
Abstract
There is a growing appreciation that membrane-bound organelles in eukaryotic cells communicate directly with one another through direct membrane contact sites. Mitochondria-associated membranes are specialized subdomains of the endoplasmic reticulum that function as membrane contact sites between the endoplasmic reticulum and mitochondria. These sites have emerged as major players in lipid metabolism and calcium signaling. More recently also autophagy and mitochondrial dynamics have been found to be regulated at ER-mitochondria contact sites. Neurons critically depend on mitochondria-associated membranes as a means to exchange metabolites and signaling molecules between these organelles. This is underscored by the fact that genes affecting mitochondrial and endoplasmic reticulum homeostasis are clearly overrepresented in several hereditary neurodegenerative disorders. Conversely, the processes affected by the contact sites between the endoplasmic reticulum and mitochondria are widely implicated in neurodegeneration. This review will focus on the most recent data addressing the structural composition and function of the mitochondria-associated membranes. In addition, the 3D morphology of the contact sites as observed using volume electron microscopy is discussed. Finally, it will highlight the role of several key proteins associated with these contact sites that are involved not only in dementias, amyotrophic lateral sclerosis and Parkinson’s disease, but also in axonopathies such as hereditary spastic paraplegia and Charcot–Marie–Tooth disease.
Similar content being viewed by others

Mitochondria-associated membranes are specialized subdomains of the ER
Compartmentalization of biochemical reactions to dedicated membrane-bound organelles allows eukaryotic cells to perform the plethora of biological processes necessary to maintain homeostasis. While this has clear advantages, it also requires transfer of metabolites and signaling molecules between organelles to maintain cell performance. Such inter-organelle communication is often achieved via vesicular transport or transcriptional pathways. In recent years, alternative pathways involving direct communication between organelles through membrane contact sites (MCSs) have gained much interest. At MCSs, the membranes of two organelles are closely apposed through the formation of protein tethers, enabling fast, direct and reciprocal signaling between both compartments. With distances between the tip of the axon and the cell body reaching up to one meter in humans, the particular morphology of neurons poses limits on vesicular transport and transcriptional pathways. Processes that require strict regulation in space and time are therefore likely to depend much more on such MCSs. We focus this review on the contact sites that are formed between mitochondria and a specialized subdomain of the endoplasmic reticulum (ER) termed Mitochondria-Associated Membranes (MAMs). Emerging evidence shows that dysfunction of the MAMs plays a prominent role in numerous neurodegenerative diseases and that genes affecting ER and mitochondrial homeostasis are clearly overrepresented in hereditary disorders.
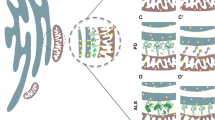
Close membrane appositions between the ER and mitochondria have been observed in electron micrographs as early as the 1950s [10, 24]. The ongoing development of subcellular fragmentation techniques repeatedly showed a resilient ER contamination in mitochondrial fractions, substantiating the existence of MCSs between the ER and mitochondria. Only in 1990, with the description of a membrane fraction ‘X’ associated with mitochondria and involved in phospholipid synthesis, the first function for this interface was identified [110]. This fraction ‘X’, now commonly referred to as the MAMs, is a specialized subdomain of the ER with a particular lipid and protein composition that is involved in the crosstalk with mitochondria. These ER-mitochondria contacts have since then been described in several organisms ranging from yeast to mammals [109]. Approximately 100 of such contacts are shown to occur in a single yeast cell and in mammalia approximately 5–20 % of the mitochondrial surface is estimated to be juxtaposed to the ER [1, 88]. Despite the ongoing development of superresolution techniques, the resolution of light microscopy is too limited for precise analysis of the morphology of ER-mitochondria contact sites, with the distance between both membranes at these sites being 10–30 nm. Transmission electron microscopy (TEM) does provide the required resolution; however, even when using tomography, it is very limited in the amount of 3D information which can be acquired. Volume electron microscopy techniques such as focused ion beam scanning electron microscopy (FIB-SEM) recently became available to obtain high-resolution 3D images of a whole cell and can thus be used as a powerful tool to gain more insight into the 3D morphology of MAMs [59]. Figure 1 and Supplementary movie 1 show the 3D reconstruction of mitochondria and their associated MAMs obtained using FIB-SEM imaging of a mouse embryonic fibroblast. These images clearly show that each mitochondrion makes multiple contacts with the ER and that all these contact sites are highly diverse in shape and size. It is currently unknown how MAM morphology relates to its function and therefore volume electron microscopy will be a crucial tool in studying the role different proteins play in regulating these contact sites.

3D morphology of ER-mitochondria contact sites. Mouse embryonic fibroblasts were imaged using a Zeiss Auriga Crossbeam focused ion beam scanning electron microscope (FIB-SEM). By automated serial block face imaging, large image stacks are generated at high resolution, allowing precise 3D reconstruction of a large cellular volume. In this dataset, over 600 10 nm z-slices were obtained resulting in a 3D dataset at 5 × 5 × 10 nm3 voxels. Mitochondria and their contact sites with the endoplasmic reticulum (ER) were manually segmented and 3D reconstructed in IMOD (http://bio3d.colorado.edu/imod/). A video of this dataset and the reconstruction can be seen in Supplementary movie 1. a The complete reconstruction of two mitochondria (transparent green) and their ER-mitochondria contact sites (magenta) is shown. A contact site is defined as a region where the ER and the mitochondrial membranes are in closer proximity than 30 nm. It is clear that a single mitochondrion makes multiple contacts with the ER and that these contacts are diverse in size, ranging from punctate sites to large patches of the outer mitochondrial membrane being juxtaposed to the ER. b–e Represent different examples of scanning electron micrographs extracted from the volume illustrating a section of the mitochondria and their contacts with the ER. The reconstructed mitochondria depicted in a are shown in transparent green. Magenta arrowheads mark the borders of the ER-mitochondria contact sites. The position of these slices is depicted in blue in a. Scale bar 200 nm
Historically, the ER-mitochondria MCSs have been associated with phospholipid exchange between the ER and mitochondria [88]. In addition, the close proximity between both membranes appears to be critical for the efficient transfer of calcium from the ER to mitochondria. Further studies have revealed additional roles for ER-mitochondria MCSs in a variety of processes ranging from mitochondrial dynamics, sterol metabolism, autophagy, cell survival to energy metabolism and protein folding. Concomitant with the growing appreciation of this signaling hub, the list of disorders associated with ER-mitochondria MCSs is extending. To date this includes cancer [108], metabolic disorders such as Wolfram syndrome [114] or GM1-gangliosidosis [91], diabetes [104], viral infection [48], obesity [7], and neurodegeneration (see Table 1; Boxes 1–4).
Several observations indicate that mitochondria-ER contact sites play crucial roles in neuronal survival and death. Mitochondria in contact with the ER can be readily observed in brain tissue, throughout neurons and at synapses [47]. Already in 2002, it has been suggested that the choreographed interplay between the ER and mitochondria is involved in the shaping of dendritic calcium signals and neuronal activity of hippocampal neurons [84]. In addition at the synapse, calcium shuttling between both compartments was suggested to be essential in determining exocytosis and synaptic activity [74]. The involvement of signaling at the MAMs is implied in several neurodegenerative disorders including Alzheimer’s dementia (AD) [93], Amyotrophic Lateral Sclerosis (ALS) and Frontotemporal Dementia (FTD) [9, 85, 97], Parkinson’s Disease (PD) [17, 42, 82] and Charcot–Marie–Tooth disease (CMT) [28] (for a comprehensive overview see Table 1; Boxes 1–4). More generally, many if not all processes linked to ER-mitochondria contacts are widely implied in neurodegenerative disorders. This review will focus on the structure and function of MAMs and how their disruption contributes to neurodegeneration.
Structural composition of the MAMs
The MAM fraction can be detached from mitochondria through proteolysis with trypsin or Proteinase K, showing that a proteinaceous tether is responsible for connecting both membranes [26]. The presence of such a protein bridge has been confirmed in electron microscopy studies [26, 97], but despite this, the identity of its components still remains largely elusive. Most proteins that have been found to be enriched at the MAMs, are unlikely to participate directly in tethering, but might be functionally involved in the processes mediated by these contacts [108, 109].
In yeast, genetic screens independently led to the discovery of the ER-Mitochondria Encounter Structure (ERMES) complex [55] and the ER membrane protein complex (EMC) [60] as functional tethers between the ER and mitochondria (see Fig. 2 lower part). The ERMES complex consists of the outer mitochondrial membrane proteins Mdm10 and Mdm34, the ER membrane protein Mmm1 and the cytosolic Mdm12. Loss of any of these proteins could be rescued by the expression of an artificial linker, establishing the structural role of ERMES. Gem1, the yeast ortholog of the mitochondrial Rho GTPases 1 and 2 (Miro1 and Miro2), and more recently also Lam6, were identified as regulators of the ERMES complex [32, 56, 78]. A second screen identified the interaction between the EMC and the outer mitochondrial membrane (OMM) translocase complex 5 (Tom5) as an interaction bridging the ER and mitochondrial membranes independent of ERMES (Fig. 2 lower part). In contrast to the ERMES complex, this contact is essential in the lipid exchange between both organelles [60]. While Lam6, Gem1 and the EMC proteins do have metazoan orthologs, it is as yet unclear whether these orthologs effectively function as regulators of interorganelle contact in mammalia.

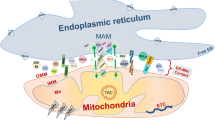
Structural components of ER-mitochondria contact sites. Upper part in mammalian cells, dimers between endoplasmic reticulum (ER)-localized Mitofusin (MFN) 2 and mitochondrial MFN1/2 were the first proposed protein tethers. Charcot–Marie–Tooth (CMT)-causing mutations in MFN2 are believed to decrease ER-mitochondrial contact, contributing to the disease. The interaction of VAMP-associated protein B and C (VAPB) in the ER membrane with protein tyrosine phosphatase-interacting protein 51 (PTPIP51) in the outer mitochondrial membrane (OMM) also contributes to anchoring the mitochondria-associated membrane (MAM) to the mitochondrial membrane. This interaction is inhibited by TAR DNA-binding protein 43 (TDP43) in a glycogen synthase kinase 3β (GSK3β) dependent manner. Both these proteins are implied in neurodegeneration (see Table 1; Boxes 1, 2). The amyotrophic lateral sclerosis (ALS)-causing P56S mutation in VAPB on the other hand increases the physical interaction between the ER and mitochondria (Table 1; Box 2). Phosphofurin acidic cluster protein 2 (PACS2) is established as an essential component of these contacts. The levels of PACS2 are found to be altered in the brains of Alzheimer’s dementia (AD) patients (see Table 1; and Box 1). A functional rather than a structural component of the MAMs is formed by a complex between ER-resident inositol 1,4,5-triphosphate (IP3) channels and the mitochondria resident voltage-dependent anion channel (VDAC), which are bridged by Grp75 and important for calcium shuttling between ER and mitochondria (see Fig. 3). Lower part in Saccharomyces cerevisiae, two tethering complexes are known: the endoplasmic reticulum (ER) mitochondria encounter structure (ERMES), composed of the mitochondrial Mdm10 and Mdm34 proteins, the ER-based Mmm1 protein and the cytosolic Mdm12 protein was the first tether to be described. A second tether important for yeast phospholipid metabolism is achieved through the interaction between the ER membrane complex (EMC) and the outer mitochondrial membrane translocate complex 5 (TOM5). The Miro GTPase Gem1 is a regulatory subunit of ERMES and also Lam6 plays a modulating role determining the extent of membrane contact. It is currently not known whether mammalian orthologs of these components play similar roles in mammalian MAMs
The first complex identified in mammalian cells that bridges the gap between the MAM and the OMM is the tripartite complex between the cytosolic chaperone Grp75, the mitochondrial voltage-dependent anion channel 1 (VDAC1) and the inositol 1,4,5-triphosphate receptor (IP3R) in the MAM [100] (Fig. 2 upper part). While knockdown of Grp75 in HeLa cells decreases the transfer of calcium from the ER to mitochondria, ER-mitochondrial linkage is unaffected in cells lacking all three IP3R isoforms [26]. Therefore, rather than representing a structural tether, this interaction is considered a functional one, promoting efficient transfer of calcium from the ER to mitochondria (see below). Other candidate protein tethers are the homodimers or heterodimers of ER-resident Mitofusin (MFN) 2, a dynamin-related GTPase, with MFN1 or MFN2 on the OMM (Fig. 2 upper part) [28]. Cells depleted of MFN2 were reported to have decreased ER-mitochondria contact and crosstalk [5, 28, 44, 82]. CMT-causing mutations in MFN2 failed to rescue the disrupted ER-mitochondria tethering (see Box 4; Table 1) [28]. MFN2 dependent ER-mitochondria crosstalk is tightly controlled by ubiquitination, further supporting its essential role [98]. Still, independent studies could not confirm the crosstalk-promoting role of MFN2 in ER-mitochondria contacts, but rather reported the opposite finding, i.e., loss of MFN2 led to an increase in MCS [25, 34]. These contradictory findings remain to be resolved, as does the effect of CMT-causing mutations in MFN2 on MAM signaling (see Table 1; Box 4).
Adding to this complexity, additional tethering interactions have been identified, which might compensate for MFN2 loss in certain conditions. One such tether is the physical interaction between the MAM protein VAMP (vesicle-associated membrane protein)-associated protein B and C (VAPB) and the mitochondrial protein tyrosine phosphatase-interacting protein 51 (PTPIP51) (Fig. 2 upper part) [29, 97]. Overexpression of either or both of these proteins increases the extent of contact while loss of either protein through siRNA-mediated knockdown significantly diminishes membrane contact and crosstalk [97]. A functional rather than a structural interaction between the ER and mitochondria has been uncovered by establishing the interaction between ER-resident BAP31 and the mitochondrial fission protein Fission 1 homolog (Fis1) [50]. The complex formed between BAP31/Fis1 constitutes an apoptosis-signaling platform needed for the recruitment of procaspase 8, the cleavage of BAP31 and a subsequent increase in Ca2+ release from the ER to the mitochondria [50]. This fits with earlier notions that a feedback ER contribution is needed to amplify mitochondrial apoptosis signals [3]. Finally, the vesicular sorting protein phosphofurin acidic cluster sorting protein 2 (PACS2) contributes to the structural integrity of the ER-mitochondria MCSs, through a stabilization mechanism that is not entirely clear [95] (Fig. 2 upper part). The siRNA-mediated knockdown of PACS2 uncouples the ER from mitochondria and induces BAP31 cleavage-dependent mitochondrial fragmentation and cell death in A7 melanoma cells [95], and similarly results in caspase-3 mediated degeneration of primary hippocampal neurons and astrocytes [47]. The complex role of PACS2 as a modulator of MAM properties is discussed further.
Although the mediators of these MCSs seem to be quite distinct between yeast and mammals, the property that different and independent linkers are involved in the coupling of both membranes is conserved. The relevance of having diverse connections is currently unclear, however, it does suggest that these could serve distinct functions. A recent study in mammalian cells indeed indicates the presence of two distinct domains in MAMs that differ in their lipid content and function [4], yet more research is needed to shed light on how distinct protein and lipid compositions can affect MAM function.
Several essential cellular processes are controlled by MAMs
As stated in the introduction, recent studies revealed several novel functions for the MAMs. In the following sections we will give a brief description of the most important MAM functions in the context of neurobiology.
Regulation of lipid metabolism
Although most membrane lipid synthesis in eukaryotic cells occurs in the ER, several key metabolic steps and lipid modifications are performed in other organelles, and thus, require transport of lipids. How hydrophobic lipids are distributed within the aqueous environment of the cell towards other organelles remains largely elusive, but recently the role of non-vesicular transport at membrane contact sites has gained a lot of interest [61, 109]. Close membrane apposition at these sites appears to allow efficient shuttling of lipids. In addition, several lipid metabolic enzymes are exclusively localized at certain MCSs, restricting the generation of these lipid species to the site where they are needed. MAMs were originally identified as a fraction highly enriched in phosphatidylserine (PS) synthase-1 and -2 (Pss1 and Pss2) and transfer of phospholipids between the ER and mitochondria was the first function ascribed to ER-mitochondria contacts [109]. Phosphatidylserine, newly synthesized in the MAM by the PS synthases, is subsequently transferred to the closely apposed mitochondrion (Fig. 3). In the inner mitochondrial membrane (IMM), decarboxylation of PS results in the production of phosphatidylethanolamine (PE). The importance of ER-mitochondria MCSs is underlined by the fact that the transfer of PS between both organelles is the rate-limiting step in PE synthesis from PS [112]. In a crude mitochondrial fraction derived from rat liver, newly synthesized PE can then be transferred back to the MAM where it is converted into phosphatidylcholine (PC) by the enzyme PE-N-methyltransferase (PEMT) [110]. Since PEMT activity is restricted to hepatocytes, it is not clear how essential MAMs are for PC synthesis in other tissues.

Lipid metabolism at MAMs. Historically, MAMs were identified as essential regions for phospholipid metabolism. For phosphatidylethanolamine (PE) synthesis, phosphatidylserine (PS) synthesized in the endoplasmic reticulum (ER) from phosphatidic acid (PA) by the PS synthetases 1 and 2 (Pss1/2) needs to be shuttled to the inner mitochondrial membrane (IMM), where it is decarboxylated to PE by the PS decarboxylase (Psd). This PE can then be shuttled back to the ER and used in further lipid metabolism, such as conversion to phosphatidylcholine (PC) by the PE-N-methyltransferase (PEMT). In addition, the synthesis of sterols requires the import of cholesterol (chol) from the ER into mitochondria
Protein identification approaches revealed that not only Pss1 and Pss2 are enriched at the MAMs, but also several other lipid synthesizing enzymes involved in cholesterol and sphingolipid biosynthesis (Fig. 3) [39, 45, 109]. This fits well with thin layer chromatography studies showing that MAMs comprise a unique lipid profile, with highly elevated levels of ceramides and cholesterol compared to the bulk ER [45]. While it is yet unclear whether the MAMs participate in trafficking of these lipids to mitochondria, this particular lipid raft-like composition plays a determining factor in the recruitment of several well-established MAM components. Both presenilin 1 and 2 (PS1 and PS2), members of the γ-secretase complex are targeted to the MAMs because of their lipid raft composition [106]. Similarly, the chaperone Sigma non-opioid intracellular receptor 1 (Sigma1R), associated with AD, ALS and FTD is strictly dependent on the presence of cholesterol and ceramide for targeting to the MAMs [45] and for performing its essential functions in calcium regulation (see Table 1; Fig. 4).

Calcium signaling at ER-mitochondria membrane contact sites. Transfer of calcium to mitochondria requires calcium hotspots, which can be achieved at mitochondria-associated membranes (MAMs) owing to the quasi-synaptic structure of this membrane contact site (MCS). Shuttling of calcium is needed during a variety of responses, as it can both stimulate ATP synthesis and promote mitophagy and apoptosis. Efficient import of calcium at MAMs is mediated by Grp75, which brings the openings of the inositol 1,4,5-triphosphate receptor (IP3R) calcium channels in the endoplasmic reticulum (ER) in close vicinity to the voltage dependent anion channel (VDAC) in the outer mitochondrial membrane (OMM). Opening of the IP3Rs is regulated by a set of proteins present at or recruited to MAMs, including calnexin (CNX), the Sigma non-opioid intracellular receptor 1 (Sigma1R), presenilin 1 and 2 (PS1 and PS2). The presence of CNX at MAMs in turn is regulated by the sorting proteins Rab32 and phosphofurin acidic cluster protein 2 (PACS2), as well as by palmitoylation of CNX. The association of Sigma1R with MAMs on the other hand depends on cholesterol levels. Dysregulation of calcium signaling at MAMs is strongly implied in neurodegenerative disorders. PS1 and PS2 play a complex role regulating both the extent of ER-mitochondrial contact and calcium signaling. The Parkinson’s disease (PD)-associated Parkin, DJ1 and α-synuclein (α-Syn) were also found to affect calcium transfer at these contacts. Finally, defects in mitofusin 2 (MFN2) or VAMP-associated protein B and C (VAPB), which contribute to the structural integrity of the MCSs, affect the calcium crosstalk between the ER and mitochondria
Regulation of calcium homeostasis at the MAMs
Already in the 19th century, the role of calcium as second messenger was established through seminal experiments on perfusion of isolated hearts. Since then, a lot of research has been devoted to understand how increases in calcium levels (so-called oscillations) are generated, decoded and translated in signaling events. Two major sources count for the rise in cytosolic calcium: the extracellular medium with a [Ca2+] of ~1 mM and the ER with a [Ca2+] >100 μM [87]. Mitochondria play a crucial role in buffering increases in cytosolic calcium, and a rise in cytosolic [Ca2+]cyt is always closely followed by a rise in [Ca2+]mito [89]. Interestingly, upon ER calcium release, mitochondrial levels can rapidly increase 10–100-fold without noticeable rises of cytosolic calcium levels [87]. In contrast to the outer mitochondrial membrane (OMM), which is permeable to calcium ions, influx of calcium into the mitochondrial matrix—driven by the electrical gradient across the IMM—occurs through the recently identified mitochondrial calcium uniporter (MCU) complex. This complex consists of the MCU protein and its regulators (MICU1, MICU2, MCUb, MCUR1, and EMRE) [72]. Paradoxically, the low calcium affinity of the MCU appears incompatible with the observed rapid calcium uptake in mitochondria at low cytosolic calcium levels. This discrepancy was solved by the observation that mitochondria are in close contact with the IP3Rs and ryanodine sensitive channels (RyRs) mediating ER store release [88]. This occurs at the MAMs and leads to the transient formation of “hotspots”, perimitochondrial microdomains in which calcium levels far exceed those in the bulk of the cytosol, enabling rapid import across the IMM [27, 40, 88].
ER-mitochondria MCSs are able to establish such microdomains required for efficient calcium transfer through (1) the close apposition of both membranes, (2) the enrichment of the calcium release channels IP3Rs and RyRs at MAMs and (3) the connection of IP3Rs to the mitochondrial VDAC1 by the chaperone Grp75 (Fig. 4). Knockdown of Grp75 abolishes functional coupling between VDAC1 and IP3R, resulting in diminished calcium transfer [100]. On the other hand, bringing the ER closer to mitochondria through an artificial linker stimulates calcium transfer [26]. Numerous studies have correlated altered ER-mitochondrial connectivity to alterations in calcium transfer between both organelles, through overexpression or knockdown of proposed tethers, including MFN2 and VAPB-PTPIP51, or regulators thereof [28, 29, 34, 97] (Fig. 4). Several modulators of IP3R channel activity are present at or recruited to MAMs under various conditions. For example, various ER-resident chaperones and oxidoreductases can affect the exchange of calcium between the ER and mitochondria [96]. Calnexin (CNX) modulates local calcium levels by acting as a calcium buffer and through regulating the activity of IP3R and the sarco/endoplasmic reticulum calcium ATPase (Serca), the main ATPase pumping cytosolic calcium into the ER [52, 68, 90]. CNX targeting to the MAM is dependent on palmitoylation [68], and on the cytosolic sorting protein PACS2 [79] and Rab32 [16] two trafficking molecules that both regulate the composition of the MAMs (Fig. 4). The ER-resident Sigma1R is released from Bip upon ER calcium release, and binds to IP3Rs at the MAMs, in this way preventing their proteasomal degradation and sustaining prolonged Ca2+ uptake by mitochondria [46] (Fig. 4). Especially in neurons, both Sigma1R and PACS2 play a crucial role in mediating calcium homeostasis and loss of Sigma1R results in neuromuscular defects [47]. This is underscored by the fact that alterations in PACS2 and Sigma1R levels are associated with the pathogenesis of AD, ALS and FTD (see Table 1; Boxes 1, 2). In addition, presenilin mutations in AD are associated with an increased IP3R calcium release (see Table 1; Box 1). Finally, in Huntington’s disease ER-mitochondrial calcium transfer appears affected as well. Mutant huntingtin, but not the wild type protein, was found to interact with IP3R and facilitate calcium transfer between the ER and mitochondria [12, 83].
Calcium transfer to mitochondria occurs under both physiological and stress conditions. A moderate mitochondrial matrix calcium increase stimulates ATP synthesis through calcium dependency of metabolic enzymes in the Krebs cycle [87]. Functional coupling of the ER and mitochondria therefore stimulates aerobic metabolism, in response to a demand of ATP-requiring processes in the cytosol as well as in the ER [96, 111]. Hence, loss of contact has adverse effects on ER homeostasis, protein folding and energy metabolism [14], and will affect cell survival and proliferation, especially in cells with high-energy demands such as neurons. Mitochondrial calcium overload on the other hand is a potent inducer of the mitochondrial permeability transition pore and can thereby lead to OMM rupture and the escape of pro-apoptotic factors such as cytochrome c into the cytosol [87]. In this respect, regulation of ER-mitochondrial calcium transfer at the MAMs is essential in the switch between cell survival and death during various stress responses. In the context of Huntington’s disease for example, the binding of mutant huntingtin to IP3R may result in a heightened susceptibility of mitochondria to activate the permeability transition pore and the subsequent release of pro-apoptotic factors [23]. Intriguingly, proteins that modulate calcium transfer between mitochondria and the ER are affected in many neurodegenerative diseases and could be tightly linked to a loss in neuronal survival [94] (see Table 1).
Regulation of mitochondrial dynamics and homeostasis
The function of both the ER and mitochondria strongly depends on a highly regulated balance in membrane dynamics. Mitochondrial dynamics include their transport along cytoskeletal tracks throughout the cell, in addition to regulated fission and fusion. Combined, these dynamics ensure optimal mitochondrial metabolism and, in neurons, the correct distribution of mitochondria to dendrites and synapses. Especially in neurons with long axons this poses a challenge, which is highlighted by the many diseases caused by defects in the proteins involved in mitochondrial dynamics [19] (see Boxes 2–4; Table 1). Mitochondrial fission is known to require the assembly of dynamin-related protein 1 (Drp1) in a helix around the OMM, followed by constriction of this ring (Fig. 5). However, it has long been unclear what signaling events precede this process and mark the site of Drp1 assembly. In 2011, Friedman et al. showed in Cos7 cells that focal accumulation of Drp1 and subsequent fission occurs at or near sites of contact with the ER, and the same holds true for the yeast Drp1 ortholog Dnm1 [37]. Mitochondrial constriction occurs in the vicinity of the ER even in the absence of Drp1 and the mitochondrial fission factor (Mff). These observations suggested that ER tubules mark the sites of mitochondrial fission and induce initial constriction, followed by the recruitment of Mff and finally Drp1 helix assembly that mediates the division [37]. Indeed, recent studies have described that actin polymerization, induced at the MCSs by an ER-bound isoform of Inverted Formin 2 (INF2), is capable and required to induce the initial constriction required for mitochondrial fission (Fig. 5) [58]. Spire1C, a newly described mitochondrial isoform of the actin-nucleating Spire proteins, interacts with INF2 to specifically nucleate actin filaments only at sites where the ER and mitochondrial membranes are closely apposed. Loss of this interaction results in long tubular mitochondria reminiscent of defective fission [70]. Mitochondrial constriction prior to Drp1 assembly depends on this actin polymerization step, however, additional force generated by Myosin II may also contribute to this process [30, 57]. How mutations in INF2, causing Charcot–Marie–Tooth disease type 2D and glomerulopathy [15] affect this process remains to be determined. Finally, it was shown in a very elegant way that Drp1 activity at the MAMs is strictly regulated by phosphorylation in a process that involves Rab32 and syntaxin 17 (STX17) [4] (Fig. 5). This process seems tightly linked to the regulation of calcium homeostasis and the initiation of autophagy (see further). In addition, SUMOylation of Drp1 was reported to affect its function at the MAM, stabilizing the ER-mitochondria contact site and thus promoting calcium crosstalk and cytochrome c release [86].

MAMs control mitochondrial dynamics. Endoplasmic reticulum (ER)-mitochondria contact sites are involved in mitochondrial fission and transport of mitochondria along cytoskeletal tracks. During fission, mitochondria-associated membranes (MAMs) determine the site of scission by contributing to the mitochondrial constriction required for dynamin-related protein 1 (Drp1) assembly. This constriction is accomplished through the actin-modulating activity of Spire1C and inverted forming 2 (INF2) at the membrane contact sites (MCSs). At least two other MAM proteins, syntaxin 17 (STX17) and Rab32, contribute to the regulation of mitochondrial fission by controlling Drp1 activity. Mitochondrial transport is primarily mediated by the mitochondrial Rho GTPases 1 and 2 (Miro1/2), which connect the mitochondria to motor proteins such as kinesins through the trafficking kinesin protein (TRAK) adaptor proteins. Miro depends on mitofusin1 and 2 (MFN1/2) to facilitate transport, and PTEN-induced putative kinase 1 (PINK1)-Parkin-dependent ubiquitination of Miro1/2 and MFN2 blocks mitochondrial movement. In zones of high calcium concentrations, such as those that occur at MAMs, calcium binding to the Miro1/2 EF-hand motifs releases the mitochondria from the cytoskeleton and halts their migration. The P56S mutation in VAMP-associated protein B and C (VAPB), which results in a higher degree of ER-mitochondria contact and calcium crosstalk, consequently results in axonal transport defects of mitochondria
In contrast to the role of Gem1p in ER-associated mitochondrial division in yeast, no such role has been described for the mammalian Gem1 orthologs Miro1 and Miro2. In mammalian cells, both Miro proteins 1 and 2 are known as essential regulators of mitochondrial motility [36, 69, 92]. These atypical Rho GTPases are anchored in the OMM and mediate anterograde and retrograde transport by connecting the mitochondria to microtubule (MT)-bound kinesin and dynein through the trafficking kinesin (TRAK1 and TRAK2) adaptor proteins (Fig. 5) [36, 69]. Neuron-specific loss of Miro1 in mice results in a neuronal disease phenotype that most closely resembles that of spastic paraplegia patients [80] (see Box 4; Table 1).
Interestingly, several observations point to a role of MAMs in Miro1/2-mediated cellular transport of mitochondria. First, as mentioned, the yeast ortholog of Miro1/2 is a regulatory subunit of the ERMES complex and Miro1 localizes at ER-mitochondria contact sites in mammalian cells [56]. Second, at sites of close contact, the ER and mitochondria move along acetylated MTs while staying attached, suggesting coordinated transport [38]. Third, in addition to coordinating fusion and tethering to the ER of the OMM, MFN2 is required for axonal transport and both MFN1 and MFN2 interact with Miro1/2 [75]. Fourth, mitochondrial transport is regulated by calcium through the EF-hand motifs in Miro1/2, which function as a calcium sensor. Calcium binding of Miro1/2 induces a conformational change that disconnects the mitochondria from the MT track it moves along, thus halting its transport [20, 69, 92]. Due to the relatively low affinity of Miro1/2 to calcium, calcium-dependent halting of mitochondrial transport is most likely to occur at calcium hotspots, such as the ER-mitochondria MCSs [20, 92]. The dependence on high calcium regions indicates a mechanism whereby the proximity with the ER can determine the redistribution of mitochondria. This provides the cell with a powerful system of targeting mitochondria based on local energy requirements. Particularly in neurons, this might be a relevant factor in determining the distribution of mitochondria to the dendrites, synapses and the nodes of Ranvier that is currently insufficiently understood. That such a system is essential for neuronal survival is underscored by the many reports of mitochondrial transport defects in neurodegenerative disorders [19]. Much research has focused on the connection between mitochondria and MTs in this context. It is becoming increasingly clear, however, that the connection with the ER is also a major determinant for axonal transport of mitochondria (see Boxes 2–4).
MAMs as a potential regulator of autophagy and mitophagy
Macroautophagy is the process whereby damaged proteins and organelles are cleared from the cell by sequestering them in a double membrane-bound vesicle termed the autophagosome. Subsequent delivery to the lysosome allows proteasomal breakdown and recycling of the substrates. Debate is still ongoing with respect to the origin of the phagophore and the identity of membrane donor sources needed for its expansion to form the mature autophagosome [62]. Axe et al. identified the omegasome, a phosphatidylinositol-3-phosphate-enriched membrane structure attached to the ER, as a membrane source for phagophore development [8]. In addition, the mitochondria, harboring many autophagy-related (ATG) proteins and regulators thereof, have been proposed as an origin for phagophore formation, based on the transfer of a fluorescently labeled mitochondrial marker to the phagosome [43]. Intriguingly, several ATG proteins have been found to accumulate specifically at ER-mitochondria MCSs in conditions of starvation, and more and more evidence is accumulating that MAMs might be the actual site of autophagosome formation, unifying both earlier models [43]. Hailey et al. first speculated on the requirement of lipid transfer from the ER to mitochondria during autophagosome biogenesis. In mouse cells lacking MFN2, autophagy induction was disturbed. The authors contributed this to a decreased lipid transfer towards mitochondria as a consequence of ER-mitochondrial uncoupling and concluded that lipids transferred from the ER accumulate in the OMM, from where they are trafficked to the expanding phagophore [43] (Fig. 6). A more direct involvement of MAMs in autophagosome biogenesis was proposed by Hamasaki and coworkers [44]. Upon starvation of mammalian cells, Vps15, Vps34, Atg14L, Beclin1—early markers of phagophore formation—and the omegasome marker double FYVE-containing protein 1 (DFCP1) accumulate in the MAM fraction (Fig. 6) [44]. This might be initiated by early translocation of STX17 [4]. In starvation conditions, STX17 translocates in an mTOR/ULK dependent manner to a domain of ER-mitochondrial contact resistant to digitonin, where it recruits ATG14L [4]. ATG14L then interacts with the phosphoinositide 3-kinase complex consisting of Beclin1, p150 and Vps34, resulting in local phosphatidylinositol-triphosphate production. This alters the local lipid composition of the MAM membrane, which leads to the recruitment of WD-repeat domain phosphoinositide interacting (WIPI) proteins, the effector proteins and the mediators of autophagosome biogenesis. It has been proposed that this could be sufficient to induce a deformation of the membrane [62]. Consistent with a role for the MAMs in autophagosome formation, knockdown of MFN2 or PACS2 abolished ATG14L puncta formation and downstream LC3 lipidation in starved cells, and caused defective STX17 localization at the MAM [44].

MAMs as sites for autophagosome biogenesis. Mitochondria-associated membranes (MAMs) were identified as a membrane source for autophagosome biogenesis. In a first instance, phosphatidylethanolamine (PE) synthesis at MAMs was believed to be a crucial lipid source for these emerging organelles. However, ER-mitochondria contact sites appear to play a more direct role. Syntaxin 17 (STX17) at MAMs recruits early autophagosome markers including Atg14L, Vps15, Vps34 and Beclin1. In addition, the omegasome marker double FYVE-containing protein 1 (DFCP1) translocates to these sites upon autophagy induction. Underscoring the relevance of ER-mitochondria membrane contact sites (MCSs), loss of phosphofurin acidic cluster sorting protein 2 (PACS2) or mitofusin 2 (MFN2) abrogates autophagosome biogenesis
In addition, during the selective autophagy of mitochondria, known as mitophagy, ER-mitochondria contact sites appear to constitute a platform promoting mitophagosome formation. In yeast, efficient mitophagy depends on coupling of mitochondria and the ER through ERMES, while bulk autophagy can apparently operate in the absence of this tether [13]. ERMES colocalizes with sites of mitophagosome biogenesis and affects the formation of the phagophore that engulfs the mitochondria destined for degradation [13, 71].
In mammalian cells, mitochondria destined for degradation recruit the E3 ubiquitin ligase Parkin to the OMM through PTEN-induced putative kinase 1 (PINK1) kinase activity. Ubiquitination of Parkin substrates subsequently recruits the autophagy machinery [31]. ER-mitochondria MCSs are implied in this process in several ways. First, focal accumulation of DFCP1 along mitochondria labeled for degradation with exogenously expressed Parkin suggests a role for the ER/MAM as a membrane source for the mitophagosome [115]. Second, Drp1-mediated fission, at least in part mediated by contacts with the ER [37], is suggested to promote mitophagy by breaking off pieces of mitochondria promoting their engulfment by the mitophagosome [105]. Lastly, depolarization of the IMM through calcium (over)loading could be involved in PINK1 and Parkin translocation to the OMM, activating mitophagy [31, 53].
While the exact mechanisms governing autophagy induction and progression are not entirely clear, several lines of evidence point to an essential role of the ER-mitochondria interface during this process. The presence at the MAMs of several important players in metabolism, such as mTORC2 [11], allows to speculate that ER-mitochondria MCSs could represent a site of crosstalk between sensors of the cells’ energetic needs and the early players in autophagy. Additionally, through interorganelle communication, dysfunctional mitochondria may be sensed at the MAMs, followed by mitophagy induction and targeted removal of these unhealthy mitochondria. Future research is needed to study these mechanisms and how they are disrupted in disease. Dysregulation of neuronal energy metabolism and mitochondrial homeostasis are important factors contributing to neurodegeneration [94]. This appears relevant in particular for PD (see Box 3), however, autophagy is a process whose dysregulation is widely implied in neurodegenerative disorders [73].
Conclusion
MCSs provide pathways of intracellular signaling that are only beginning to be unraveled. Although many questions remain as to how ER-mitochondria MCSs are maintained and regulated, it is clear that many different pathways intertwine at these signaling hubs. Whether all the different functions described here occur simultaneously at the same MCS, or whether specialized ER-mitochondria MCSs exist that mediate a subset of these processes is currently unclear. Gaining further insight into the protein tethers that connect both organelles will be crucial to shed more light on the regulation of MAM functions.
As these functions become uncovered, also the relevance of ER-mitochondria contact sites in disease becomes apparent. Neuronal cells in particular appear to be very vulnerable to insults affecting the balance in ER-mitochondrial communication. Indeed, many of the processes that require these contact sites are implied in neurodegeneration. As such, resolving the role of MAMs in the pathomechanisms leading to these disorders may unify several of the previously identified defects that occur during neurodegeneration, including imbalances in calcium or lipid signaling, mitochondrial dynamics and autophagy pathways. Since tipping the balance in either direction appears detrimental for neuronal survival, uncovering the different players that maintain this delicate equilibrium will be crucial to understand the role that MCSs play in these disorders and possibly lead to novel strategies to reverse ER-mitochondrial homeostasis defects in patients.
Abbreviations
- AD:
-
Alzheimer’s dementia
- ALS:
-
Amyotrophic lateral sclerosis
- APP:
-
Amyloid precursor protein
- ATG:
-
Autophagy-related
- ATL1/3:
-
Atlastin 1/3
- CMT:
-
Charcot–Marie–Tooth disease
- CNX:
-
Calnexin
- DFCP1:
-
Double FYVE-containing protein 1
- DJ-1:
-
Protein deglycase DJ-1
- Drp1:
-
Dynamin-related protein 1
- EMC:
-
Endoplasmic reticulum membrane complex
- ER:
-
Endoplasmic reticulum
- ERMES:
-
Endoplasmic reticulum-mitochondria encounter structure
- FAD:
-
Familial Alzheimer’s dementia
- FIB-SEM:
-
Focused ion beam scanning electron microscopy
- Fis1:
-
Fission 1 homolog
- FTD:
-
Frontotemporal dementia
- GSK3β:
-
Glycogen synthase kinase-3β
- HSAN:
-
Hereditary sensory and autonomous neuropathy
- HSP:
-
Hereditary spastic paraplegia
- IMM:
-
Inner mitochondrial membrane
- INF2:
-
Inverted formin 2
- IP3R:
-
Inositol 1,4,5-triphosphate receptor
- MAM:
-
Mitochondria-associated membrane
- MCS:
-
Membrane contact site
- MCU:
-
Mitochondrial calcium uniporter
- Mff:
-
Mitochondrial fission factor
- MFN1/2:
-
Mitofusin 1/2
- miro1/2:
-
Mitochondrial Rho GTPases 1/2
- MT:
-
Microtubule
- OMM:
-
Outer mitochondrial membrane
- PA:
-
Phosphatidic acid
- PACS2:
-
Phosphofurin acidic cluster sorting protein 2
- PC:
-
Phosphatidylcholine
- PD:
-
Parkinson’s disease
- PE:
-
Phosphatidylethanolamine
- PEMT:
-
Phosphatidylcholine-N-methyltransferase
- PINK1:
-
PTEN-induced putative kinase 1
- PS1/2:
-
Presenilin 1/2
- PS:
-
Phosphatidylserine
- Pss1/2:
-
Phosphatidylserine synthase 1/2
- PTPIP51:
-
Protein tyrosine phosphatase-interacting protein 51
- REEP1/2:
-
Receptor expression-enhancing protein 1/2
- RTN2/4b:
-
Reticulon 2/4b
- RyR:
-
Ryanodine receptor
- SAD:
-
Sporadic Alzheimer’s dementia
- Sigma1R:
-
Sigma non-opioid intracellular receptor 1
- STX17:
-
Syntaxin 17
- TDP43:
-
TAR DNA-binding protein 43
- TEM:
-
Transmission electron microscopy
- Tom5:
-
Outer mitochondrial membrane translocase complex 5
- TRAK1/2:
-
Trafficking kinesin protein 1/2
- VAPB:
-
Vesicle-associated membrane protein-associated protein B
- VDAC1:
-
Voltage-dependent anion channel 1
- α-Syn:
-
Alpha-synuclein
References
Achleitner G, Gaigg B, Krasser A, Kainersdorfer E, Kohlwein SD, Perktold A, Zellnig G, Daum G (1999) Association between the endoplasmic reticulum and mitochondria of yeast facilitates interorganelle transport of phospholipids through membrane contact. Eur J Biochem FEBS 264:545–553. doi:10.1046/j.1432-1327.1999.00658.x
Al-Saif A, Al-Mohanna F, Bohlega S (2011) A mutation in sigma-1 receptor causes juvenile amyotrophic lateral sclerosis. Ann Neurol 70:913–919. doi:10.1002/ana.22534
Alirol E, James D, Huber D, Marchetto A, Vergani L, Martinou J-CC, Scorrano L (2006) The mitochondrial fission protein hFis1 requires the endoplasmic reticulum gateway to induce apoptosis. Mol Biol Cell 17:4593–4605. doi:10.1091/mbc.E06-05-0377
Arasaki K, Shimizu H, Mogari H, Nishida N, Hirota N, Furuno A, Kudo Y, Baba M, Baba N, Cheng J et al (2015) A role for the ancient SNARE syntaxin 17 in regulating mitochondrial division. Dev Cell 32:304–317. doi:10.1016/j.devcel.2014.12.011
Area-Gomez E, Castillo M, Tambini M, Guardia-Laguarta C, Groof A, Madra M, Ikenouchi J, Umeda M, Bird T, Sturley S et al (2012) Upregulated function of mitochondria-associated ER membranes in Alzheimer disease. EMBO J. doi:10.1038/emboj.2012.202
Area-Gomez E, de Groof AJC, Boldogh I, Bird TD, Gibson GE, Koehler CM, Yu WH, Duff KE, Yaffe MP, Pon LA et al (2009) Presenilins are enriched in endoplasmic reticulum membranes associated with mitochondria. Am J Pathol 175:1810–1816. doi:10.2353/ajpath.2009.090219
Arruda AP, Pers BM, Parlakgül G, Güney E, Inouye K, Hotamisligil GS (2014) Chronic enrichment of hepatic endoplasmic reticulum-mitochondria contact leads to mitochondrial dysfunction in obesity. Nat Med 20:1427–1435. doi:10.1038/nm.3735
Axe EL, Walker SA, Manifava M, Chandra P, Roderick HL, Habermann A, Griffiths G, Ktistakis NT (2008) Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol. doi:10.1083/jcb.200803137
Bernard-Marissal N, Medard JJ, Azzedine H, Chrast R (2015) Dysfunction in endoplasmic reticulum-mitochondria crosstalk underlies SIGMAR1 loss of function mediated motor neuron degeneration. Brain. doi:10.1093/brain/awv008
Bernhard W, Rouiller C (1956) Close topographical relationship between mitochondria and ergastoplasm of liver cells in a definite phase of cellular activity. J Biophys Biochem Cytol 2:73–78
Betz C, Stracka D, Prescianotto-Baschong C, Frieden M, Demaurex N, Hall MN (2013) mTOR complex 2-Akt signaling at mitochondria-associated endoplasmic reticulum membranes (MAM) regulates mitochondrial physiology. Proc Natl Acad Sci 110:12526–12534. doi:10.1073/pnas.1302455110
Bezprozvanny I, Hayden MR (2004) Deranged neuronal calcium signaling and Huntington disease. Biochem Biophys Res Commun 322:1310–1317. doi:10.1016/j.bbrc.2004.08.035
Böckler S, Westermann B (2014) Mitochondrial ER contacts are crucial for mitophagy in yeast. Dev Cell. doi:10.1016/j.devcel.2014.01.012
Bouman L, Schlierf A, Lutz AK, Shan J, Deinlein A, Kast J, Galehdar Z, Palmisano V, Patenge N, Berg D et al (2011) Parkin is transcriptionally regulated by ATF4: evidence for an interconnection between mitochondrial stress and ER stress. Cell Death Differ 18:769–782. doi:10.1038/cdd.2010.142
Boyer O, Nevo F, Plaisier E, Funalot B, Gribouval O, Benoit G, Huynh Cong E, Arrondel C, Tete MJ, Montjean R et al (2011) INF2 mutations in Charcot–Marie–Tooth disease with glomerulopathy. N Engl J Med 365:2377–2388. doi:10.1056/NEJMoa1109122
Bui M, Gilady SY, Fitzsimmons REB, Benson MD, Lynes EM, Gesson K, Alto NM, Strack S, Scott JD, Simmen T (2010) Rab32 modulates apoptosis onset and mitochondria-associated membrane (MAM) properties. J Biol Chem 285:31590–31602. doi:10.1074/jbc.M110.101584
Calì T, Ottolini D, Negro A, Brini M (2013) Enhanced parkin levels favor ER-mitochondria crosstalk and guarantee Ca(2+) transfer to sustain cell bioenergetics. Biochim Biophys Acta 1832:495–508. doi:10.1016/j.bbadis.2013.01.004
Calì T, Ottolini D, Negro A, Brini M (2012) α-Synuclein controls mitochondrial calcium homeostasis by enhancing endoplasmic reticulum-mitochondria interactions. J Biol Chem. doi:10.1074/jbc.M111.302794
Chan DC (2006) Mitochondria: dynamic organelles in disease, aging, and development. Cell 125:1241–1252. doi:10.1016/j.cell.2006.06.010
Chang KT, Niescier RF, Min K-TT (2011) Mitochondrial matrix Ca2+ as an intrinsic signal regulating mitochondrial motility in axons. Proc Natl Acad Sci USA 108:15456–15461. doi:10.1073/pnas.1106862108
Cheung K-HH, Mei L, Mak D-ODO, Hayashi I, Iwatsubo T, Kang DE, Foskett JK (2010) Gain-of-function enhancement of IP3 receptor modal gating by familial Alzheimer’s disease-linked presenilin mutants in human cells and mouse neurons. Sci Signal. doi:10.1126/scisignal.2000818
Cheung K-HH, Shineman D, Müller M, Cárdenas C, Mei L, Yang J, Tomita T, Iwatsubo T, Lee VM, Foskett JK (2008) Mechanism of Ca2+ disruption in Alzheimer’s disease by presenilin regulation of InsP3 receptor channel gating. Neuron 58:871–883. doi:10.1016/j.neuron.2008.04.015
Choo YS, Johnson GV, MacDonald M, Detloff PJ, Lesort M (2004) Mutant huntingtin directly increases susceptibility of mitochondria to the calcium-induced permeability transition and cytochrome c release. Hum Mol Genet 13:1407–1420. doi:10.1093/hmg/ddh162
Copeland DE, Dalton AJ (1959) An association between mitochondria and the endoplasmic reticulum in cells of the pseudobranch gland of a teleost. J Biophys Biochem Cytol 5:393–396
Cosson P, Marchetti A, Ravazzola M, Orci L (2012) Mitofusin-2 independent juxtaposition of endoplasmic reticulum and mitochondria: an ultrastructural study. PLos One. doi:10.1371/journal.pone.0046293
Csordás G, Renken C, Várnai P, Walter L, Weaver D, Buttle KF, Balla T, Mannella CA, Hajnóczky G (2006) Structural and functional features and significance of the physical linkage between ER and mitochondria. J Cell Biol. doi:10.1083/jcb.200604016
Csordás G, Thomas AP, Hajnóczky G (1999) Quasi-synaptic calcium signal transmission between endoplasmic reticulum and mitochondria. EMBO J 18:96–108. doi:10.1093/emboj/18.1.96
de Brito OM, Scorrano L (2008) Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 456:605–610. doi:10.1038/nature07534
De Vos KJ, Mórotz GM, Stoica R, Tudor EL, Lau K-FF, Ackerley S, Warley A, Shaw CE, Miller CC (2012) VAPB interacts with the mitochondrial protein PTPIP51 to regulate calcium homeostasis. Hum Mol Genet 21:1299–1311. doi:10.1093/hmg/ddr559
DuBoff B, Götz J, Feany MB (2012) Tau promotes neurodegeneration via DRP1 mislocalization in vivo. Neuron. doi:10.1016/j.neuron.2012.06.026
Eiyama A, Okamoto K (2015) PINK1/Parkin-mediated mitophagy in mammalian cells. Curr Opin Cell Biol 33:95–101. doi:10.1016/j.ceb.2015.01.002
Elbaz-Alon Y, Eisenberg-Bord M, Shinder V, Stiller SB, Shimoni E, Wiedemann N, Geiger T, Schuldiner M (2015) Lam6 regulates the extent of contacts between organelles. Cell rep 12:7–14. doi:10.1016/j.celrep.2015.06.022
Ferreira IL, Ferreiro E, Schmidt J, Cardoso JM, Pereira CMF, Carvalho AL, Oliveira CR, Rego ACR (2015) Aβ and NMDAR activation cause mitochondrial dysfunction involving ER calcium release. Neurobiol Aging 36:680692. doi:10.1016/j.neurobiolaging.2014.09.006
Filadi R, Greotti E, Turacchio G, Luini A, Pozzan T, Pizzo P (2015) Mitofusin 2 ablation increases endoplasmic reticulum-mitochondria coupling. Proc Natl Acad Sci USA 112:81. doi:10.1073/pnas.1504880112
Fortin DL, Troyer MD, Nakamura K, Kubo S, Anthony MD, Edwards RH (2004) Lipid rafts mediate the synaptic localization of alpha-synuclein. J Neurosci 24:6715–6723. doi:10.1523/JNEUROSCI.1594-04.2004
Fransson S, Ruusala A, Aspenström P (2006) The atypical Rho GTPases Miro-1 and Miro-2 have essential roles in mitochondrial trafficking. Biochem Biophys Res Commun 344:500–510. doi:10.1016/j.bbrc.2006.03.163
Friedman JR, Lackner LL, West M, DiBenedetto JR, Nunnari J, Voeltz GK (2011) ER tubules mark sites of mitochondrial division. Science 334:358–362. doi:10.1126/science.1207385
Friedman JR, Webster BM, Mastronarde DN, Verhey KJ, Voeltz GK (2010) ER sliding dynamics and ER–mitochondrial contacts occur on acetylated microtubules. J Cell Biol. doi:10.1083/jcb.200911024
Fujimoto M, Hayashi T, Su T-P (2012) The role of cholesterol in the association of endoplasmic reticulum membranes with mitochondria. Biochem Biophys Res Commun 417:635–639. doi:10.1016/j.bbrc.2011.12.022
Giacomello M, Drago I, Bortolozzi M, Scorzeto M, Gianelle A, Pizzo P, Pozzan T (2010) Ca2+ hot spots on the mitochondrial surface are generated by Ca2+ mobilization from stores, but not by activation of store-operated Ca2+ channels. Mol Cell. doi:10.1016/j.molcel.2010.04.003
Golpich M, Amini E, Hemmati F, Ibrahim NM, Rahmani B, Mohamed Z, Raymond AA, Dargahi L, Ghasemi R, Ahmadiani A (2015) Glycogen synthase kinase-3 beta (GSK-3beta) signaling: implications for Parkinson’s disease. Pharmacol Res 97:16–26. doi:10.1016/j.phrs.2015.03.010
Guardia-Laguarta C, Area-Gomez E, Rüb C, Liu Y, Magrané J, Becker D, Voos W, Schon EA, Przedborski S (2014) α-Synuclein is localized to mitochondria-associated ER membranes. J Neurosci 34:249–259. doi:10.1523/jneurosci.2507-13.2014
Hailey DW, Rambold AS, Satpute-Krishnan P, Mitra K, Sougrat R, Kim PK, Lippincott-Schwartz J (2010) Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell. doi:10.1016/j.cell.2010.04.009
Hamasaki M, Furuta N, Matsuda A, Nezu A, Yamamoto A, Fujita N, Oomori H, Noda T, Haraguchi T, Hiraoka Y et al (2013) Autophagosomes form at ER-mitochondria contact sites. Nature 495:389–393. doi:10.1038/nature11910
Hayashi T, Fujimoto M (2010) Detergent-resistant microdomains determine the localization of sigma-1 receptors to the endoplasmic reticulum-mitochondria junction. Mol Pharmacol 77:517–528. doi:10.1124/mol.109.062539
Hayashi T, Su T-P (2007) Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca2+ signaling and cell survival. Cell. doi:10.1016/j.cell.2007.08.036
Hedskog L, Pinho CM, Filadi R, Rönnbäck A, Hertwig L, Wiehager B, Larssen P, Gellhaar S, Sandebring A, Westerlund M et al (2013) Modulation of the endoplasmic reticulum–mitochondria interface in Alzheimer’s disease and related models. Proc Natl Acad Sci 110:7916–7921. doi:10.1073/pnas.1300677110
Horner SM, Liu HM, Park HS, Briley J, Gale M Jr (2011) Mitochondrial-associated endoplasmic reticulum membranes (MAM) form innate immune synapses and are targeted by hepatitis C virus. Proc Natl Acad Sci USA 108:14590–14595. doi:10.1073/pnas.1110133108
Hubner CA, Kurth I (2014) Membrane-shaping disorders: a common pathway in axon degeneration. Brain 137:3109–3121. doi:10.1093/brain/awu287
Iwasawa R, Mahul-Mellier AL, Datler C, Pazarentzos E, Grimm S (2011) Fis1 and Bap31 bridge the mitochondria-ER interface to establish a platform for apoptosis induction. EMBO J 30:556–568. doi:10.1038/emboj.2010.346
Jin J, Li GJ, Davis J, Zhu D, Wang Y, Pan C, Zhang J (2007) Identification of novel proteins associated with both alpha-synuclein and DJ-1. Mol Cell Proteomics 6:845–859. doi:10.1074/mcp.M600182-MCP200
Joseph SK, Boehning D, Bokkala S, Watkins R, Widjaja J (1999) Biosynthesis of inositol trisphosphate receptors: selective association with the molecular chaperone calnexin. Biochem J 342(Pt 1):153–161
Kaufman RJ, Malhotra JD (2014) Calcium trafficking integrates endoplasmic reticulum function with mitochondrial bioenergetics. Biochim Biophys Acta 1843:2233–2239. doi:10.1016/j.bbamcr.2014.03.022
Kornak U, Mademan I, Schinke M, Voigt M, Krawitz P, Hecht J, Barvencik F, Schinke T, Gießelmann S, Beil TF et al (2014) Sensory neuropathy with bone destruction due to a mutation in the membrane-shaping atlastin GTPase 3. Brain 137:683–692. doi:10.1093/brain/awt357
Kornmann B, Currie E, Collins SR, Schuldiner M, Nunnari J, Weissman JS, Walter P (2009) An ER-mitochondria tethering complex revealed by a synthetic biology screen. Science 325:477–481. doi:10.1126/science.1175088
Kornmann B, Osman C, Walter P (2011) The conserved GTPase Gem1 regulates endoplasmic reticulum–mitochondria connections. Proc Natl Acad Sci 108:14151–14156. doi:10.1073/pnas.1111314108
Korobova F, Gauvin TJ, Higgs HN (2014) A role for myosin II in mammalian mitochondrial fission. Curr Biol. doi:10.1016/j.cub.2013.12.032
Korobova F, Ramabhadran V, Higgs HN (2013) An actin-dependent step in mitochondrial fission mediated by the ER-associated formin INF2. Science 339:464–467. doi:10.1126/science.1228360
Kremer A, Lippens S, Bartunkova S, Asselbergh B, Blanpain C, Fendrych M, Goossens A, Holt M, Janssens S, Krols M et al (2015) Developing 3D SEM in a broad biological context. J Microsci 259:80–96. doi:10.1111/jmi.12211
Lahiri S, Chao JT, Tavassoli S, Wong AKO, Choudhary V, Young BP, Loewen CJR, Prinz WA (2014) A conserved endoplasmic reticulum membrane protein complex (EMC) facilitates phospholipid transfer from the ER to mitochondria. PLoS Biol. doi:10.1371/journal.pbio.1001969
Lahiri S, Toulmay A, Prinz WA (2015) Membrane contact sites, gateways for lipid homeostasis. Curr Opin Cell Biol 33:82–87. doi:10.1016/j.ceb.2014.12.004
Lamb CA, Yoshimori T, Tooze SA (2013) The autophagosome: origins unknown, biogenesis complex. Nat Rev Mol Cell Biol 14:759–774. doi:10.1038/nrm3696
Lattante S, Rouleau GA, Kabashi E (2013) TARDBP and FUS mutations associated with amyotrophic lateral sclerosis: summary and update. Hum Mutat 34:812–826. doi:10.1002/humu.22319
Lim Y, Cho I-TT, Schoel LJ, Cho G, Golden JA (2015) Hereditary spastic paraplegia-linked reep1 modulates ER-mitochondria contacts. Ann Neurol. doi:10.1002/ana.24488
Liu S, Sawada T, Lee S, Yu W, Silverio G, Alapatt P, Millan I, Shen A, Saxton W, Kanao T et al (2012) Parkinson’s disease-associated kinase PINK1 regulates Miro protein level and axonal transport of mitochondria. PLoS Genet. doi:10.1371/journal.pgen.1002537
Llorens-Martín M, Jurado J, Hernández F, Avila J (2014) GSK-3β, a pivotal kinase in Alzheimer disease. Front Mol Neurosci 7:46. doi:10.3389/fnmol.2014.00046
Luty AA, Kwok JBJ, Dobson-Stone C, Loy CT, Coupland KG, Karlström H, Sobow T, Tchorzewska J, Maruszak A, Barcikowska M et al (2010) Sigma nonopioid intracellular receptor 1 mutations cause frontotemporal lobar degeneration—motor neuron disease. Ann Neurol 68:639–649. doi:10.1002/ana.22274
Lynes EM, Bui M, Yap MC, Benson MD, Schneider B, Ellgaard L, Berthiaume LG, Simmen T (2012) Palmitoylated TMX and calnexin target to the mitochondria-associated membrane. EMBO J 31:457–470. doi:10.1038/emboj.2011.384
Macaskill AF, Rinholm JE, Twelvetrees AE, Arancibia-Carcamo IL, Muir J, Fransson A, Aspenstrom P, Attwell D, Kittler JT (2009) Miro1 is a calcium sensor for glutamate receptor-dependent localization of mitochondria at synapses. Neuron 61:541–555. doi:10.1016/j.neuron.2009.01.030
Manor U, Bartholomew S, Golani G, Christenson E, Kozlov M, Higgs H, Spudich J, Lippincott-Schwartz J (2015) A mitochondria-anchored isoform of the actin-nucleating Spire protein regulates mitochondrial division. eLife 4. doi:10.7554/eLife.08828
Mao K, Wang K, Liu X, Klionsky DJ (2013) The Scaffold protein Atg11 recruits fission machinery to drive selective mitochondria degradation by autophagy. Dev Cell. doi:10.1016/j.devcel.2013.05.024
Marchi S, Pinton P (2014) The mitochondrial calcium uniporter complex: molecular components, structure and physiopathological implications. J Physiol 592:829–839. doi:10.1113/jphysiol.2013.268235
Martinez-Vicente M (2015) Autophagy in neurodegenerative diseases: from pathogenic dysfunction to therapeutic modulation. Semin Cell Dev Biol 40:115–126. doi:10.1016/j.semcdb.2015.03.005
Mironov SL, Symonchuk N (2006) ER vesicles and mitochondria move and communicate at synapses. J Cell Sci 119:4926–4934. doi:10.1242/jcs.03254
Misko A, Jiang S, Wegorzewska I, Milbrandt J, Baloh RH (2010) Mitofusin 2 is necessary for transport of axonal mitochondria and interacts with the Miro/Milton complex. J Neurosci 30:4232–4240. doi:10.1523/JNEUROSCI.6248-09.2010
Misko AL, Sasaki Y, Tuck E, Milbrandt J, Baloh RH (2012) Mitofusin2 mutations disrupt axonal mitochondrial positioning and promote axon degeneration. J Neurosci 32:4145–4155. doi:10.1523/JNEUROSCI.6338-11.2012
Mórotz GM, De Vos KJ, Vagnoni A, Ackerley S, Shaw CE, Miller CC (2012) Amyotrophic lateral sclerosis-associated mutant VAPBP56S perturbs calcium homeostasis to disrupt axonal transport of mitochondria. Hum Mol Genet 21:1979–1988. doi:10.1093/hmg/dds011
Murley A, Sarsam RD, Toulmay A, Yamada J, Prinz WA, Nunnari J (2015) Ltc1 is an ER-localized sterol transporter and a component of ER-mitochondria and ER-vacuole contacts. J Cell Biol 209:539–548. doi:10.1083/jcb.201502033
Myhill N, Lynes EM, Nanji JA, Blagoveshchenskaya AD, Fei H, Carmine Simmen K, Cooper TJ, Thomas G, Simmen T (2008) The subcellular distribution of calnexin is mediated by PACS-2. Mol Biol Cell 19:2777–2788. doi:10.1091/mbc.E07-10-0995
Nguyen TT, Oh SS, Weaver D, Lewandowska A, Maxfield D, Schuler M-HH, Smith NK, Macfarlane J, Saunders G, Palmer CA et al (2014) Loss of Miro1-directed mitochondrial movement results in a novel murine model for neuron disease. Proc Natl Acad Sci USA 111:40. doi:10.1073/pnas.1402449111
Nishimura AL, Mitne-Neto M, Silva HC, Richieri-Costa A, Middleton S, Cascio D, Kok F, Oliveira JR, Gillingwater T, Webb J et al (2004) A mutation in the vesicle-trafficking protein VAPB causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am J Hum Genet 75:822–831. doi:10.1086/425287
Ottolini D, Calì T, Negro A, Brini M (2013) The Parkinson disease-related protein DJ-1 counteracts mitochondrial impairment induced by the tumour suppressor protein p53 by enhancing endoplasmic reticulum–mitochondria tethering. Hum Mol Genet 22:2152–2168. doi:10.1093/hmg/ddt068
Panov AV, Gutekunst C-AA, Leavitt BR, Hayden MR, Burke JR, Strittmatter WJ, Greenamyre JT (2002) Early mitochondrial calcium defects in Huntington’s disease are a direct effect of polyglutamines. Nat Neurosci 5:731–736. doi:10.1038/nn884
Pivovarova NB, Pozzo-Miller LD, Hongpaisan J, Andrews SB (2002) Correlated calcium uptake and release by mitochondria and endoplasmic reticulum of CA3 hippocampal dendrites after afferent synaptic stimulation. J Neurosci 22:10653–10661
Prause J, Goswami A, Katona I, Roos A, Schnizler M, Bushuven E, Dreier A, Buchkremer S, Johann S, Beyer C et al (2013) Altered localization, abnormal modification and loss of function of Sigma receptor-1 in amyotrophic lateral sclerosis. Hum Mol Genet 22:1581–1600. doi:10.1093/hmg/ddt008
Prudent J, Zunino R, Sugiura A, Mattie S, Shore GC, McBride HM (2015) MAPL sumoylation of drp1 stabilizes an ER/mitochondrial platform required for cell death. Mol Cell 59:941–955. doi:10.1016/j.molcel.2015.08.001
Rizzuto R, De Stefani D, Raffaello A, Mammucari C (2012) Mitochondria as sensors and regulators of calcium signalling. Nat Rev Mol Cell Biol 13:566–578. doi:10.1038/nrm3412
Rizzuto R, Pinton P, Carrington W, Fay FS, Fogarty KE, Lifshitz LM, Tuft RA, Pozzan T (1998) Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 280:1763–1766. doi:10.1126/science.280.5370.1763
Rizzuto R, Simpson AW, Brini M, Pozzan T (1992) Rapid changes of mitochondrial Ca2+ revealed by specifically targeted recombinant aequorin. Nature 358:325–327. doi:10.1038/358325a0
Roderick HL, Lechleiter JD, Camacho P (2000) Cytosolic phosphorylation of calnexin controls intracellular Ca2+ oscillations via an interaction with SERCA2b. J Cell Biol 149:1235–1247. doi:10.1083/jcb.149.6.1235
Sano R, Annunziata I, Patterson A, Moshiach S, Gomero E, Opferman J, Forte M, d’Azzo A (2009) GM1-ganglioside accumulation at the mitochondria-associated ER membranes links ER stress to Ca(2+)-dependent mitochondrial apoptosis. Mol Cell 36:500–511. doi:10.1016/j.molcel.2009.10.021
Saotome M, Safiulina D, Szabadkai G, Das S, Fransson A, Aspenstrom P, Rizzuto R, Hajnóczky G (2008) Bidirectional Ca2+ -dependent control of mitochondrial dynamics by the Miro GTPase. Proc Natl Acad Sci USA 105:20728–20733. doi:10.1073/pnas.0808953105
Schon EA, Area-Gomez E (2013) Mitochondria-associated ER membranes in Alzheimer disease. Mol Cell Neurosci 55:26–36. doi:10.1016/j.mcn.2012.07.011
Schon EA, Przedborski S (2011) Mitochondria: the next (neurode)generation. Neuron 70:1033–1053. doi:10.1016/j.neuron.2011.06.003
Simmen T, Aslan JE, Blagoveshchenskaya AD, Thomas L, Wan L, Xiang Y, Feliciangeli SF, Hung CH, Crump CM, Thomas G (2005) PACS-2 controls endoplasmic reticulum–mitochondria communication and Bid-mediated apoptosis. EMBO J 24:717–729. doi:10.1038/sj.emboj.7600559
Simmen T, Lynes EM, Gesson K, Thomas G (2010) Oxidative protein folding in the endoplasmic reticulum: tight links to the mitochondria-associated membrane (MAM). Biochim Biophys Acta. doi:10.1016/j.bbamem.2010.04.009
Stoica R, De Vos KJ, Paillusson S, Mueller S, Sancho RM, Lau K-FF, Vizcay-Barrena G, Lin W-LL, Xu Y-FF, Lewis J et al (2014) ER-mitochondria associations are regulated by the VAPB-PTPIP51 interaction and are disrupted by ALS/FTD-associated TDP-43. Nat Commun 5:3996. doi:10.1038/ncomms4996
Sugiura A, Nagashima S, Tokuyama T, Amo T (2013) MITOL regulates endoplasmic reticulum-mitochondria contacts via mitofusin2. Mol Cell. doi:10.1016/j.molcel.2013.04.023
Sutendra G, Dromparis P, Wright P, Bonnet S, Haromy A, Hao Z, McMurtry MS, Michalak M, Vance JE, Sessa WC et al (2011) The role of Nogo and the mitochondria-endoplasmic reticulum unit in pulmonary hypertension. Sci Transl Med. doi:10.1126/scitranslmed.3002194
Szabadkai G, Bianchi K, Várnai P, De Stefani D, Wieckowski MR, Cavagna D, Nagy AI, Balla T, Rizzuto R (2006) Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J Cell Biol. doi:10.1083/jcb.200608073
Tambini MD, Pera M, Kanter E, Yang H, Guardia-Laguarta C, Holtzman D, Sulzer D, Area-Gomez E, Schon EA (2015) ApoE4 upregulates the activity of mitochondria-associated ER membranes. EMBO Rep. doi:10.15252/embr.201540614
Tanaka A, Cleland MM, Xu S, Narendra DP, Suen DF, Karbowski M, Youle RJ (2010) Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol 191:1367–1380. doi:10.1083/jcb.201007013
Timmerman V, Clowes VE, Reid E (2013) Overlapping molecular pathological themes link Charcot–Marie–Tooth neuropathies and hereditary spastic paraplegias. Exp Neurol 246:14–25. doi:10.1016/j.expneurol.2012.01.010
Tubbs E, Theurey P, Vial G, Bendridi N, Bravard A, Chauvin M-A, Ji-Cao J, Zoulim F, Bartosch B, Ovize M et al (2014) Mitochondria-associated endoplasmic reticulum membrane (MAM) integrity is required for insulin signaling and is implicated in hepatic insulin resistance. Diabetes 63:3279–3294. doi:10.2337/db13-1751
Twig G, Elorza A, Molina AJA, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G et al (2008) Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J 27:433–446. doi:10.1038/sj.emboj.7601963
Urano Y, Hayashi I, Isoo N, Reid PC, Shibasaki Y, Noguchi N, Tomita T, Iwatsubo T, Hamakubo T, Kodama T (2005) Association of active gamma-secretase complex with lipid rafts. J Lipid Res 46:904–912. doi:10.1194/jlr.M400333-JLR200
Van Laar VS, Roy N, Liu A, Rajprohat S, Arnold B, Dukes AA, Holbein CD, Berman SB (2014) Glutamate excitotoxicity in neurons triggers mitochondrial and endoplasmic reticulum accumulation of Parkin, and in the presence of N-acetyl cysteine, mitophagy. Neurobiol Dis. doi:10.1016/j.nbd.2014.11.015
van Vliet AR, Verfaillie T, Agostinis P (2014) New functions of mitochondria associated membranes in cellular signaling. Biochim Biophys Acta 1843:2253–2262. doi:10.1016/j.bbamcr.2014.03.009
Vance JE (2014) MAM (mitochondria-associated membranes) in mammalian cells: lipids and beyond. Biochim Biophys Acta. doi:10.1016/j.bbalip.2013.11.014
Vance JE (1990) Phospholipid synthesis in a membrane fraction associated with mitochondria. J Biol Chem 265:7248–7256
Vishnu N, Jadoon Khan M, Karsten F, Groschner LN, Waldeck-Weiermair M, Rost R, Hallström S, Imamura H, Graier WF, Malli R (2014) ATP increases within the lumen of the endoplasmic reticulum upon intracellular Ca2+ release. Mol Biol Cell 25:368–379. doi:10.1091/mbc.E13-07-0433
Voelker DR (1989) Phosphatidylserine translocation to the mitochondrion is an ATP-dependent process in permeabilized animal cells. Proc Natl Acad Sci USA 86:9921–9925
Wang X, Winter D, Ashrafi G, Schlehe J, Wong YL, Selkoe D, Rice S, Steen J, LaVoie MJ, Schwarz TL (2011) PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell 147:893–906. doi:10.1016/j.cell.2011.10.018
Wiley SE, Andreyev AY, Divakaruni AS, Karisch R, Perkins G, Wall EA, van der Geer P, Chen Y-FF, Tsai T-FF, Simon MI et al (2013) Wolfram Syndrome protein, Miner1, regulates sulphydryl redox status, the unfolded protein response, and Ca2+ homeostasis. EMBO Mol Med 5:904–918. doi:10.1002/emmm.201201429
Yang J-Y, Yang WY (2013) Bit-by-bit autophagic removal of parkin-labelled mitochondria. Nat Commun. doi:10.1038/ncomms3428
Zhu P-PP, Denton KR, Pierson TM, Li X-JJ, Blackstone C (2014) Pharmacologic rescue of axon growth defects in a human iPSC model of hereditary spastic paraplegia SPG3A. Hum Mol Genet 23:5638–5648. doi:10.1093/hmg/ddu280
Acknowledgments
M. K. is supported by a PhD fellowship of the Fund for Scientific Research (FWO-Flanders, Belgium). The research of V.T. is supported by the FWO, University of Antwerp, the Association Belge contre les Maladies Neuro-Musculaires (ABMM) and the EU FP7/2007–2013 under Grant Agreement Number 2012-305121 (NEUROMICS). The research of S. J. is supported by the FWO, University of Ghent and GROUP-ID. The Zeiss Auriga was acquired through a clem grant from Minister Ingrid Lieten to the VIB Bio Imaging Core. Special thanks to Sonia Bartunkova for technical support and Benjamin Pavie for advise in image processing.
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
401_2015_1528_MOESM1_ESM.avi
Supplementary movie 1: 3D morphology of ER-mitochondria contact sites. The same dataset and reconstruction as described in Fig. 1 are shown. A part of the original electron micrographs obtained using the Zeiss Auriga Crossbeam microscope is shown by moving along the Z-axis. Next, the volume is shown by the reverse movement along the Z-axis and the simultaneous rotation around the X-axis while the rendered reconstruction of mitochondria (green) and their associated endoplasmic reticulum (ER) (magenta) appear. Next, an XZ-projected image appears while zooming in on one ER-mitochondria contact site. This contact site is shown in 3D as the image stack is digitally resliced along its Y-axis. IMOD was used for image registration, manual segmentation, 3D rendering and animation. (AVI 102184 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Krols, M., van Isterdael, G., Asselbergh, B. et al. Mitochondria-associated membranes as hubs for neurodegeneration. Acta Neuropathol 131, 505–523 (2016). https://doi.org/10.1007/s00401-015-1528-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-015-1528-7