacles in front of the DNA replication fork was further strengthened by the discovery of WSS1 and its role in the removal of covalently bound Topoisomerase 1 (Top1) to DNA, known as Top1-cc cleavage complex (Stingele et al. 2014). In addition, WSS1-depleted yeast cells are hypersensitive to formaldehyde that induces the covalent attachment of proteins to DNA, known as DNA-protein crosslinks (DPCs). DPCs are highly mutagenic and cytotoxic lesions, and if not removed from DNA, these proteins present a severe block to the progression of the DNA replication and RNA transcription machineries (Tretyakova et al. 2013). Toxic DPCs arise from various products of metabolism such as formaldehyde and acetaldehyde or by exogenous sources, such as UV-light. These sources (e.g. aldehydes or UV-light) can potentially crosslink any protein in the vicinity of DNA. Also, DPCs can arise from the enzymatic action of proteins that form transient covalent interactions with DNA like DNA polymerases, DNA methyltranferases or topoisomerases. In general, the interaction between protein and DNA is transient, but in some circumstances, the enzymes can be trapped, thus causing the accumulation of DPCs (Stingele and Jentsch 2015). Very little is known about how DPCs are removed ahead of DNA replication forks, but also how cells avoid DPC-induced cytotoxicity. Recent work from two independent groups demonstrated that WSS1 is a DNA-dependent metalloprotease important for the removal of covalently trapped Top1-cc, an enzymatically induced DNA-protein crosslink complex (DPCs) (Balakirev et al. 2015; Stingele et al. 2014). Yeast lacking both WSS1 and tyrosyl-DNA-phosphodiesterase (Tdp1; the enzyme that removes the remaining fragment peptide on DNA after proteasome-dependent degradation of Top1-cc) accumulate Top1-cc. WSS1 cleaves Top1 but only in the presence of DNA, and WSS1 proteolytic activity is inhibited by adding EDTA to the reaction, suggesting WSS1 is a DNA-dependent metalloprotease. Further cellular characterisation of different yeast mutants suggests that WSS1, in combination with p97/Cdc48, is involved in the removal of toxic-DPCs during S-phase DNA synthesis, but not bulky cellular DPCs. So, it seems that p97/Cdc48-WSS1 plays a central role in the proteolysis of DNA-replication related DPCs and thus prevents DNA replication fork stalling at sites of DPCs and consequently DNA fork collapse (Fig. 4b), which would lead to increased levels of recombination and genomic instability induced by gross chromosomal rearrangements (Stingele et al. 2014). On top of the role of p97/Cdc48-WSS1 in proteolytic removal of Top1-cc, it was also shown that p97/Cdc48-WSS1 and another p97 cofactor Doa1 form a novel p97 complex (p97/Cdc48-Wss1-Doa1) that is recruited to SUMOylated substrates on chromatin after DNA damage (Balakirev et al. 2015). Once recruited to chromatin, WSS1 possesses an intrinsic E3-SUMO ligase activity that further catalyses SUMO-chain extension on chromatin substrates. This consequently activates the WSS1 intrinsic protease activity that cleaves and removes SUMOylated substrates from chromatin. So it seems that the p97/Cdc48-WSS1-Doa1 complex with the intrinsic E3-SUMO ligase and proteolytic activities of WSS1 and the segregase activity of p97 regulates the progression of DNA replication when the DNA replication forks approaches DNA-protein blocks that can either be DPCs or even non-covalently, but tightly bound, proteins to DNA. It has also been reported that the removal of DPCs is essential for the progression of the DNA replication fork in Xenopus egg extract (Duxin et al. 2014). However, the enzyme(s) which remove(s) DPCs in metazoans during DNA replication is/are still not identified. SPRTN is a metazoan cofactor of p97/Cdc48 with domain structure similar to that of WSS1 (Stingele et al. 2015). Both WSS1 and SPRTN contain N-terminal protease (WSS1-protease in WSS1) or putative protease domain (SprT) and p97-interacting domain (SHP and VIM in WSS1 and SHP in SPRTN). In contrast to SPRTN, which contains two ubiquitin-binding domains and a PIP box important for SPRTN recruitment to sites of stalled DNA replication fork, WSS1 contains two SUMO-interacting motifs. Thus, it would be interesting to investigate whether SPRTN is also a protease which is important for the removal of DNA-protein blocks in front of the DNA replication fork, and whether similar mechanisms for the removal of DPCs in front of DNA replication fork exist in metazoans and especially in mammals.
Termination of DNA replication
Termination of the DNA replication machinery occurs when replication forks arising from two neighbouring origins meet (Bailey et al. 2015; Dewar et al. 2015). In that moment, the replisome, which is usually tightly bound to DNA, must be disassembled. Although replisome loading and its activation have been extensively studied, replisome termination was for many years, somehow, neglected. Recently, independent work from two laboratories demonstrated an essential role for p97 in replisome disassembly/termination (Fig. 4e) in yeast and Xenopus laevis cell-free extracts, thus encircling another role of p97 in DNA replication (Maric et al. 2014; Moreno et al. 2014). In budding yeast, the CMG helicase complex is ubiquitinated in late S-phase, specifically on the MCM7 subunits, by the SCF-Dia2 ubiquitin ligase complex (Maric et al. 2014). This result suggested that ubiquitination of the CMG helicases occurs at the point of replisome termination. Interestingly, disassembly of the CMG helicase complex at the end of chromosomal replication strongly depends on the segregase activity of p97/Cdc48, but not on the proteasome (Maric et al. 2014; Moreno et al. 2014). p97/Cdc48 binds SCF-Dia2 K48-ubiquitinated MCM7 and, using its ATPase activity, removes the whole CMG complex. Inactivation of p97/Cdc48, either by expressing temperature sensitive yeast mutants or by removal of endogenous p97/Cdc48 using an inducible degron system, leads to hyperaccumulation of chromatin bound CMG helicase with ubiquitinated MCM7, and the retention of the whole CMG complex on chromatin until the next G1 phase of the cell cycle (Maric et al. 2014). Similar results were obtained biochemically in Xenopus egg extracts (Moreno et al. 2014); inactivation of p97 function by complementation with a p97/VCP ATPase inactive mutant led to the accumulation of the ubiquitinated CMG complex on chromatin. However, the eukaryotic E3-ubiqutin ligase which ubiquitinates MCM7 at sites of DNA replication fork termination has not yet been identified. The work from both labs (Maric et al. 2014; Moreno et al. 2014) clearly showed the essential role of p97 in segregating the active replisome at the end of replication by removing ubiquitinated MCM7 in a proteasome independent manner. However, the p97 cofactors and E3-ubiquitin ligase for ubiquitination of metazoan MCM7 for replisome disassembly are still to be identified.
Disturbance of the p97 system in DNA replication leads to cancer and premature ageing in humans
SPARTAN syndrome
Direct evidence for the role of the p97 system in human diseases related to DNA replication defects has just recently begun to emerge (Fig. 5). It has just been discovered that biallelic and monogenic mutations in the p97 cofactor SPRTN causes a new human syndrome characterised by chromosomal instability, early-onset hepatocellular carcinoma and premature ageing (Lessel et al. 2014). The syndrome was officially named Ruijs-Aalfs Syndrome (RJALS) according to the authors who described the first patient (family A patient 1; A-1) as an atypical Werner Syndrome (Ruijs et al. 2003). Our discovery of the second family with two additional patients (family B patients 2 and 3; B-2 and B-3), and whole exome sequencing of all three patients confirmed this is a new monogenic human syndrome caused by biallelic mutations in the SPRTN gene (Fig. 6) (Lessel et al. 2014). As RJALS syndrome is, amongst scientists, much better known as SPARTAN (due to mutations in the SPRTN gene) syndrome (Hiom 2014), we propose that both names could be used for this syndrome.

The role of replication related p97 functions in ageing and cancer in humans. Mutations in the p97-adaptor SPRTN cause SPARTAN syndrome characterised by premature ageing and early onset hepatocellular carcinoma. p97 regulates Werner protein (WRN) localisation, and mutations in WRN cause Werner Syndrome characterised by premature ageing and various cancer types. Loss or down-regulation of the p97 adaptor FAF1 has been observed in various human cancers

Schematic representation of the SPRTN mutations identified in SPARTAN syndrome patients. The patient from family A contains two truncated alleles of SPRTN where the C-terminal part is missing (ΔC). The patients from family B contain one allele similar to family A (ΔC), and one full-length allele with a single missense mutation (Y117C) in the putative metalloprotease domain SprT. aa amino acid. Colour code: MIU-domain motif interacting with ubiquitin, SprT-domain SprT-like zinc metallopeptidase, SHP-box (Shp1, eye-closed, p47), PIP-box PCNA interacting peptide, UBZ-domain ubiquitin-binding zinc finger. Please note that the parents and siblings of index patients bearing one SPRTN wild-type allele together with one mutated allele WT/ Y117C or WT/ΔC are unaffected individuals (not shown)
All three patients, family A patient no. 1 (A-1) and family B patients no. 2 and no. 3 (B-2 and B-3) developed hepatocellular carcinoma at the age of 13 years, two of which died of hepatocellular carcinoma complications. The third patient (B-3) was cured by liver transplantation and is currently stable and in good health 2 years post-operation (David Amor and Paul Lockhart, personal communication).
The C-terminal region of SPRTN contains the p97-interacting motif (SHP), PCNA interacting box (PIP) and ubiquitin-binding domain (UBZ domain). Therefore, the pathogenicity of the SPRTN-ΔC mutation found in the A-1 patient could easily be explained by its inability to be recruited to the sites of stalled and collapsed DNA replication forks, due to its missing PIP-box, and an inability to recruit p97, due to its missing SHP-domain, to segregate substrates at the fork. Interestingly, the two patients from family B (B-2 and B-3) have a similar mutation to patient A-1 on one allele, SPRTN-ΔC; however, on the other allele, they only have a single amino acid change (Y117C) in SprT-domain, a putative metalloprotease domain (Fig. 6). As the parents and healthy siblings of the affected patients contain one SPRTN wild-type allele and one mutated allele, and do not develop any signs of this syndrome, we concluded that the missense mutation in SprT domain must be pathogenic in order for patients B1 and B2 to develop the disease, and that this domain has an essential role in genome stability and cancer protection in humans. Molecular analysis of SPRTN and disease-related mutations has revealed that SPRTN plays an essential role in DNA replication fork progression and stability, as well as in guarding G2/M checkpoint function (Lessel et al. 2014).
These results suggest that the putative metalloprotease domain of SPRTN plays an essential role in DNA replication and the prevention of genome instability in humans. This raises the question as to why a single missense mutation in the SprT domain is pathogenic and whether the SprT domain indeed possesses essential metalloprotease activity.
Similarly to SPRTN-mutated patients, haploinsufficiency of SPRTN in mice causes chromosomal instability, cellular senescence and progeroid phenotypes characterised by cataracts, lodrokyphosis and cachexia at a young age (Maskey et al. 2014). However, complete loss of SPRTN (−/−) causes early embryonic lethality (before embryonic day 7).
In both human and mouse studies, it has been demonstrated that the putative metalloprotease SprT-domain of SPRTN plays an essential role in DNA replication and cell proliferation (Lessel et al. 2014; Maskey et al. 2014). Interestingly, a SPRTN-like protein in yeast, WSS1, was recently shown to function as a DNA-dependent metalloprotease, which in combination with p97/Cdc48, cleaves Top1-cc (Stingele et al. 2014). Thus, a further in-depth biochemical investigation of SPRTN, and its potential protease activity, is required to support our hypothesis that human SPRTN is also a metalloprotease essential for the removal of Top1-cc or even other and not yet identified DPCs in front of the DNA replication fork.
FAF1-mutated/low expressed cancers
In addition to SPRTN mutations, it was recently demonstrated that mutations or low expression levels of FAF1, another p97 cofactor, are associated with several cancer types (Liu et al. 2012; Menges et al. 2009; Weber 2015). FAF1 is essential for the stability of DNA replication forks in worms and human cell lines (Franz et al. 2016). Therefore, the role of FAF1 in carcinogenesis could be directly linked to the function of FAF1 in DNA replication. The p97-FAF1 complex (CDC-48-UBXN-3) binds and facilitates the proteasome-dependent degradation of the licensing factor CDT-1/Cdt1 and the components of the main DNA replication helicase CMG (Cdc45 and GINS) and thus allowing proper DNA synthesis. Elevated levels of Cdt1 and CMG helicase were shown to cause DNA replication stress manifested by re-replication and severe genomic instability that could lead to cancer (Karavias et al. 2016; Seo et al. 2005; Srinivasan et al. 2013). Moreover, it was also suggested that the ubiquitin-binding domain of FAF1 is essential for its function in protection from carcinogenesis (Lee et al. 2012). Therefore, the main role of FAF1 in protection from carcinogenesis could be its function in DNA replication. Hence, it would be worthwhile checking whether FAF1-mutated/low expressed cancers contain elevated level of Cdt1, Cdc45 and GINS.
Werner syndrome
The role of the p97 system in ageing and cancer is further emphasised by the role of p97 in Werner protein (WRN) mobility after DNA replication stress (Indig et al. 2004; Partridge et al. 2003). Mutations in Werner gene/protein cause Werner syndrome (WS), one of best characterised human premature ageing syndromes resembling physiological ageing at an early age (Coppede 2012). In contrast to usual ageing, WS is characterised by multiple cancers, and these include disproportional number of sarcomas, affecting middle-aged patients (Friedrich et al. 2010). WS is an autosomal recessive genetic disease caused by mutations in WRN. WRN is a member of the RecQ DNA helicase family and has 3′-5′ DNA helicase and 3′-5′ exonuclease activities (Rossi et al. 2010). WRN has multifunctional roles in the maintenance of genome stability, including its role in DNA replication, DNA repair and RNA transcription. The role of WRN in DNA replication is not completely clear. However, WRN physically associates with components of the DNA replication fork as it interacts with replicative and TLS DNA polymerases δ and η and RPA (Hyun et al. 2012; Kamath-Loeb et al. 2000, 2007). It was also shown that WRN increases DNA pol δ processivity as well as its localisation in the nucleolus. It has been suggested that WRN helicase activity resolves difficult DNA structures such as G-quadruplex and Holliday junctions in front of the DNA replication fork, and that the 3′-5′ exonuclease activity of WRN degrades alternative DNA structures and assists DNA polymerase δ in high fidelity DNA synthesis (Kamath-Loeb et al. 2012) (Croteau et al. 2014). p97 physically interacts with WRN in nucleoli (Partridge et al. 2003). This interaction is direct and carried out via the conserved RecQ domain of WRN (Indig et al. 2004). The p97-WRN complex is disrupted by a Top1 inhibitor camptothecin and WRN is released from nucleoli. The molecular mechanism of the p97-WRN interaction is not yet clear, but the aforementioned results suggest that p97 regulates WRN release from nucleoli during DNA replication stress, and thus, p97 might facilitate WRN recruitment to sites of stalled and/or collapsed DNA replication fork. The role of p97 in WRN dynamics is an additional example of how the disturbance of the p97 system related to processes involved in DNA replication might directly cause premature ageing and cancer.
Concluding Remarks
This review article aims to emphasise the essential role of the ubiquitin-dependent molecular segregase p97 (VCP or Cdc48) in DNA replication. Here, we present a body of evidence that suggests a central role for p97 and its cofactors (p97 system) in DNA replication. There is more and more evidence that DNA replication stress is one of the main causes of various types of cancer, both sporadic and hereditary, but also tissue degeneration and ageing (Khurana and Oberdoerffer 2015; Lessel et al. 2014; Macheret and Halazonetis 2015). Hence, we feel that generating new knowledge and awareness of every aspect of DNA replication is essential for biology and medicine. From the aforementioned facts, it is clear that molecular insights into p97 during DNA replication are essential, and most probably tightly coupled with the proteasome degradation machinery. It is highly probable that p97 regulates many other not yet discovered substrates throughout the DNA replication processes. Understanding the role of p97 in DNA replication is therefore paramount for our understanding of ageing-related diseases, cancerogenesis and cancer therapy. Disturbance of the p97 system, such as by mutations in SPRTN or FAF1, severely affect the progression of the DNA replication fork and causes DNA replication stress in proliferating cells. Altogether, we conclude that p97, along with its cofactors Ufd1, Npl4, SPRTN, WSS1 and FAF1/UBXN-3, plays a strategic and conserved role in DNA replication from yeast to humans. Accordingly, molecular insights into p97, its cofactors and substrates during DNA replication are essential to our understanding of genome stability. As the components of the ubiquitin-proteasome system and DNA replication have already been used for the treatment of various malignancies, the function of the p97 system in DNA replication accentuates additional complexities, but also gives excellent diagnostic and therapeutic opportunities for ageing-related diseases such as cancer and tissue degeneration. The role of p97 in DNA replication should be especially considered for cancer therapy and for therapeutics that directly affect DNA replication, such as topoisomerase 1 and 2 inhibitors (Chazin Ede et al. 2014; Chen et al. 2015; Pommier 2013). Topoisomerase inhibitors trap the topoisomerase 1 and 2 cleavage complex on DNA, consequently blocking the progression of DNA replication and transcription, thus inhibiting cell proliferation (Ashour et al. 2015). As p97/Cdc48-complexes in yeast (Stingele et al. 2014) are able to remove toxic topoisomerase cleavage complexes from DNA (Stingele et al. 2015), cancer cells with elevated level of p97 (Yamamoto et al. 2003a, b, 2004a, b, 2005) might be completely resistant to topoisomerase inhibitors; whether this mechanism is also relevant in mammalian cells needs to be further addressed. In addition, a proteasome inhibitor bortezomib (Velcade®) is used for the treatment of B cell malignancies such as mantle cell lymphoma and myeloma (Robak et al. 2015). However, the mechanistic insight of how bortezomib inhibits cancer cell growth and killing remains unclear (McCormack 2015). The ATPase activity of p97 remodels and segregates many ubiquitinated substrates in tight coordination with the proteasome (Barthelme et al. 2014, 2015; Sha and Goldberg 2014). Therefore, inhibiting proteasomal activity could also have an effect on p97 function in DNA replication. p97 is a new and potential target for cancer therapy, and the development of p97 inhibitors has recently generated a lot of attention (Chapman et al. 2015a, b; Chou et al. 2011; Deshaies 2014; Magnaghi et al. 2013). Furthermore, there are currently several p97 inhibitors in clinical trials for cancer therapy (http://www.cleavebio.com/pipeline/p97.aspx). For that reason, the understanding of the precise molecular mechanism of p97 and its substrates during DNA replication will be of a direct benefit for cancer therapy.
We believe that this review highlights the role of p97 in genome stability, cancer and ageing and establishes the p97-system as an integral part of the DNA replication machinery.
References
Acs K, Luijsterburg MS, Ackermann L, Salomons FA, Hoppe T, Dantuma NP (2011) The AAA-ATPase VCP/p97 promotes 53BP1 recruitment by removing L3MBTL1 from DNA double-strand breaks. Nat Struct Mol Biol 18:1345–1350. doi:10.1038/nsmb.2188
Alexandru G, Graumann J, Smith GT, Kolawa NJ, Fang R, Deshaies RJ (2008) UBXD7 binds multiple ubiquitin ligases and implicates p97 in HIF1alpha turnover. Cell 134:804–816. doi:10.1016/j.cell.2008.06.048
Alver RC, Chadha GS, Blow JJ (2014) The contribution of dormant origins to genome stability: from cell biology to human genetics. DNA Repair (Amst) 19:182–189. doi:10.1016/j.dnarep.2014.03.012
Ashour ME, Atteya R, El-Khamisy SF (2015) Topoisomerase-mediated chromosomal break repair: an emerging player in many games. Nat Rev Cancer 15:137–151. doi:10.1038/nrc3892
Bailey R, Priego Moreno S, Gambus A (2015) Termination of DNA replication forks: “Breaking up is hard to do”. Nucleus 6:187–196. doi:10.1080/19491034.2015.1035843
Balakirev MY et al. (2015) Wss1 metalloprotease partners with Cdc48/Doa1 in processing genotoxic SUMO conjugates. Elife 4 doi:10.7554/eLife.06763
Barlow JH, Nussenzweig A (2014) Replication initiation and genome instability: a crossroads for DNA and RNA synthesis. Cell Mol Life Sci 71:4545–4559. doi:10.1007/s00018-014-1721-1
Barthelme D, Chen JZ, Grabenstatter J, Baker TA, Sauer RT (2014) Architecture and assembly of the archaeal Cdc48*20S proteasome. Proc Natl Acad Sci U S A 111:E1687–1694. doi:10.1073/pnas.1404823111
Barthelme D, Jauregui R, Sauer RT (2015) An ALS disease mutation in Cdc48/p97 impairs 20S proteasome binding and proteolytic communication. Protein Sci 24:1521–1527. doi:10.1002/pro.2740
Beli P, Jackson SP (2015) Ubiquitin regulates dissociation of DNA repair factors from chromatin. Oncotarget 6:14727–14728. doi:10.18632/oncotarget.4410
Bergink S, Jentsch S (2009) Principles of ubiquitin and SUMO modifications in DNA repair. Nature 458:461–467. doi:10.1038/nature07963
Blow JJ, Dutta A (2005) Preventing re-replication of chromosomal DNA. Nat Rev Mol Cell Biol 6:476–486. doi:10.1038/nrm1663
Boos D, Frigola J, Diffley JF (2012) Activation of the replicative DNA helicase: breaking up is hard to do. Curr Opin Cell Biol 24:423–430. doi:10.1016/j.ceb.2012.01.011
Brambati A, Colosio A, Zardoni L, Galanti L, Liberi G (2015) Replication and transcription on a collision course: eukaryotic regulation mechanisms and implications for DNA stability. Front Genet 6:166. doi:10.3389/fgene.2015.00166
Buchberger A, Schindelin H, Hanzelmann P (2015) Control of p97 function by cofactor binding. FEBS Lett 589:2578–2589. doi:10.1016/j.febslet.2015.08.028
Burhans WC, Weinberger M (2007) DNA replication stress, genome instability and aging. Nucleic Acids Res 35:7545–7556. doi:10.1093/nar/gkm1059
Burhans WC, Vassilev LT, Caddle MS, Heintz NH, DePamphilis ML (1990) Identification of an origin of bidirectional DNA replication in mammalian chromosomes. Cell 62:955–965
Caruso ME et al (2008) GTPase-mediated regulation of the unfolded protein response in Caenorhabditis elegans is dependent on the AAA+ ATPase CDC-48. Mol Cell Biol 28:4261–4274. doi:10.1128/MCB.02252-07
Centore RC, Yazinski SA, Tse A, Zou L (2012) Spartan/C1orf124, a reader of PCNA ubiquitylation and a regulator of UV-induced DNA damage response. Mol Cell 46:625–635. doi:10.1016/j.molcel.2012.05.020
Chapman E, Maksim N, de la Cruz F, La Clair JJ (2015a) Correction: Chapman, E. et al. Inhibitors of the AAA+ chaperone p97. Molecules 2015, 20, 3027–3049. Molecules 20:4357–4358. doi:10.3390/molecules20034357
Chapman E, Maksim N, de la Cruz F, La Clair JJ (2015b) Inhibitors of the AAA+ chaperone p97. Molecules 20:3027–3049. doi:10.3390/molecules20023027
Chazin Ede L, Reis Rda R, Junior WT, Moor LF, Vasconcelos TR (2014) An overview on the development of new potentially active camptothecin analogs against cancer. Mini Rev Med Chem 14:953–962
Chen J, Bozza W, Zhuang Z (2011) Ubiquitination of PCNA and its essential role in eukaryotic translesion synthesis. Cell Biochem Biophys 60:47–60. doi:10.1007/s12013-011-9187-3
Chen SH, Chan NL, Hsieh TS (2013) New mechanistic and functional insights into DNA topoisomerases. Annu Rev Biochem 82:139–170. doi:10.1146/annurev-biochem-061809-100002
Chen T, Sun Y, Ji P, Kopetz S, Zhang W (2015) Topoisomerase IIalpha in chromosome instability and personalized cancer therapy. Oncogene 34:4019–4031. doi:10.1038/onc.2014.332
Chou TF et al (2011) Reversible inhibitor of p97, DBeQ, impairs both ubiquitin-dependent and autophagic protein clearance pathways. Proc Natl Acad Sci U S A 108:4834–4839. doi:10.1073/pnas.1015312108
Coppede F (2012) Premature aging syndrome. Adv Exp Med Biol 724:317–331. doi:10.1007/978-1-4614-0653-2_24
Crossan GP, Patel KJ (2012) The Fanconi anaemia pathway orchestrates incisions at sites of crosslinked DNA. J Pathol 226:326–337. doi:10.1002/path.3002
Croteau DL, Popuri V, Opresko PL, Bohr VA (2014) Human RecQ helicases in DNA repair, recombination, and replication. Annu Rev Biochem 83:519–552. doi:10.1146/annurev-biochem-060713-035428
Daigaku Y, Davies AA, Ulrich HD (2010) Ubiquitin-dependent DNA damage bypass is separable from genome replication. Nature 465:951–955. doi:10.1038/nature09097
Dantuma NP, Hoppe T (2012) Growing sphere of influence: Cdc48/p97 orchestrates ubiquitin-dependent extraction from chromatin. Trends Cell Biol 22:483–491. doi:10.1016/j.tcb.2012.06.003
Dantuma NP, Acs K, Luijsterburg MS (2014) Should I stay or should I go: VCP/p97-mediated chromatin extraction in the DNA damage response. Exp Cell Res 329:9–17. doi:10.1016/j.yexcr.2014.08.025
Da-Re C, Halazonetis TD (2015) DNA replication stress as an Achilles’ heel of cancer. Oncotarget 6:1–2
Davis EJ, Lachaud C, Appleton P, Macartney TJ, Nathke I, Rouse J (2012) DVC1 (C1orf124) recruits the p97 protein segregase to sites of DNA damage. Nat Struct Mol Biol 19:1093–1100. doi:10.1038/nsmb.2394
Deichsel A, Mouysset J, Hoppe T (2009) The ubiquitin-selective chaperone CDC-48/p97, a new player in DNA replication. Cell Cycle 8:185–190
Deshaies RJ (2014) Proteotoxic crisis, the ubiquitin-proteasome system, and cancer therapy. BMC Biol 12:94. doi:10.1186/s12915-014-0094-0
Dewar JM, Budzowska M, Walter JC (2015) The mechanism of DNA replication termination in vertebrates. Nature. doi:10.1038/nature14887
Duxin JP, Dewar JM, Yardimci H, Walter JC (2014) Repair of a DNA-protein crosslink by replication-coupled proteolysis. Cell 159:346–357. doi:10.1016/j.cell.2014.09.024
Fragkos M, Ganier O, Coulombe P, Mechali M (2015) DNA replication origin activation in space and time. Nat Rev Mol Cell Biol 16:360–374. doi:10.1038/nrm4002
Franz A et al (2011) CDC-48/p97 coordinates CDT-1 degradation with GINS chromatin dissociation to ensure faithful DNA replication. Mol Cell 44:85–96. doi:10.1016/j.molcel.2011.08.028
Franz A et al (2016) Chromatin-associated degradation is defined by UBXN-3/FAF1 to safeguard DNA replication fork progression. Nat Commun 7:10612. doi:10.1038/ncomms10612
Friedrich K et al (2010) WRN mutations in Werner syndrome patients: genomic rearrangements, unusual intronic mutations and ethnic-specific alterations. Hum Genet 128:103–111. doi:10.1007/s00439-010-0832-5
Gaillard H, Garcia-Muse T, Aguilera A (2015) Replication stress and cancer. Nat Rev Cancer 15:276–289. doi:10.1038/nrc3916
Garaycoechea JI, Patel KJ (2014) Why does the bone marrow fail in Fanconi anemia? Blood 123:26–34. doi:10.1182/blood-2013-09-427740
Ghosal G, Leung JW, Nair BC, Fong KW, Chen J (2012) Proliferating cell nuclear antigen (PCNA)-binding protein C1orf124 is a regulator of translesion synthesis. J Biol Chem 287:34225–34233. doi:10.1074/jbc.M112.400135
Gibbs-Seymour I et al (2015) Ubiquitin-SUMO circuitry controls activated Fanconi anemia ID complex dosage in response to DNA damage. Mol Cell 57:150–164. doi:10.1016/j.molcel.2014.12.001
Goren A, Cedar H (2003) Replicating by the clock. Nat Rev Mol Cell Biol 4:25–32. doi:10.1038/nrm1008
Halawani D, Latterich M (2006) p97: the cell’s molecular purgatory? Mol Cell 22:713–717. doi:10.1016/j.molcel.2006.06.003
Heller RC, Kang S, Lam WM, Chen S, Chan CS, Bell SP (2011) Eukaryotic origin-dependent DNA replication in vitro reveals sequential action of DDK and S-CDK kinases. Cell 146:80–91. doi:10.1016/j.cell.2011.06.012
Hiom K (2014) SPRTN is a new player in an old story. Nat Genet 46:1155–1157. doi:10.1038/ng.3125
Hoppe T (2005) Multiubiquitylation by E4 enzymes: ‘one size’ doesn’t fit all. Trends Biochem Sci 30:183–187. doi:10.1016/j.tibs.2005.02.004
Hu J, McCall CM, Ohta T, Xiong Y (2004) Targeted ubiquitination of CDT1 by the DDB1-CUL4A-ROC1 ligase in response to DNA damage. Nat Cell Biol 6:1003–1009. doi:10.1038/ncb1172
Huang C, Li G, Lennarz WJ (2012) Dynamic flexibility of the ATPase p97 is important for its interprotomer motion transmission. Proc Natl Acad Sci U S A 109:9792–9797. doi:10.1073/pnas.1205853109
Hyun M et al (2012) Physical and functional interactions of Caenorhabditis elegans WRN-1 helicase with RPA-1. Biochemistry 51:1336–1345. doi:10.1021/bi200791p
Indig FE, Partridge JJ, von Kobbe C, Aladjem MI, Latterich M, Bohr VA (2004) Werner syndrome protein directly binds to the AAA ATPase p97/VCP in an ATP-dependent fashion. J Struct Biol 146:251–259. doi:10.1016/j.jsb.2003.11.009
Jo U, Kim H (2015) Exploiting the Fanconi anemia pathway for targeted anti-cancer therapy. Mol Cell 38:669–676. doi:10.14348/molcells.2015.0175
Jorgensen S et al (2007) The histone methyltransferase SET8 is required for S-phase progression. J Cell Biol 179:1337–1345. doi:10.1083/jcb.200706150
Jorgensen S et al (2011) SET8 is degraded via PCNA-coupled CRL4(CDT2) ubiquitylation in S phase and after UV irradiation. J Cell Biol 192:43–54. doi:10.1083/jcb.201009076
Juhasz S, Balogh D, Hajdu I, Burkovics P, Villamil MA, Zhuang Z, Haracska L (2012) Characterization of human Spartan/C1orf124, an ubiquitin-PCNA interacting regulator of DNA damage tolerance. Nucleic Acids Res 40:10795–10808. doi:10.1093/nar/gks850
Kamath-Loeb AS, Johansson E, Burgers PM, Loeb LA (2000) Functional interaction between the Werner Syndrome protein and DNA polymerase delta. Proc Natl Acad Sci U S A 97:4603–4608
Kamath-Loeb AS, Lan L, Nakajima S, Yasui A, Loeb LA (2007) Werner syndrome protein interacts functionally with translesion DNA polymerases. Proc Natl Acad Sci U S A 104:10394–10399. doi:10.1073/pnas.0702513104
Kamath-Loeb AS, Shen JC, Schmitt MW, Loeb LA (2012) The Werner syndrome exonuclease facilitates DNA degradation and high fidelity DNA polymerization by human DNA polymerase delta. J Biol Chem 287:12480–12490. doi:10.1074/jbc.M111.332577
Karavias D, Maroulis I, Papadaki H, Gogos C, Kakkos S, Karavias D, Bravou V (2016) Overexpression of CDT1 is a predictor of poor survival in patients with hepatocellular carcinoma. J Gastrointest Surg 20:568–579. doi:10.1007/s11605-015-2960-7
Khurana S, Oberdoerffer P (2015) Replication stress: a lifetime of epigenetic change. Genes (Basel) 6:858–877. doi:10.3390/genes6030858
Kim H, D’Andrea AD (2012) Regulation of DNA cross-link repair by the Fanconi anemia/BRCA pathway. Genes Dev 26:1393–1408. doi:10.1101/gad.195248.112
Kim MS, Machida Y, Vashisht AA, Wohlschlegel JA, Pang YP, Machida YJ (2013) Regulation of error-prone translesion synthesis by Spartan/C1orf124. Nucleic Acids Res 41:1661–1668. doi:10.1093/nar/gks1267
Koller KJ, Brownstein MJ (1987) Use of a cDNA clone to identify a supposed precursor protein containing valosin. Nature 325:542–545. doi:10.1038/325542a0
Kondo T et al (2004) Rapid degradation of Cdt1 upon UV-induced DNA damage is mediated by SCFSkp2 complex. J Biol Chem 279:27315–27319. doi:10.1074/jbc.M314023200
Lee JJ, Kim YM, Jeong J, Bae DS, Lee KJ (2012) Ubiquitin-associated (UBA) domain in human Fas associated factor 1 inhibits tumor formation by promoting Hsp70 degradation. PLoS One 7:e40361. doi:10.1371/journal.pone.0040361
Lehmann AR (2006) Translesion synthesis in mammalian cells. Exp Cell Res 312:2673–2676. doi:10.1016/j.yexcr.2006.06.010
Lessel D et al (2014) Mutations in SPRTN cause early onset hepatocellular carcinoma, genomic instability and progeroid features. Nat Genet 46:1239–1244. doi:10.1038/ng.3103
Li G, Huang C, Zhao G, Lennarz WJ (2012) Interprotomer motion-transmission mechanism for the hexameric AAA ATPase p97. Proc Natl Acad Sci U S A 109:3737–3741. doi:10.1073/pnas.1200255109
Liu AQ, Ge LY, Ye XQ, Luo XL, Luo Y (2012) Reduced FAF1 Expression and Helicobacter Infection: Correlations with Clinicopathological Features in Gastric Cancer. Gastroenterol Res Pract 2012:153219 doi:10.1155/2012/153219
Longerich S, Li J, Xiong Y, Sung P, Kupfer GM (2014) Stress and DNA repair biology of the Fanconi anemia pathway. Blood 124:2812–2819. doi:10.1182/blood-2014-04-526293
Macheret M, Halazonetis TD (2015) DNA replication stress as a hallmark of cancer. Annu Rev Pathol 10:425–448. doi:10.1146/annurev-pathol-012414-040424
Machida Y, Kim MS, Machida YJ (2012) Spartan/C1orf124 is important to prevent UV-induced mutagenesis. Cell Cycle 11:3395–3402. doi:10.4161/cc.21694
Magnaghi P et al (2013) Covalent and allosteric inhibitors of the ATPase VCP/p97 induce cancer cell death. Nat Chem Biol 9:548–556. doi:10.1038/nchembio.1313
Maiorano D, Moreau J, Mechali M (2000) XCDT1 is required for the assembly of pre-replicative complexes in Xenopus laevis. Nature 404:622–625. doi:10.1038/35007104
Maric M, Maculins T, De Piccoli G, Labib K (2014) Cdc48 and a ubiquitin ligase drive disassembly of the CMG helicase at the end of DNA replication. Science 346:1253596. doi:10.1126/science.1253596
Masai H, Matsumoto S, You Z, Yoshizawa-Sugata N, Oda M (2010) Eukaryotic chromosome DNA replication: where, when, and how? Annu Rev Biochem 79:89–130. doi:10.1146/annurev.biochem.052308.103205
Maskey RS et al (2014) Spartan deficiency causes genomic instability and progeroid phenotypes. Nat Commun 5:5744. doi:10.1038/ncomms6744
McCormack PL (2015) Bortezomib: a review in mantle cell lymphoma in previously untreated patients unsuitable for stem-cell transplantation. BioDrugs 29:207–214. doi:10.1007/s40259-015-0131-8
Meerang M et al (2011) The ubiquitin-selective segregase VCP/p97 orchestrates the response to DNA double-strand breaks. Nat Cell Biol 13:1376–1382. doi:10.1038/ncb2367
Mehanna A, Diffley JF (2012) Pre-replicative complex assembly with purified proteins. Methods 57:222–226. doi:10.1016/j.ymeth.2012.06.008
Menges CW, Altomare DA, Testa JR (2009) FAS-associated factor 1 (FAF1): diverse functions and implications for oncogenesis. Cell Cycle 8:2528–2534
Meyer HH (2005) Golgi reassembly after mitosis: the AAA family meets the ubiquitin family. Biochim Biophys Acta 1744:481–492
Meyer H, Weihl CC (2014) The VCP/p97 system at a glance: connecting cellular function to disease pathogenesis. J Cell Sci 127:3877–3883. doi:10.1242/jcs.093831
Meyer H, Bug M, Bremer S (2012) Emerging functions of the VCP/p97 AAA-ATPase in the ubiquitin system. Nat Cell Biol 14:117–123. doi:10.1038/ncb2407
Mirkin EV, Mirkin SM (2007) Replication fork stalling at natural impediments. Microbiol Mol Biol Rev 71:13–35. doi:10.1128/MMBR.00030-06
Moir D, Stewart SE, Osmond BC, Botstein D (1982) Cold-sensitive cell-division-cycle mutants of yeast: isolation, properties, and pseudoreversion studies. Genetics 100:547–563
Moreno SP, Gambus A (2015) Regulation of unperturbed DNA replication by ubiquitylation. Genes (Basel) 6:451–468. doi:10.3390/genes6030451
Moreno SP, Bailey R, Campion N, Herron S, Gambus A (2014) Polyubiquitylation drives replisome disassembly at the termination of DNA replication. Science 346:477–481. doi:10.1126/science.1253585
Mosbech A et al (2012) DVC1 (C1orf124) is a DNA damage-targeting p97 adaptor that promotes ubiquitin-dependent responses to replication blocks. Nat Struct Mol Biol 19:1084–1092. doi:10.1038/nsmb.2395
Mouysset J, Deichsel A, Moser S, Hoege C, Hyman AA, Gartner A, Hoppe T (2008) Cell cycle progression requires the CDC-48UFD-1/NPL-4 complex for efficient DNA replication. Proc Natl Acad Sci U S A 105:12879–12884. doi:10.1073/pnas.0805944105
Moyer SE, Lewis PW, Botchan MR (2006) Isolation of the Cdc45/Mcm2-7/GINS (CMG) complex, a candidate for the eukaryotic DNA replication fork helicase. Proc Natl Acad Sci U S A 103:10236–10241. doi:10.1073/pnas.0602400103
Muller JM, Deinhardt K, Rosewell I, Warren G, Shima DT (2007) Targeted deletion of p97 (VCP/CDC48) in mouse results in early embryonic lethality. Biochem Biophys Res Commun 354:459–465. doi:10.1016/j.bbrc.2006.12.206
Niedzwiedz W, Mosedale G, Johnson M, Ong CY, Pace P, Patel KJ (2004) The Fanconi anaemia gene FANCC promotes homologous recombination and error-prone DNA repair. Mol Cell 15:607–620. doi:10.1016/j.molcel.2004.08.009
Nishitani H, Lygerou Z (2002) Control of DNA replication licensing in a cell cycle. Genes Cell 7:523–534
Nishitani H, Lygerou Z, Nishimoto T, Nurse P (2000) The Cdt1 protein is required to license DNA for replication in fission yeast. Nature 404:625–628. doi:10.1038/35007110
Nishitani H et al (2006) Two E3 ubiquitin ligases, SCF-Skp2 and DDB1-Cul4, target human Cdt1 for proteolysis. EMBO J 25:1126–1136. doi:10.1038/sj.emboj.7601002
Partridge JJ, Lopreiato JO Jr, Latterich M, Indig FE (2003) DNA damage modulates nucleolar interaction of the Werner protein with the AAA ATPase p97/VCP. Mol Biol Cell 14:4221–4229. doi:10.1091/mbc.E03-02-0111
Petermann E, Helleday T (2010) Pathways of mammalian replication fork restart. Nat Rev Mol Cell Biol 11:683–687. doi:10.1038/nrm2974
Peters JM, Walsh MJ, Franke WW (1990) An abundant and ubiquitous homo-oligomeric ring-shaped ATPase particle related to the putative vesicle fusion proteins Sec18p and NSF. EMBO J 9:1757–1767
Plosky BS, Woodgate R (2004) Switching from high-fidelity replicases to low-fidelity lesion-bypass polymerases. Curr Opin Genet Dev 14:113–119. doi:10.1016/j.gde.2004.02.002
Pommier Y (2013) Drugging topoisomerases: lessons and challenges. ACS Chem Biol 8:82–95. doi:10.1021/cb300648v
Puumalainen MR, Lessel D, Ruthemann P, Kaczmarek N, Bachmann K, Ramadan K, Naegeli H (2014) Chromatin retention of DNA damage sensors DDB2 and XPC through loss of p97 segregase causes genotoxicity. Nat Commun 5:3695. doi:10.1038/ncomms4695
Ramadan K (2012) p97/VCP- and Lys48-linked polyubiquitination form a new signaling pathway in DNA damage response. Cell Cycle 11:1062–1069. doi:10.4161/cc.11.6.19446
Ramadan K, Bruderer R, Spiga FM, Popp O, Baur T, Gotta M, Meyer HH (2007) Cdc48/p97 promotes reformation of the nucleus by extracting the kinase Aurora B from chromatin. Nature 450:1258–1262. doi:10.1038/nature06388
Raman M, Havens CG, Walter JC, Harper JW (2011) A genome-wide screen identifies p97 as an essential regulator of DNA damage-dependent CDT1 destruction. Mol Cell 44:72–84. doi:10.1016/j.molcel.2011.06.036
Richly H, Rape M, Braun S, Rumpf S, Hoege C, Jentsch S (2005) A series of ubiquitin binding factors connects CDC48/p97 to substrate multiubiquitylation and proteasomal targeting. Cell 120:73–84. doi:10.1016/j.cell.2004.11.013
Robak T et al (2015) Bortezomib-based therapy for newly diagnosed mantle-cell lymphoma. N Engl J Med 372:944–953. doi:10.1056/NEJMoa1412096
Romero J, Lee H (2008) Asymmetric bidirectional replication at the human DBF4 origin. Nat Struct Mol Biol 15:722–729. doi:10.1038/nsmb.1439
Rossi ML, Ghosh AK, Bohr VA (2010) Roles of Werner syndrome protein in protection of genome integrity. DNA Repair (Amst) 9:331–344. doi:10.1016/j.dnarep.2009.12.011
Ruijs MW, van Andel RN, Oshima J, Madan K, Nieuwint AW, Aalfs CM (2003) Atypical progeroid syndrome: an unknown helicase gene defect? Am J Med Genet A 116A:295–299. doi:10.1002/ajmg.a.10730
Sclafani RA, Holzen TM (2007) Cell cycle regulation of DNA replication. Annu Rev Genet 41:237–280. doi:10.1146/annurev.genet.41.110306.130308
Seo J, Chung YS, Sharma GG, Moon E, Burack WR, Pandita TK, Choi K (2005) Cdt1 transgenic mice develop lymphoblastic lymphoma in the absence of p53. Oncogene 24:8176–8186. doi:10.1038/sj.onc.1208881
Sha Z, Goldberg AL (2014) Proteasome-mediated processing of Nrf1 is essential for coordinate induction of all proteasome subunits and p97. Curr Biol 24:1573–1583. doi:10.1016/j.cub.2014.06.004
Spivak G, Hanawalt PC (2015) Photosensitive human syndromes. Mutat Res 776:24–30. doi:10.1016/j.mrfmmm.2014.11.003
Srinivasan SV, Dominguez-Sola D, Wang LC, Hyrien O, Gautier J (2013) Cdc45 is a critical effector of myc-dependent DNA replication stress. Cell Rep 3:1629–1639. doi:10.1016/j.celrep.2013.04.002
Stingele J, Jentsch S (2015) DNA-protein crosslink repair. Nat Rev Mol Cell Biol 16:455–460. doi:10.1038/nrm4015
Stingele J, Schwarz MS, Bloemeke N, Wolf PG, Jentsch S (2014) A DNA-dependent protease involved in DNA-protein crosslink repair. Cell 158:327–338. doi:10.1016/j.cell.2014.04.053
Stingele J, Habermann B, Jentsch S (2015) DNA-protein crosslink repair: proteases as DNA repair enzymes. Trends Biochem Sci 40:67–71. doi:10.1016/j.tibs.2014.10.012
Tonddast-Navaei S, Stan G (2013) Mechanism of transient binding and release of substrate protein during the allosteric cycle of the p97 nanomachine. J Am Chem Soc 135:14627–14636. doi:10.1021/ja404051b
Toueille M, Hubscher U (2004) Regulation of the DNA replication fork: a way to fight genomic instability. Chromosoma 113:113–125. doi:10.1007/s00412-004-0303-7
Tretyakova NY et al (2013) DNA-reactive protein monoepoxides induce cell death and mutagenesis in mammalian cells. Biochemistry 52:3171–3181. doi:10.1021/bi400273m
Ulrich HD (2014) Two-way communications between ubiquitin-like modifiers and DNA. Nat Struct Mol Biol 21:317–324. doi:10.1038/nsmb.2805
Vaz B, Halder S, Ramadan K (2013) Role of p97/VCP (Cdc48) in genome stability. Front Genet 4:60. doi:10.3389/fgene.2013.00060
Weber GF (2015) Molecular Analysis of a Recurrent Sarcoma Identifies a Mutation in FAF1. Sarcoma 2015:839182 doi:10.1155/2015/839182
Wohlschlegel JA, Dwyer BT, Dhar SK, Cvetic C, Walter JC, Dutta A (2000) Inhibition of eukaryotic DNA replication by geminin binding to Cdt1. Science 290:2309–2312. doi:10.1126/science.290.5500.2309
Wojcik C, Yano M, DeMartino GN (2004) RNA interference of valosin-containing protein (VCP/p97) reveals multiple cellular roles linked to ubiquitin/proteasome-dependent proteolysis. J Cell Sci 117:281–292. doi:10.1242/jcs.00841
Yamamoto S et al (2003a) Expression level of valosin-containing protein is strongly associated with progression and prognosis of gastric carcinoma. J Clin Oncol 21:2537–2544. doi:10.1200/JCO.2003.12.102
Yamamoto S et al (2003b) Elevated expression of valosin-containing protein (p97) in hepatocellular carcinoma is correlated with increased incidence of tumor recurrence. J Clin Oncol 21:447–452
Yamamoto S et al (2004a) Expression level of valosin-containing protein (p97) is associated with prognosis of esophageal carcinoma. Clin Cancer Res 10:5558–5565. doi:10.1158/1078-0432.CCR-0723-03
Yamamoto S et al (2004b) Valosin-containing protein (p97) and Ki-67 expression is a useful marker in detecting malignant behavior of pancreatic endocrine neoplasms. Oncology 66:468–475. doi:10.1159/000079501
Yamamoto S et al (2005) Increased expression of valosin-containing protein (p97) is correlated with disease recurrence in follicular thyroid cancer. Ann Surg Oncol 12:925–934. doi:10.1245/ASO.2005.07.002
Yardimci H, Walter JC (2014) Prereplication-complex formation: a molecular double take? Nat Struct Mol Biol 21:20–25. doi:10.1038/nsmb.2738
Ye Y (2006) Diverse functions with a common regulator: ubiquitin takes command of an AAA ATPase. J Struct Biol 156:29–40. doi:10.1016/j.jsb.2006.01.005
Yeeles JT, Deegan TD, Janska A, Early A, Diffley JF (2015) Regulated eukaryotic DNA replication origin firing with purified proteins. Nature 519:431–435. doi:10.1038/nature14285
Yekezare M, Gomez-Gonzalez B, Diffley JF (2013) Controlling DNA replication origins in response to DNA damage—inhibit globally, activate locally. J Cell Sci 126:1297–1306. doi:10.1242/jcs.096701
Yeung HO, Kloppsteck P, Niwa H, Isaacson RL, Matthews S, Zhang X, Freemont PS (2008) Insights into adaptor binding to the AAA protein p97. Biochem Soc Trans 36:62–67. doi:10.1042/BST0360062
Yurov YB, Vorsanova SG, Iourov IY (2011) The DNA replication stress hypothesis of Alzheimer’s disease. Sci World J 11:2602–2612. doi:10.1100/2011/625690
Zhong W, Feng H, Santiago FE, Kipreos ET (2003) CUL-4 ubiquitin ligase maintains genome stability by restraining DNA-replication licensing. Nature 423:885–889. doi:10.1038/nature01747
Acknowledgments
We are grateful to Prof Thorsten Hoppe and Dr Andre Franz (Institute for Genetics, University Cologne, Germany) for their discussion on DNA replication, critical comments and useful suggestions for the final preparation of this manuscript. We thank the reviewers for their helpful comments and suggestions. We apologise to the authors whose work was not cited.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This work is supported by the MRC programme grant (MC_PC_12001/1) and CRUK/MRC Oxford Institute for Radiation Oncology, University of Oxford core funding to KR, MRC student scholarship to KW and Goodger scholarship to SH.
Conflict of interest
The authors declare no conflict of interest.
Ethical approval
This is a review article and describes only published literature. The authors in this article did not perform any studies with human participants or animal experiments.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Ramadan, K., Halder, S., Wiseman, K. et al. Strategic role of the ubiquitin-dependent segregase p97 (VCP or Cdc48) in DNA replication. Chromosoma 126, 17–32 (2017). https://doi.org/10.1007/s00412-016-0587-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00412-016-0587-4