
Abstract
DNA-damaging agents can induce clustered lesions or multiply damaged sites (MDSs) on the same or opposing DNA strands. In the latter, attempts to repair MDS can generate closely opposed single-strand break intermediates that may convert non-lethal or mutagenic base damage into double-strand breaks (DSBs). We constructed a diploid S. cerevisiae yeast strain with a chromosomal context targeted by integrative DNA fragments carrying different damages to determine whether closely opposed base damages are converted to DSBs following the outcomes of the homologous recombination repair pathway. As a model of MDS, we studied clustered uracil DNA damages with a known location and a defined distance separating the lesions. The system we describe might well be extended to assessing the repair of MDSs with different compositions, and to most of the complex DNA lesions induced by physical and chemical agents.
Similar content being viewed by others

Introduction
The DNA molecule contains the genetic instructions used for the development and functioning of all living organisms. Exposure of DNA to ionizing radiation (IR) entails multiple damages, including oxidized bases, abasic (AP) sites and single-strand DNA breaks (SSBs) (Sutherland et al. 2000, 2002). The close proximity of such lesions, either on the same or opposing strands, within a short stretch of DNA (one or two helical turns) forms a cluster of DNA damages (Ward 1994), also known as multiply damaged sites (MDSs).
Since oxidized base damage and AP sites are repaired predominantly by base excision repair (BER), and the initiation of repair produces an SSB-repair intermediate (reviewed by Demple and Harrison 1994), MDSs are considered of high biological risk because repairing them may yield de novo DSBs, the most cytotoxic DNA lesions (Ho et al. 2007).
Several in vivo and in vitro studies analyzed the repair dynamics of MDSs with different compositions of damaged-bases and known distances separating the lesions. Some authors reported a hierarchy in the repair of closely spaced lesions that limited DSB formation. For instance, the presence of an AP site or a SSB drastically reduces the excision/incision efficiency of base damage or AP sites separated by 1–3 bp (Chaudhry and Weinfeld 1997; Harrison et al. 1999; David-Cordonnier et al. 2001, 2002; Eot-Houllier et al. 2005). Such repair-resistant MDSs were proven to have high mutagenic potential in comparison with single lesions. Bacterial plasmid-based assays revealed a significantly higher than normal mutation frequency for clustered dihydrothymine (DHT) and 8-oxo-7, 8 dihydroguanine (8-oxoG) lesions, both in wild-type and glycosylase-deficient E. coli strains (Shikazono et al. 2006; Malyarchuk et al. 2004). Generally, the frequency of mutation declined as the distance between the lesions rose (0–14 bp).
Alternatively, DSBs can result from a lower rate of repair or its inhibition in a MDS, as well as in repair-processing intermediates. The in vitro reconstitution of the E.coli BER repair pathway demonstrated that a MDS, consisting of an 8-oxoG and a SSB > 1 bp apart, is converted into a DSB (Harrison et al. 1999). A complementary approach, employing E.coli lacking the three oxidative DNA glycosylases/AP lyases (nth nei fpg), showed a significant increase in the radioresistance of this triple mutant compared to wild-type cells. This increase was ascribed to the lesser formation of DSBs post-irradiation, substantiating the idea that in wild-type cells the DNA glycosylases create DSBs during attempted but abortive repair at sites of cluster damages (Blaisdell and Wallace 2001). Moreover, a study in E.coli on the MDS repair of uracil (U) lesions on opposite strands revealed DSB formation for lesions ≤7 bp apart only in wild-type cells and not in the mutant of the uracil DNA glycosylase (UNG) (D’souza and Harrison 2003). Stable human TK6 B-lymphoblastoid cell lines inducibly overexpressing either one or two of the human DNA glycosylases/AP lyases, hNTH1 and hOGG1, yielded similar findings. After exposing these cells to gamma-rays, the overexpressing clones were more radiosensitive than the non-induced controls, showing a post-irradiation increase in DSB formation and mutation frequency (Yang et al. 2004).
Two distinct pathways, highly conserved throughout evolution, compete in DSB repair after their formation: Homologous recombination (HR), and non-homologous end-joining (NHEJ). Both are essential to maintaining the integrity of the genome, and defects in either one, or their failure to coordinate with the cell cycle leads to genetic instability and tumorigenesis (Delacote and Lopez 2008). Recently, Kosmin et al., in a study in haploid strains of S. cerevisiae combining the use of the integrative and replicative plasmids housing different MDS, confirmed the generality of the type of clusters that result in DSBs. Their plasmid viability assay indirectly indicated that the BER repair process of MDSs composed of opposed U and/or AP sites entails the extensive formation of DSBs.
Our work aimed to extend the in vivo MDS repair studies using the yeast S. cerevisiae as a model to determine the importance of clustered DNA damages as a source of DSBs. We directly monitored the dynamics of repair following DSB formation and evaluated the mutagenic potential of complex DNA lesions. We developed a system that consists of a tester yeast diploid strain (27.6.1) and a “warehouse” vector (pMM-25) housing a specific yeast integrative sequence (cassette) prone to modification by commercial modified bases. We chose to consider single- and clustered-uracil DNA damages (U) base-paired with adenine residues to build different uracil-containing DNA cassettes. The latter were designed to be transferred into yeast, selectively target only one chromosome XV, in the diploid strain 27.6.1, and define a sequence replacement. The gene targeting events often are explained to result from two crossovers at the ends of the transforming fragment (reviewed in Paques and Haber 1999), such that the effect of the non-conservative insertion of the cassette is to place uracil residue(s) into the recipient chromosome.
In S. cerevisiae, the base excision repair (BER) repairs U residues. The UNG1 gene encodes the major DNA glycosylase (UDG) that excises uracil residues from DNA, thus generating an apyrimidinic site (AP); thereafter, the phosphodiester bonds are hydrolyzed by the AP endonuclease APN1, resulting in a SSB with 3′ hydroxyl and 5′ deoxyribose phosphate termini. The latter are removed, in a limiting step, by the activity of a DNA deoxyribophosphodiesterase (dRpase), and subsequently, a DNA polymerase fills in the resulting single-nucleotide gaps. The covalent integrity of the DNA strands finally is restored by a DNA ligase (Wang et al. 1997).
Should the repair of clustered U residues evolve into DSBs, originating from the simultaneous presence of SSB on opposite strands, the DNA ends of the lesion might undergo base-pairing with the sequence of the homologous chromosome, thus starting the first step of the HR repair pathway. The recovery of the recombinatorial outcomes can be quickly performed via complementation on selective media and/or by PCR-based profiling of the cassette’s integration region.
In our diploid system, employing the HR pathway is furthered by the down-regulation of the NHEJ (Valencia et al. 2001) that is thought to inaccurately repair the clustered lesions converted to a DSB in chromosomal DNA (Malyarchuk et al. 2008). According to recent evidence, the HR pathway has a significant role in repairing IR-induced breaks during the late S/G2 phases of the mammalian cell cycle (Rothkamm et al. 2003; Frankenberg-Schwager et al. 2009).
Our system was set and tested with uracil-containing molecules, but the versatility of the vector pMM-25 will support extending it to studying the repair of MDSs of different compositions, and to most of the complex DNA lesions induced by physical and chemical agents.
Materials and methods
Strains, plasmids, and media
The yeast diploid strain 27.6.1 used for the transformation experiments is derived from BY 4741 (MATa; his3Δ1; leu2Δ0; met15Δ0; ura3Δ0), and BY 4742 (MATα; his3Δ1; leu2Δ0; lys2Δ0; ura3Δ0). Both haploid strains were obtained from Euroscarf (Frankfurt, Germany).
Two independent ADE2 partial deletion mutants were produced in a haploid background using the one-step gene replacement protocol (Burke et al. 2000) and then mated. The ADE2 locus, a 1,716-bp ORF, maps on chromosome XV spanning the genetic and physic coordinates 566192–564477 (http://www.yeastgenome.org/cgi-bin/locus.fpl?locus=ADE2).
In strain BY4741, the LEU2 marker was inserted in position 565631-566372 thus generating an ade2(Δ1-561) null allele; in strain BY4742, we inserted the HIS3 marker in position 565419-564398 yielding an ade2(Δ773-1716) null allele (Fig. 1a). Colonies were selected on supplemented minimal SD-His or-Leu agar plates. All ten His+ or Leu+ tested transformants were Ade−.

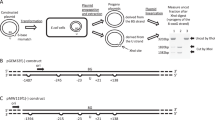
Tester strain building: diploid 27.6.1. a Partial map of the ADE2 locus and surrounding regions on chromosome XV. Coordinate positions relevant to the work presented here are shown. In the haploid strain BY4741, the LEU2 selectable marker (red segment) was inserted between pos. 565631 and 566372, generating the partial deletion allele ade2(Δ1-561). Similarly, in the haploid strain BY4742, an ade2(Δ773-1716) allele was generated by inserting the HIS3 selectable marker (green segment) between the pos. 564398 and 565419. Both these ade2 allelic forms are null (the numbers that follow the Δ symbol indicate the deleted residues in the ADE2 ORF). The position of the oligos A and H is noted since they were used to characterize, by PCR, the profile of the wild-type strain (Supplementary Fig. 1). b Schematic of the crossing of the ade2 mutant to generate the diploid strain 27.6.1. The two haploids, BY4741 ade2(Δ1-561), and BY4742 ade2(Δ773-1716) were crossed and the diploids formed were selected by complementation. Noticeably, the chromosomes of couple XV have a region of shared homology in the ADE2 locus, spanning coordinates pos. 565419 to 565631. Further, the position of the oligos A and H is no more the one reported for the wild-type strain, but is in accord with the insertion of the selectable markers, HIS3 and LEU2
The correct integration of the selectable markers HIS3 and LEU2 was verified by PCR positioning the A and H primers, respectively, 145 bp (pos. 564332) and 310 bp (pos. 566502) apart from the stop and start codons of the ADE2 gene. Using the high-processivity Taq enzyme (Phusion F-540S, Finnzymes, Espoo, Finland), the reaction gave a product of 2,171 bp in both the Ade+ wild-type strains BY 4741 and BY 4742, 2380 bp in BY 4742 ade2(Δ773-1716) His+, and 3750 bp in BY 4741 ade2(Δ1-561) Leu+.
We crossed the two PCR-confirmed haploids BY4741 ade2(Δ1-561) Leu+ and BY4742 ade2(Δ773-1716) His+ (Fig. 1b). The diploids formed were identified by complementation, PCR on Mat locus (not shown) (Huxley et al. 1990), and confirmed by the profile of the ADE2 chromosomal context with primer mix A/H (Supplementary Fig. 1). Noticeably, in this strain, the two chromosomes of the couple XV share a region in the ADE2 ORF ranging from position 565419 to 565631.
For the ADE2 partial gene replacement, the selectable markers HIS3 and LEU2, respectively, were PCR-amplified from the vectors pRS303 and pRS305 (ATCC 77138 and 77140). The oligonucleotides were purchased as PAGE or HPLC-purified species from IDT (Coralville, USA; Sequences in Supplementary Table 1).
Growth conditions and medium preparation were standard (Burke et al. 2000).
Building and modifications of the integrative fragments
“Warehouse” plasmid pMM-25 was constructed as follows: we performed a PCR-amplification on diploid strain 27.6.1 using primers selectively targeting the chromosome XV having the ade2(Δ773-1716) allele (Supplementary Fig. 2A). Amplification was performed with adapter primers spanning the promoter region of the ADE2 gene (Pos.566346, oligo C1), and part of the HIS3 ORF (Pos.565092, oligo C2). The reaction originated a 1,343-bp product with a SmaI restriction site at both of its extremities, while simultaneously introducing two additional bases (CG) that mapped in the ORF of the HIS3 gene. The plasmid pMM-25, was constructed by inserting the amplification product as a SmaI fragment (1,305 bp) into SmaI site in the MCS of plasmid pBluescript II SK ± (2,961 bp, Fermentas), thus generating a 4,266-bp vector (Supplementary Fig. 2B).
The vector pMM-25, amplified in E. coli DH5α competent cells and sequenced, was used as the sole source in all our experiments. Sequencing revealed the orientation of the fragment in the MCS, confirmed the only presence of two additional bases (CG) in the HIS3 ORF and no further mutations toward the chromosomal template sequence. The sequence of pMM-25 is available at GeneBank, with the accession # GQ988388.
The plasmid pMM-25 was cut with SmaI (blunt cut) and the 1305-bp band purified. Noticeably, the two endonucleases BanI and BglII (both generate sticky ends) have one site, respectively, in position 539 and 576. The double enzymatic digestion generates three fragments: Two of them, 539 and 729 bp were gel-purified using the Nucleospin ExtractII columns (Machery Nagel, Bethlehem, USA) with minor modifications of the manufacturer’s protocol and the 37 bp one was discarded (Fig. 2). In all preparative steps, the DNA fragments were shielded from UV photodamage.

Linear cassette(s) building from vector pMM-25. The “warehouse” vector pMM-25 was SmaI-cut, and the 1.3-Kb cassette purified by agarose gel electrophoresis using Nucleospin ExtractII columns (Machery Nagel, Bethlehem, USA). A further double enzymatic digestion (BglII/BanI) of the 1.3-Kb cassette generated three products (37, 539, and 729 bp), the last two of which were purified and used in the ligase reactions for building the cassettes
Polyacrylamide gel-purified 5′-end phosphate oligonucleotides were used to generate double-stranded DNA molecules containing no damage, a single uracil or two closely opposed uracil residues (Supplementary Table 2). The 4-nt overhanging extremities of the annealed 37 bp products represent half BanI and BglII restriction sites, and their sequences match that of the 37-bp fragment removed in the digestion described earlier (Supplementary Table 2). Equimolar amounts of complementary oligonucleotides resuspended in 50 mM Tris pH 7.4 were mixed and placed in a thermocycler (Biometra, Goettingen, Germany), then the samples were first heated to 90°C over 10 min, and then cooled to 4°C at a rate of 0.5°C/min. The check of the duplexes formation and their purification was performed on a preparative 12% polyacrylamide gel [29:1, acrylamide/bisacrylamide (w/w), 100 mM Tris borate, 1 mM EDTA (pH 8)].
In this study, five different 37 bp duplexes (cold control cassette (CCC), cluster damage +1 (Clu +1), cluster damage −1 (Clu −1), single-strand damage 1 (SSD1) and single-strand damage 2 (SSD2)) were ligated, in separate reactions, with the 539-bp fragment described earlier. The resulting 576-bp products were gel-purified and ligated with the 729-bp fragment. The conditions of the ligase reaction were 120 U of T4 DNA ligase, 10 mM ATP, and 2 μM total DNA. After heating the samples at 37°C, they were cooled at 0.5°C/min to 16°C before adding the enzyme. The reactions were incubated 18 h at 16°C; thereafter, the verification of the successful ligations (≥80% product yield) and their purification were performed on preparative 5% polyacrylamide gel for the 576-bp fragments and on 1.5% agarose gel for the 1,305-bp products (Supplementary Fig. 3A).
The central 37-bp part of the resulting 1,305-bp products (cassettes) differs, before using them to transform the yeast, aliquots were treated with UDG/Ape1 to confirm the distribution of the uracil residues. Treatment was performed on 100 ng of DNA in replicate reactions with (+) and without (−) the enzymes. Briefly, a half-hour incubation of the 1,305-bp cassettes with 40 units of UDG was followed by 15 min incubation with 250 pg of hApe1. Individual reactions were precipitated by ethanol treatment and analyzed on a 1.5% denaturing agarose gel stained with SybrGreenII dye (Supplementary Fig. 3B).
All enzymes described were purchased from New England Biolabs (Ipswich, USA), except hApe1, a kind gift from Dr. David Wilson.
Yeast transformation and selection
Yeast growth, media and protocols are as described (Burke et al. 2000). Yeast cells (107) were transformed with 1 μg of 1,305 bp ligase product (cassette) with minor modifications of the lithium acetate method (Gietz and Woods 2002). In parallel, two different selections were performed, one for His−, and a second one for Ade+ transformants. Briefly, an aliquot of the transformed cells was plated in the absence of selection (on master plates), and 4 days later about 2 × 104 colonies were replica-plated on minimal supplemented media SD–His. Three days later, the replica plates (SD–His) were examined for differential growth of the colonies. Cells from the appropriate colonies on the master plates (candidate His− transformants) were tested again on selective media, and used to start the pure cultures we used in this study. ADE+ transformants were selected directly by complementation on SD–Ade by plating a 2 × 104 cell aliquot.
Sequencing and PCR screening of transformants
Transformants were analyzed for wild-type-sized PCR products to elucidate the outcomes. For this purpose, a complete PCR-based analysis was performed on all transformants (His−) using the oligo mix A/H that overlaps the entire target region of cassette integration. Yeast colony PCRs gave product sizes ranging from 2,170 to 3,970 bp and were resolved on 0.8% agarose gel.
The His− transformants from Cold Control Cassette (CCC), SSD1 and SSD2 experiments also were sequenced in the region spanning the positions of the uracil residues and the additional CG bases. The I/L 669-bp PCR product was cloned in pCR®4Blunt-TOPO vector (Invitrogen, Carlsbad, USA) and sequenced according to the manufacturer’s specifications.
Results
Transfer of the 1.3-kb integrative cassettes in the tester strain 27.6.1
The sequence of the five cassettes synthesized and described (Supplementary Fig. 3) spans the chromosome XV coordinates pos. 565055–566360. Our goal was to transfer the molecules in the diploid test strain 27.6.1 to gene target one of the two chromosomes XV.
The chromosome XV targeted is the one with the allelic form ade2(Δ773-1716) since it was used as the template in the PCR to amplify the chromosomal fragment to be cloned in the vector pMM-25. The selective strategy for cassette integration was based on histidine auxotrophy induced by two additional bases (CG) in the sequence of the transforming DNA, compared to the wild-type target sequence of the HIS3 ORF (Fig. 3a).

Transformation experiments with linear cassettes. a Schematic representation of the chromosomal context targeted by the linear cassette with the location of the oligos used to characterize the outcomes. The oligo mix A/H was used for PCR-profiling the transformants, and the mix I/L was used to amplify and clone the sequence spanning a relevant part of the insertion point. In the diploid tester strain, 27.6.1, the PCR with oligo mix A/H gives 2,380 and 3,750 bp bands, while the mix I/L yields a 669 bp product. b Summary of PCR reactions with oligo mix A/H on His− transformants. Total reactions were loaded on 0.8% agarose gels and the run (5 V/cm, 3 h) was followed by ethidium bromide staining. Samples were loaded as indicated above each lane. In lanes L and T were loaded the molecular weight marker (1 Kb Promega, MD, USA) and the untransformed 27.6.1 strain, respectively. Left: all tested transformants from the experiment with control undamaged cassette (CCC) and single-strand damaged ones (SSD1 and SSD2) exhibit the parental profile of the untransformed tester strain 27.6.1 (2,380, 3,750 bp bands). Right: all tested transformants from the experiments with linear cassettes having cluster damages (Cluster +1 and Cluster −1) gave four non-parental profiles (indicated as α, β, γ, and δ) that are compatible with the predicted HR repair outcomes (Supplementary Fig. 4)
The mean transformation efficiency is similar for all five cassettes described in this study, and ranges from (4.6–8.5) × 103 colonies per μg DNA and 107 early log cells. The value is slightly higher if we consider the recovery of Ade+ transformants, presumably because of the different selection protocols (see “Materials and methods”); in fact, it encompasses the value (1.3–1.8) × 104 colonies per μg DNA and 107 early log cells. The one way analysis of variance (ANOVA, not shown) for the transformation efficiency values among these experiments revealed that the observed differences are statistically non-significant (P = 0.491). This tends to lend support to the concept that the integration of the cassette in the chromosome is the transformation efficiency-limiting step rather than the reparability of the lesion.
Each transformation was performed in duplicate (series 1 and 2) in two independent experiments (experiments 1 and 2) (Table 1). The transformation experiments with cassettes that do not have any uracil residues (cassette CCC) or have only one (cassettes SSD1 and SSD2) describe a different scenario (both by complementation and by molecular profiling) compared to those that arose by transformation with clusters of uracil damages (cassettes Cluster +1 and Cluster −1).
Single uracil residue transformation experiments (cassettes SSD1 and SSD2)
Two separate experiments were performed in duplicate, and the transformants were selected for histidine auxotrophy (His−) and adenine prototrophy (Ade+) as described (see “Materials and methods”). As shown in Table 1, no Ade+ transformants were recovered in the experiments with the CCC, SSD1, and SSD2 cassettes.
The molecular characterization of the target region performed by PCR with oligo mix A-H revealed the parental amplification pattern (2,380 and 3,750 bp bands) of strain 27.6.1 in all the assayed His− colonies (Fig. 3b, left). As expected, sequence analysis of the integration site revealed the additional bases, CG, in the HIS3 gene that engendered a frameshift in the ORF, and the His− phenotype of transformants. Interestingly, the repair of the uracil residues (originally base-paired with an A) was not mutagenic; they were properly repaired by the placement of a T residue in the SSD1 and SSD2 cassette transformants (sequencing report in Table 2).
Cluster of uracil residues transformation experiments (cassettes Cluster +1 and Cluster −1)
Two independent experiments, in duplicate, were performed. As shown in Table 1, coupled with Histidine auxotrophy, some transformants showed adenine prototrophy, with comparable frequency (1.3 vs. 1.8 × 104) in Cluster +1 and Cluster −1 experiments, respectively. The transformants of the two phenotypic classes (His−/Ade− and His−/Ade+), contrary to the single-uracil molecule transformation experiments described above, had lost the chromosomal structure of the 27.6.1 strain as assessed by PCR using the oligo mix A/H. Two molecular profiles for each phenotypic class were detected and frequencies are reported in Table 3. In detail, the His−/Ade− class exhibits a dual-band profile of 3,750/3,970 bp, and a single-band profile of 3,750 bp. The His−/Ade+ class has two dual-band profiles of 2,170/3,750 and 2,170/3,970 bp. A summary of the described classes of these two experiments was resolved on a 0.8% agarose gel (Fig. 3b, right).
The uracil residues in the cassettes map in the portion of shared homology between the two homolog chromosomes (coordinates 565419–565631, Figs. 1b, 3a). If attempting the repair of the cluster damage a DSB is formed, it could be repaired by the HR repair pathway, whose predicted outcomes in that defined region are adenine prototrophy and leucine homozygosity (Supplementary Fig. 4). The four HR repair outcomes were identified as alpha (3750/3970 bp, α), beta (3750 bp, β), gamma (2170/3970 bp, γ), and delta (2170/3750 bp, δ). In a t test analysis, the differences in the frequency of each outcome in the two experiments were found statistically non-significant (P values in Table 3). The PCR profiles arising by transformation with both configurations of cluster damages are compatible with the reconstitution of the wild type ADE2 gene sequence (2,170 bp, as in γ and δ), and with the relocation of the selectable marker LEU2 which became attached to HIS3 (3,970 bp, as in α and γ). Further, the 3,750 bp band is expected to reflect ade2(Δ1-561) sequence conservation (as in α and δ), and conforms to the one-band profile of the homozygous Leu2 (as in β). Taken together, the PCR-based verifications of the transformants confirmed that a DSB was generated and that it was repaired efficiently via an HR-mediated repair pathway.
Discussion
A multiply damaged site (MDS) may be mutagenic, cytotoxic, or both, and generate double-strand breaks (DSBs) that are lethal unless repaired (Pfeiffer et al. 2000).
In the present study, we aimed to develop an in vivo system to monitor the repair of MDSs on chromosomal sequences, determine their significance as a source of DSBs, and estimate their mutagenic potential. We exploited both the highly recombinogenic nature of DSB that, in the general model of the Homologous Recombination (HR), primes the exchange of genetic information between homologous sequences (Szostak et al. 1983), and the high gene targeting efficiency in the yeast S. cerevisiae (Petes and Symington 1991). This organism, genetically well characterized, has proven an appropriate paradigm for DNA repair in all eukaryotic cells (Friedberg 1991).
The early studies on MDS repair were performed in vitro, testing purified repair enzymes on custom-synthesized damaged DNA duplexes, and in vivo, mostly in bacteria, using damaged plasmid DNA (see Introduction). Chaudhry and Weinfeld examined the capacity of the major human- and E.coli-AP endonucleases to generate DBSs in substrates containing close bi-stranded base lesions. Such approaches, with purified enzymes, elicited a marked variation in response, which depended on the distance between the lesions, their type, and orientation to each other. For instance, most configurations of opposing AP sites (separated up to 7 bp) generated DSBs (the exception was on a cluster +1 that showed a SSB and a cleavage-resistant AP site) (Chaudhry and Weinfeld 1997). However, on substrates containing opposite DHTs set apart by 1 and 3 bp, resulted in only one SSB. Thus, it was inferred that the glycosylase activity of endonuclease III, but not the AP lyase one, is inhibited by the presence of a closely positioned break in the opposite strand (Chaudhry and Weinfeld 1995). In a related study (Dianov et al. 1991), repair of a plasmid DNA construct containing opposite uracil residues spaced by 12 bp (and flanked by directed repeats) entailed in DSB formation both in vitro, by treatment with E. coli cell-free lysate, and in vivo by measuring the deletion frequency in the transformed population. However, DSB production depended on the expression of the E. coli uracil-DNA glycosylase (Ung), thus implicating the formation of double AP site-containing intermediates. A more extensive analysis of the conversion to DSBs of closely opposed uracil residues (combinations from 0–33 bp apart) utilized the firefly luciferase reporter in wild-type and DNA repair-deficient bacterial cells, in the absence of DNA replication (D’souza and Harrison 2003). The MDS combinations generated DSBs, measured as a reduction in luciferase activity, when the two uracils were separated by <13 bp, confirmed the dependence on Ung, and the susceptibility to cleavage of the AP sites. In contrast, the same system adapted and later applied to eukaryotic cells (Malyarchuk and Harrison 2005), revealed that the repair of two closely opposed uracil residues prevents the generation of DSB. Probably, in eukaryotes, the BER pathways evolved to limit the formation of clustered AP sites, to reduce the chance of producing DSB. Consistently, eliminating the step of uracil (or base) removal using clusters of two furan residues (AP site analogs), the repair process in mouse fibroblast readily converted the AP sites to DSBs (Malyarchuk et al. 2008). The cleavage inhibition of a damaged base (8-oxoG) during the repair of complex MDSs was demonstrated also to largely depend on surrounding SSBs and nucleotide gap (Lomax et al. 2004; Eot-Houllier et al. 2005; Eot-Houllier et al. 2007).
Recently, Kozmin et al., described a strategy to study MDSs with complex compositions in the yeast, S. cerevisiae. Since the sequences of different MDSs display no homology in yeast genome, this prevented any DSBs formed from being repaired by HR, thereby narrowing the scope of repair to only illegitimate repair pathways. In the present study, we deemed that to quantify the DSBs formed by a MDS lesion, the repair should be monitored in a population of cells, wherein each has suffered a lesion that can be repaired stably by either non-homologous and HR repair pathways. As a first approximation of this, the system was designed to have MDS mapping in a sequence with a chromosomal counterpart because, in the HR repair pathway, if a DSB occurs the undamaged homolog sequence is used as template to retrieve lost genetic information. In yeast and mammalian cells, sister chromatids are preferred templates for homologous repair of DSB, but when unavailable, homologs, and sequence repeats on heterologs, efficiently substitute as templates (Kadyk and Hartwell 1992; Moynahan and Jasin 1997; Richardson et al. 1998). The hallmarks of HR are Rad50/Mre11/Xrs2-mediated end processing, Rad52-, Rad54-, Rad55- and Rad57-assisted formation of Rad51-complexed nucleoprotein filaments and strand invasion into the homologous donor duplex, DNA synthesis and, finally, resolution of the intermediate (reviewed by Paques and Haber 1999).
Alternatively, DSB repair can proceed through the potentially error-prone NHEJ, which simply pieces together the broken DNA ends, sometimes after limited processing. NHEJ in S. cerevisiae is mediated by the DNA double-stranded end-recognizing and end-binding activities of the Yku70/Yku80 heterodimer (Doherty and Jackson 2001), followed by the endbridging function of the Rad50/Mre11/Xrs2 protein complex (Chen et al. 2001). It is finalized by the action of DNA ligase IV, consisting of the catalytic subunit Dnl4p, and the regulatory subunit Lif1p (Lewis and Resnick 2000). Since the homologs of all these proteins in both pathways are known in mammals, our system allows a generalization of the outcomes of complex DNA lesions and their biological significance.
The tester strain (27.6.1) transformation offers direct evidence about repair because the integration in the chromosome of the DNA molecule housing the lesion is coupled with histidine auxotrophy that marks the transformed clones (see Material and Methods). The following characterization of the chromosomal context of each His− clone represents the main difference compared to previous approaches based on the concept of differential transformation efficiency, and on the reduced activity of fluorescent reporters of whole transformed populations to have estimation on the DSBs formed by MDS repair (Kozmin et al. 2009; D’souza and Harrison 2003). Moreover, although transformation with the cassette has a low efficiency, as described for “ends-out” integrative recombination (Hastings et al. 1993), the observed values are consistent and comparable for all the experiments described in this study (see Table 1). Hence, the frequency of lesions that evolve into DSBs or generate mutant scenarios can be quantified. Also the system’ specificity for DSB formation is verified by the appearance of the recombinatorial products only after transformation with cassette housing damaged bases on opposite strands (Cluster +1 and Cluster −1, see Fig. 3b). It was expected, since several converging reports demonstrated DSB formation during the repair of uracil MDS with an inter-lesion distance less than 13 bp (see Introduction). Using the vector pMM-25 and replacing the BglII/BanI 37 bp fragment with a commercial duplex housing DNA damages, it is possible the synthesis of an extremely broad set of cassettes with different and more complex MDS. An extensive MDS repair analysis is in progress (Moscariello in preparation) and future directions are focused on clarifying the effect of chemoterapics or physical agents that concomitantly damage both DNA strands.
The tester strain (27.6.1) can be engineered in key genes involved in DNA repair (e.g., BER, HR, and NHEJ), and by isolating suppressor genes, and performing complementation analysis, to verify the presence of redundant or backup MDS repair pathways. Further, the overexpression of specific factors might allow the testing of the dose-dependent repair efficiency of MDS and isolating mutants. In parallel, it is possible to develop a system based on similar strategies in higher eukaryotes since have been developed selection and screening procedures that eliminate unwanted random integrants (Mansour et al. 1988). Also high gene targeting efficiency can be achieved with embryonic stem cells (ES) as well as in somatic human cell lines by hRad51 overexpression (Yanez and Porter 1999).
References
Blaisdell J, Wallace SS (2001) Abortive base-excision repair of radiation-induced clustered DNA lesions in Escherichia coli. Proc Natl Acad Sci USA 98(13):7426–7430
Burke DT, Dawson D, Searns T (2000) Laboratory course manual for methods in yeast genetics. Cold Spring Harbour Laboratory Press, Cold Spring Harbour
Chaudhry MA, Weinfeld M (1995) The action of Escherichia coli endonuclease III on multiply damaged sites in DNA. J Mol Biol 249(5):914–922
Chaudhry MA, Weinfeld M (1997) Reactivity of human apurinic/apyrimidinic endonuclease and Escherichia coli Exonuclease III with Bistranded Abasic Sites in DNA. J Biol Chem 272(25):15650–15655
Chen L, Trujillo K, Ramos W, Sung P, Tomkinson AE (2001) Promotion of Dnl4-catalyzed DNA end-joining by the Rad50/Mre11/Xrs2 and Hdf1/Hdf2 complexes. Mol Cell 8(5):1105–1115
D’souza DI, Harrison L (2003) Repair of clustered uracil DNA damages in Escherichia coli. Nucleic Acids Res 31(15):4573–4581
David-Cordonnier MH, Boiteux S, O’Neill P (2001) Efficiency of excision of 8-Oxo-guanine within DNA clustered damage by XRS5 nuclear extracts and purified human OGG1 protein. Biochemistry 40(39):11811–11818
David-Cordonnier MH, Cunniffe SM et al (2002) Efficiency of incision of an AP site within clustered DNA damage by the major human AP endonuclease. Biochemistry 41(2):634–642
Delacote F, Lopez BS (2008) Importance of the cell cycle phase for the choice of the appropriate DSB repair pathway, for genome stability maintenance: the trans-S double-strand break repair model. Cell Cycle 7(1):33–38
Demple B, Harrison L (1994) Repair of oxidative damage to DNA: enzymology and biology. Annu Rev Biochem 63(1):915–948
Dianov GL, Timchenko TV, Sinitsina OI, Kuzminov AV, Medvedev OA, Salganik RI (1991) Repair of uracil residues closely spaced on the opposite strands of plasmid DNA results in double-strand break and deletion formation. Mol Gen Genet 225(3):448–452
Doherty AJ, Jackson SP (2001) DNA repair: how Ku makes ends meet. Curr Biol 11(22):R920–R924
Eot-Houllier GS, Eon-Marchais S, Gasparutto D, Sage E (2005) Processing of a complex multiply damaged DNA site by human cell extracts and purified repair proteins. Nucleic Acids Res 33(1):260–271
Eot-Houllier GS, Gonera M, Gasparutto D, Giustranti C, Sage E (2007) Interplay between DNA N-glycosylases/AP lyases at multiply damaged sites and biological consequences. Nucleic Acids Res 35(10):3355–3366
Frankenberg-Schwager M, Gebauer A, Koppe C, Wolf H, Pralle E, Frankenberg D (2009) Single-strand annealing, conservative homologous recombination, nonhomologous DNA end joining, and the cell cycle-dependent repair of DNA double-strand breaks induced by sparsely or densely ionizing radiation. Radiat Res 171(3):265–273
Friedberg EC (1991) Eukaryotic DNA repair: glimpses through the yeast Saccharomyces cerevisiae. Bioessays 13(6):295–302
Gietz RD, Woods RA (2002) Transformation of yeast by lithium acetate/single-stranded carrier DNA/polyethylene glycol method. Methods Enzymol 350:87–96
Harrison L, Hatahet Z, Wallace SS (1999) In vitro repair of synthetic ionizing radiation-induced multiply damaged DNA sites. J Mol Biol 290(3):667–684
Hastings PJ, McGill C, Shafer B, Strathern JN (1993) Ends-in vs. ends-out recombination in yeast. Genetics 135(4):973–980
Ho EL, Parent M, Satoh MS (2007) Induction of base damages representing a high risk site for double-strand DNA break formation in genomic DNA by exposure of cells to DNA damaging agents. J Biol Chem 282(30):21913–21923
Huxley C, Green ED, Dunham I (1990) Rapid assessment of S. cerevisiae mating type by PCR. Trends Genet 6(8):236
Kadyk LC, Hartwell LH (1992) Sister chromatids are preferred over homologs as substrates for recombinational repair in Saccharomyces cerevisiae. Genetics 132(2):387–402
Kozmin SG, Sedletska Y, Reynaud-Angelin A, Gasparutto D, Sage E (2009) The formation of double-strand breaks at multiply damaged sites is driven by the kinetics of excision/incision at base damage in eukaryotic cells. Nucleic Acids Res 37(6):1767–1777
Lewis LK, Resnick MA (2000) Tying up loose ends: nonhomologous end-joining in Saccharomyces cerevisiae. Mutat Res 451(1–2):71–89
Lomax ME, Cunniffe S et al (2004) 8-OxoG retards the activity of the ligase III/XRCC1 complex during the repair of a single-strand break, when present within a clustered DNA damage site. DNA Repair (Amst) 3(3):289–299
Malyarchuk S, Harrison L (2005) DNA repair of clustered uracils in HeLa cells. J Mol Biol 345(4):731–743
Malyarchuk S, Brame KL, Youngblood R, Shi R, Harrison L (2004) Two clustered 8-oxo-7, 8-dihydroguanine (8-oxodG) lesions increase the point mutation frequency of 8-oxodG, but do not result in double strand breaks or deletions in Escherichia coli. Nucleic Acids Res 32(19):5721–5731
Malyarchuk S, Castore R, Harrison L (2008) DNA repair of clustered lesions in mammalian cells: involvement of non-homologous end-joining. Nucleic Acids Res 36(15):4872–4882
Mansour SL, Thomas KR, Capecchi MR (1988) Disruption of the proto-oncogene int-2 in mouse embryo-derived stem cells: a general strategy for targeting mutations to non-selectable genes. Nature 336(6197):348–352
Moynahan M, Jasin M (1997) Loss of heterozygosity induced by a chromosomal double-strand break. Proc Natl Acad Sci USA 94(17):8988–8993
Paques F, Haber JE (1999) Multiple Pathways of Recombination Induced by Double-Strand Breaks in Saccharomyces cerevisiae. Microbiol Mol Biol Rev 63(2):349–404
Petes T, Symington L (1991) Molecular and Cellular Biology of the Yeast Saccharomyces (pp. 407–521). Plainview, NY
Pfeiffer P, Goedecke W, Obe G (2000) Mechanisms of DNA double-strand break repair and their potential to induce chromosomal aberrations. Mutagenesis 15(4):289–302
Richardson C, Moynahan ME, Jasin M (1998) Double-strand break repair by interchromosomal recombination: suppression of chromosomal translocations. Genes Dev 12(24):3831–3842
Rothkamm K, Kruger I, Thompson LH, Lobrich M (2003) Pathways of DNA double-strand break repair during the mammalian cell cycle. Mol Cell Biol 23(16):5706–5715
Shikazono N, Pearson C, O’Neill P, Thacker J (2006) The roles of specific glycosylases in determining the mutagenic consequences of clustered DNA base damage. Nucleic Acids Res 34(13):3722–3730
Sutherland BM, Bennett PV, Sidorkina O, Laval J (2000) Clustered damages and total lesions induced in DNA by ionizing radiation: oxidized bases and strand breaks. Biochemistry 39(27):8026–8031
Sutherland BM, Bennett PV, Sutherland JC, Laval J (2002) Clustered DNA damages induced by X rays in human cells. Radiat Res 157(6):611–616
Szostak JW, Orr-Weaver TL, Rothstein RJ, Stahl FW (1983) The double-strand-break repair model for recombination. Cell 33(1):25–35
Valencia M, Bentele M, Vaze MB, Herrmann G, Kraus E, Lee SE, Schar P, Haber JE (2001) NEJ1 controls non-homologous end joining in. Saccharomyces cerevisiae 414(6864):666–669
Wang Z, Wu X, Friedberg EC (1997) Molecular mechanism of base excision repair of Uracil-containing DNA in yeast cell-free extracts. J Biol Chem 272(38):24064–24071
Ward JF (1994) The complexity of DNA damage: relevance to biological consequences. Int J Radiat Biol 66(5):427–432
Yanez RJ, Porter AC (1999) Gene targeting is enhanced in human cells overexpressing hRAD51. Gene Ther 6(7):1282–1290
Yang N, Galick H, Wallace SS (2004) Attempted base excision repair of ionizing radiation damage in human lymphoblastoid cells produces lethal and mutagenic double strand breaks. DNA Repair (Amst) 3(10):1323–1334
Acknowledgments
This research was supported by a Human Research Program of the Exploration Systems Mission Directorate of the National Aeronautics and Space Administration (Grant NNJ07HC731) and Low Radiation Dose Program of the Office of Biological and Environmental Research of the U.S. Department of Energy (Grant DOE: BO-089) to BMS. The authors gratefully acknowledge Dr. S. Tafrov for helpful suggestions, and J. Jardine and W. Abele for technical support.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
This manuscript is based on a contribution given at the “Heavy Ions in Therapy and Space Symposium 2009,” July 6–10, 2009, Cologne, Germany.
A week before the submission of this article, Betsy Sutherland passed away. The Biology Department of Brookhaven National Laboratory and the worldwide community of scientists who are focused on DNA damage and repair have lost a friend and a colleague, a critical discussion partner, and an innovative thinker. Her loss leaves a void that it will take a while to fill.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Moscariello, M., Sutherland, B. Saccharomyces cerevisiae-based system for studying clustered DNA damages. Radiat Environ Biophys 49, 447–456 (2010). https://doi.org/10.1007/s00411-010-0303-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00411-010-0303-3