
Abstract
The intermediate filament protein desmin is an essential component of the extra-sarcomeric cytoskeleton in muscle cells. This three-dimensional filamentous framework exerts central roles in the structural and functional alignment and anchorage of myofibrils, the positioning of cell organelles and signaling events. Mutations of the human desmin gene on chromosome 2q35 cause autosomal dominant, autosomal recessive, and sporadic myopathies and/or cardiomyopathies with marked phenotypic variability. The disease onset ranges from childhood to late adulthood. The clinical course is progressive and no specific treatment is currently available for this severely disabling disease. The muscle pathology is characterized by desmin-positive protein aggregates and degenerative changes of the myofibrillar apparatus. The molecular pathophysiology of desminopathies is a complex, multilevel issue. In addition to direct effects on the formation and maintenance of the extra-sarcomeric intermediate filament network, mutant desmin affects essential protein interactions, cell signaling cascades, mitochondrial functions, and protein quality control mechanisms. This review summarizes the currently available data on the epidemiology, clinical phenotypes, myopathology, and genetics of desminopathies. In addition, this work provides an overview on the expression, filament formation processes, biomechanical properties, post-translational modifications, interaction partners, subcellular localization, and functions of wild-type and mutant desmin as well as desmin-related cell and animal models.
Similar content being viewed by others

General introduction
Desminopathies (synonyms: desmin-related myopathy, desmin myopathy, desmin storage myopathy, and others [166]) belong to the clinically and genetically heterogeneous group of myofibrillar myopathies (MFM), which are morphologically characterized by the presence of desmin-positive protein aggregates and degenerative changes of the myofibrillar apparatus [160, 164]. Since the first description of desmin and αB-crystallin mutations causing MFM in 1998 [58, 187], an increasing number of MFM disease genes coding for sarcomeric and extra-sarcomeric proteins, i.e., BAG-3, FHL1, filamin-C, myotilin, plectin, titin, and ZASP, have been identified [126, 138, 160, 164]. In addition, protein aggregate myopathies due to mutations in the DNAJB6 and VCP genes share part of their morphological features with MFM [84, 151]. The precise molecular pathways leading from an individual MFM gene defect to a shared myopathological disease manifestation are still unclear.
Desmin is a member of the intermediate filament (IF) protein gene family, which comprises 70 members [78]. Up to now, the IF gene family represents one of the most highly mutated groups of related genes in the human genome, accounting for at least 94 different disease entities [132, 176]. IF proteins are expressed in a tissue- and development-specific manner, e.g., keratins in epithelial tissues, GFAP in astrocytes, and neurofilament proteins in neurons. As a particular unique property, the IF gene family harbors three genes that code for proteins localizing to the nuclear envelope within the cell nucleus, i.e., lamin A and its smaller splice form lamin C, lamin B1, and lamin B2. According to the degree of sequence identity, IF proteins have been grouped into six sequence homology classes (SHC): acidic keratins (SHC I); basic keratins (SHC II); desmin, vimentin and other mesenchymal IF proteins such as GFAP (SHC III); neurofilament proteins (SHC IV); lamins (SHC V); and an orphan group harboring the lens-specific IF proteins phakinin and filensin [72]. Human IF-related diseases range from skin blistering (keratins), Alexander’s disease (GFAP), Charcot-Marie-Tooth disease (neurofilament proteins), and Hutchinson-Gilbert progeria (lamin A/C) to cataracts (phakinin). Notably, and subject of this review, mutations of desmin, the major class III IF protein in striated and smooth muscle cells, causes progressive myopathy, cardiomyopathy, cardiac conduction defects, and arrhythmias [160, 183]. Information on the expression, localization, interaction, and function of IFs in general and desmin in particular is a prerequisite for the understanding of human desminopathies. In the present article we will summarize the essential data on desmin and desminopathies derived from numerous clinical and molecular studies.
Clinical phenotypes
Epidemiology
Since detailed epidemiological studies on MFM are not available, the incidence and prevalence of desmin myopathy and/or cardiomyopathy are currently unclear. Interpretation of the thus far published data allows the assumption that desminopathies fulfill the definition of a rare disease with no more than 5 affected individuals in 10,000. In a study on the prevalence of desmin mutations in a cohort of 116 families and 309 additional patients with pure dilatative cardiomyopathy, desmin mutations accounted for up to 2 % of disease manifestation [178]. Desmin mutations also seem to be one of the more frequently encountered gene defects in the MFM group. In a cohort of 53 patients from 35 Spanish MFM families, myotilin mutations were the predominant cause affecting 18 families followed by desmin mutations in 11 families [130]. In earlier studies reporting on the Mayo MFM cohort of 63 patients, 6 carried myotilin and 4 carried desmin mutations [165, 166]. Desminopathies have been reported in diverse ethnic groups and affect both female and male patients. Gender effects have been reported in two studies, in which male heterozygous mutation carriers were more prone to cardiac disease manifestations [5, 183]. The disease manifestation is highly variable with an age of onset ranging from the 1st to the 8th decade of life. In rare recessive forms the disease manifests in the 1st decade of life [31, 58, 121, 140]. In the more frequently encountered familial and sporadic cases due to heterozygous desmin mutations, a disease onset ranging from the 2nd to the 4th decade of life has been reported in the majority of patients [160, 183].
Skeletal muscle disease
Initially, desminopathies have been associated with a progressive distal myopathy phenotype starting in the lower legs. However, subsequent studies reported the association between desmin mutations and limb girdle, scapuloperoneal, and generalized myopathy phenotypes [9, 35, 191]. A meta-analysis based on the interpretation of published data from 159 patients carrying 40 different heterozygous desmin mutations provided highly valuable insights into this complex issue [183]. Signs of combined distal and proximal muscular weakness were found in two thirds (67 %; 71/106) of mutation carriers, whereas true distal and proximal myopathy phenotypes were only present in 27 % (29/106) and 6 % (6/106), respectively. In this study, 74 % (110/148) of carriers had signs of skeletal muscle disease. Isolated skeletal muscle disease was reported in 22 % (31/141) of carriers. A combination of signs of skeletal muscle and cardiac pathology was found in 49 % (67/137).
Creatine kinase (CK) levels in desmin mutation carriers are of limited diagnostic value; 57 % (62/109) of mutation carriers had elevated CK levels (91 % displayed a ≤4-fold increase). Remarkably, 30 % (25/83) of patients with manifest skeletal muscle disease were reported to have normal CK levels [183]. Needle electromyography (EMG) typically reveals a myopathic pattern with short duration, polyphasic, and low amplitude motor unit potentials. In addition, positive sharp waves, fibrillation potentials, and pseudomyotonic/myotonic discharges have frequently been documented in desminopathy patients [160]. Sensory and motor nerve conduction studies usually give normal results [59]. Muscle MRI studies pointed out that signal alterations in the gluteus maximus, semitendinosus, sartorius, gracilis, and peroneal muscles are early and predominant signs of desminopathies [48, 156].
Cardiac disease
Cardiac disease manifestations in desminopathies, which may precede, coincide with, or succeed skeletal muscle weakness, comprise true cardiomyopathy as well as various forms of cardiac conduction defects (CCD) and arrhythmias [92, 173, 178, 183, 185]. The above-cited meta-analysis revealed the presence of cardiological signs in 74 % (105/141) of desmin mutation carriers [183]. Isolated cardiological signs were reported in 22 % (34/152) of carriers. Out of 67 patients with verified cardiomyopathy (49 %; 67/138), 23 were classified as dilatative, 18 as unspecified, 16 as restrictive, 8 as hypertrophic, and 2 as arrhythmogenic right ventricular cardiomyopathy. Desmin mutations frequently lead to clinically symptomatic and asymptomatic ECG abnormalities (62 %; 83/133). In this group, CCD seems far more frequent (39 %; 52/133) than isolated arrhythmias (5 %; 6/133). A combination of both was found in 19 % (25/133) of carriers. A more detailed characterization of 77 patients with CCD revealed that atrioventricular block (47) and right bundle branch block (14) were the most prevalent manifestations. In 31 patients with arrhythmias, atrial fibrillation (9), ventricular premature beats (8), and ventricular tachycardia (7) were the most frequently detected ECG abnormalities. Thus, a basic electrophysiological workup should include a 12-lead surface ECG and a 24-h Holter ECG. The cardiac function is routinely assessed by transthoracic echocardiography. However, cardiac MRI has been proposed as a more sensitive diagnostic tool to detect focal cardiac pathology in early and clinically asymptomatic stages of desminopathy [173].
Pulmonary and miscellaneous disease manifestations
Patients with desmin mutations are at risk to develop respiratory problems. In the desminopathy meta-analysis 26 % (29/110) of carriers were reported to suffer from respiratory insufficiency [183]. Thus, blood gas analysis and spirometry are mandatory in patients with desminopathies. Cataracts, swallowing difficulties, intestinal pseudo-obstruction, and repetitive episodes of diarrhea and constipation have been reported as miscellaneous or putative disease-related symptoms [6, 58, 129, 183].
Disease progression and mortality
Desmin myopathies and cardiomyopathies are characterized by a progressive course and may change their initial clinical presentation. In a case report, the transformation of an initially hypertrophic cardiomyopathy into a restrictive and finally dilated cardiomyopathy has been documented [65]. A further study reported on a 10-year follow-up of 28 patients from 19 families with desmin mutations [190]. In 11 patients, primary skeletal muscle involvement was followed by cardiac disease after 6.3 ± 3.5 years, whereas in 16 patients with a primary cardiac disease manifestation skeletal muscle problems occurred after 6.8 ± 4.1 years. Out of the latter group, six patients presented with major cardiac complications. A progression from mild conduction defects to high degree conduction blocks requiring permanent pacing was observed in 8 out of 19 patients. In this cohort of 28 patients, 5 died at a mean age of 58.0 ± 6.5 years, accounting for a mortality rate of 17.8 % [190]. In the desminopathy meta-analysis, however, a mortality rate of 26 % (27/104) of desmin mutation carriers with a mean age 49 ± 9.3 years was reported [183]. Documented causes of death in both studies were sudden cardiac death, heart failure, respiratory insufficiency, chest infection, and iatrogenic complications of cardiac treatment. Both studies conclusively underline that the cardiac disease manifestation is the major cause of premature death in desminopathies.
Muscle biopsy findings
Light microscopy
Diagnostic skeletal muscle biopsies from patients with desminopathies usually show mild to severe signs of a degenerative myopathy with rounding of muscle fibers, fiber splitting, internalization of myonuclei, and increased connective and fat tissue. Pathological protein aggregates, the hallmark of MFM, generally emerge as subsarcolemmal and/or sarcoplasmic inclusions. In addition, sarcoplasmic bodies as well as rimmed and non-rimmed vacuoles may be present. The typical pathology of MFM is best visualized in H&E and modified Gomori trichrome (GT) stains (Fig. 1a, b). Enzymatic stains (NADH-TR, SDH, and COX) may show further characteristic oxidative enzyme and mitochondrial abnormalities comprising rubbed-out fibers and core-like lesions (Fig. 1c). The stage of disease progression in individual muscles often mirrors the severity of the observed myopathological changes. However, one should keep in mind that the myopathological picture of desminopathies is highly variable. The myopathological findings in genetically proven desminopathies range from no overt pathology over subtle myopathic changes with or without pathological protein aggregates to the picture of a vacuolar myopathy [35, 160, 161].

Protein aggregation pathology and mitochondrial abnormalities in desminopathies. Arrows and double arrows in a and b denote pathological protein aggregates in the sarcoplasm and in the subsarcolemmal region, respectively. Arrowhead and double arrowhead in c highlight a rubbed-out lesion and a core-like lesion, respectively
Immunodetection
Desmin immunostaining is mandatory to depict desmin-positive pathological protein aggregates in the subsarcolemmal and/or sarcoplasmic region. In addition to the characteristic immunoreactivity to desmin (Fig. 2a), the pathological protein aggregates are positively stained by a wide variety of antibodies directed against cytoskeletal proteins (e.g., dystrophin, F-actin, filamin C, myotilin, plectin, synemin), heat shock proteins (e.g., αB-crystallin, Hsp27), ubiquitin, Alzheimer-related proteins (e.g., β-APP), and cyclin-dependent kinases (e.g., CDK2, p21). Out of this long list, stains for αB-crystallin (Fig. 2b), filamin-C, and myotilin are sensitive diagnostic tools to depict pathological protein aggregation in desminopathies and other forms of MFM [161, 166].

Indirect immunofluorescence labeling of desmin and αB-crystallin in a desminopathy. Note the presence of sarcoplasmic and subsarcolemmal pathological protein aggregates
Electron microscopy
Granulofilamentous material in the subsarcolemmal region and/or between neighboring myofibrils is the classical ultrastructural hallmark of desminopathies (Fig. 3). However, granulofilamentous material is not specific for this disease entity as it has been described in other forms of MFM, in particular in αB crystallinopathies. Other features of pathological protein aggregation are cytoplasmic bodies and autophagic vacuoles. These changes are found in conjunction with signs of myofibrillar degeneration comprising Z-disc alterations (streaming, irregularities, loss, and rods), myofibrillar remnants, and core and core-like lesions. Typical signs of concomitant mitochondrial pathology are areas with accumulation or depletion of mitochondria with normal or abnormal morphology.

Electron microscopy findings in desminopathies. Asterisks denote the presence of granulofilamentous material in the subsarcolemmal region. White arrows depict electron-dense granular deposits; black arrows highlight filamentous material. ECM extracellular matrix, M mitochondrion
Cardiac pathology
Desmin-positive protein aggregates as well as granulofilamentous and electron-dense amorphous materials are also the morphological hallmarks of desmin cardiomyopathies. Pathological aggregates have been demonstrated in subsarcolemmal, intermyofibrillar, and perinuclear regions. Further reported pathological findings were myocyte hypertrophy, disarray of the myocytes, mis-shaped myonuclei, cytoplasmic vacuolar degeneration, focally lysed myofibrils, various degrees of diffuse interstitial fibrosis, and mitochondrial abnormalities [3, 4, 20]. With respect to CDC and arrhythmias, pathological changes were noted in the cardiac conduction system of two autopsy cases. In one, calcifications at the bundle of His and calcium deposits at the left and right bundle branches were noted [199], whereas in the other extensive fibrosis in the terminal portion of the branching bundle and the initial segments of the left and right bundles were described [20]. The shape of intercalated discs has been reported to be abnormal with convoluted, elongated, and zigzag patterns [185]. Furthermore, decreased immunoreactivities of the desmosomal proteins desmoplakin and plakophilin-2 and the ventricular gap junctional protein connexin-43 have been described [133]. Remarkably, smooth muscle cells of cardiac blood vessels seem not to contain desmin-positive aggregates [4]. As in skeletal muscle biopsy, the extent of typical pathology is highly variable in cardiac specimens from desminopathies.
Differential diagnosis
The differential diagnosis of desminopathies is complex and depends on the initial clinical disease presentation. Patients with a highly indicative phenotype of desminopathy (combined skeletal muscle and cardiac symptoms, no extra-muscular signs, autosomal dominant inheritance, disease onset between the 2nd and 4th decade of life) represent only a minority. In cases with isolated cardiac problems, a broad spectrum of acquired and hereditary conditions has to be taken into consideration. In cases of progressive skeletal muscle weakness without cardiac involvement, the differential diagnosis ranges from primary distal myopathies, limb girdle muscular dystrophies, and scapuloperoneal syndromes to generalized myopathies. A diagnostic muscle biopsy in these cases often provides the first clue to the diagnosis of MFM. Careful interpretation of the clinical data, including sex, age of onset, mode of inheritance, and presence or absence of extramuscular signs such as cataracts (αB-crystallinopathy), early respiratory failure (titinopathy), blistering skin (plectinopathy), and rigid spine and scoliosis (FHL1opathy, BAG-3opathy) is essential to differentiate specific MFM subtypes. In MFM patients with a disease onset beyond the 4th decade of life, mutations in genes coding for myotilin, ZASP, and filamin-C should initially be considered [160, 164].
Genetics of desminopathies
Human desmin gene
The human desmin gene (DES) on chromosome 2q35 is a single copy gene that spans over a length of approximately 8.3 kb and comprises nine exons coding for a 470-amino acid protein with a molecular weight of 53.5 kDa [106]. DES belongs to a gene cluster further comprising APEG1 (synonym SPEG or striated muscle preferentially expressed protein kinase) and CHPF. This gene cluster is most likely regulated by the DES 5′ locus control region (LCR), which has been identified 9–18 kb upstream of DES [177]. The currently available information on the epigenetic regulation of DES is still limited. The DES promoter contains a CpG-island also covering the DES transcription start site and exon 1. It has been described that the DES promoter is non-methylated regardless of its expression status. The expression of desmin in muscle cells seems to be activated by acetylation of histones H3 and H4 as well as methylation of histone H3 at lysine residue 4 (H3K4me2 and me3) around the transcription start site of DES. In non-muscle cells, the DES promoter is silenced by methylation of histone H3 lysine residue 27 (H3K27me3) [108].
Desmin mutation spectrum
Disease-causing mutations spread over the entire DES gene. They significantly cluster in exon 6, which encodes the C-terminal half of the coil 2 domain (Fig. 4). Mutations in this domain have primarily been associated with a skeletal muscle phenotype, whereas mutations residing in the head and tail domains of the desmin protein seem more frequently associated with a cardiac phenotype [183]. As of June 2012, 67 disease-causing mutations of the DES gene have been published. These mutations may lead to the expression of 61 different mutant forms of desmin (Table 1).

Structural organization of the human desmin molecule and molecular model of its dimeric rod domain. a The black boxes represent α-helical segments designated “coil.” Segments of unknown structure connecting coil 1A and coil 1B as well as coil 1B and coil 2 are termed linkers L1 and L12, respectively. Non-structured amino- (“head”) and carboxy- (“tail”) terminal domains are depicted as colored bars. Numbers indicate the amino acid position of the domain borders. b The molecular model of the dimeric desmin coiled coil domain is based on its high structural homology to the corresponding vimentin domain [33]. The three α-helical segments (coil 1A, coil 1B and coil 2) are shown as black ribbons. The linkers L1 and L12 are in grey. The first segment (coil 1A) has only a weak tendency to form coiled coils [33]. Locations of pathogenic mutations are mapped in orange (missense mutations), purple (deletions), and cyan (truncations). For clarity, mutation sites in only one chain of the dimer are marked. The mutations within the unstructured head and tail domains are also listed; see also Table 1
Autosomal dominant inheritance
The vast majority of familial desminopathies follow an autosomal dominant mode of inheritance. The most frequent DES mutations are missense mutations leading to single amino acid substitutions. Splice site mutations causing the loss of exon 3 (p.Asp214_Glu245del), small in-frame deletions of one, three or seven codons, and frame shift mutations, which may lead to the expression of truncated desmin protein species, have been reported only in a small number of patients (Table 1). For the p.Arg350Pro mutation, the most frequently encountered pathogenic desmin missense mutation in Germany, a founder allele has been established [191].
Autosomal recessive inheritance
Five families with an autosomal recessive mode of inheritance have been published thus far. In one family, the homozygous Arg16Cys missense mutation was observed [4]. In two families, a small in-frame deletion has been identified leading to p.Arg173_Glu179del [121, 140]. In another family, a 22 base pair deletion in exon 6 causing a premature stop codon associated with a virtually complete lack of desmin protein has been reported [31]. In the fifth family, the disease manifestation has been attributed to a compound heterozygote DES mutation (p.Ala360Pro/p.Asn393Ile [58]) (Table 1).
Sporadic forms
In addition to these familial cases, an increasing number of sporadic desminopathies have been published. These sporadic disease manifestations are due to missense, splice site, or frame-shift mutations (Table 1).
Desmin protein
Protein structure and filament assembly
Human desmin is a 470-amino acid protein ([79, 153]; UniProtKB/Swiss-Prot database entry P17661) with a calculated molecular weight of 53.5 kDa. Like all IF proteins, it is fibrous in nature, exhibiting a tripartite structure with a central, mostly α-helical domain of conserved size, i.e., ~45 nm. This so-called “rod” is flanked by non-α-helical amino-terminal (“head”) and carboxy-terminal (“tail”) domains of highly varying amino acid number [73] (Fig. 4a). The “rod” domain comprises two continuous α-helical segments, coil 1 and coil 2, which are connected by a “linker” (L12) of unknown structure. The current structural model of desmin is derived from recent structural analyses of the closely related class III IF protein vimentin [33] (Fig. 4b). Coil 1 consists of two α-helical subdomains, i.e., coil 1A and coil 1B. They are separated by linker L1, which could also accommodate α-helical conformation. While coil 1B forms a stable dimeric coiled coil, the coil 1A segment has only a weak coiled-coil forming capacity. In addition, the current model omits the previously used separation into coil 2A and 2B domains, but demonstrates it to be a continuous α-helix [123].
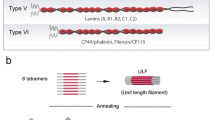
Desmin is a highly insoluble protein and hence it can be kept in solution as a monomer and purified only under highly denaturing conditions as provided by buffers containing 8 M urea. For assembly, it is usually first re-natured by dialysis into buffers of low salt such as 2 mM sodium phosphate (pH 7.5). The assembly process starts actually during the course of re-naturation, and two molecules dimerize by coiled-coil formation of the central α-helical rod domains of two desmin monomers in parallel orientation [77]. A so-called heptad repeat pattern, which is characteristic for a coiled-coil structure [26], drives the supercoiling of the two helices, yielding the dimeric, elongated structure. Two of these coiled-coil dimers further associate in a half-staggered, anti-parallel fashion to a tetramer of ~60 nm length [77, 107] (Fig. 5a). These tetramers are able to spontaneously assemble into highly ordered long filaments upon increase of the ionic strength to physiological values. The assembly process can be described to occur in three phases: (1) The lateral parallel assembly of tetramers yielding full-width, 60-nm-long filaments that have been termed unit-length filaments or ULFs [71, 77] (Fig. 5b). (2) The formation of extended intermediate filaments by serial longitudinal annealing of ULFs and further longitudinal annealing of short filaments [71] (Fig. 5c1,c2,c3,d). (3) After a few minutes of assembly, filaments undergo a final maturation step characterized by a radial compaction process [76, 79].

Schematic model of cytoplasmic IF assembly. a Two dimers associate in an anti-parallel, half-staggered fashion by overlap of the two coiled coils via their coil1, i.e., coil 1A and coil 1B (brown segments). b Upon initiation of assembly conditions the tetramers associate laterally into a unit-length filament (ULF). In vimentin, a ULF counts eight tetramers on the average [76]. c Individual ULFs may longitudinally anneal with another ULF (c1) or with a filament consisting of two annealed ULFs (c2); eventually, two short filaments may longitudinally anneal to a longer filament (c3). d Upon extended incubation, long filaments will spontaneously reduce their diameter by radial compaction to form a mature IF [76]. The figure is redrawn and modified after [79]
Protein expression
Desmin is the most abundant IF protein in striated and smooth muscle cells. Immunoblotting after one-dimensional SDS-PAGE with desmin-specific antibodies shows a single band corresponding to an apparent molecular weight of 58 kDa. In addition to muscle, desmin expression has been described in a wide variety of normal and diseased cells, for example, pericytes [24], hepatic stellate cells [124], myoid stromal cells of placenta [170], Sertoli cells [148], decidual cells [67], injured glomerular podocytes [70], and mesotelioma cells [85].
Desmin is one of the earliest markers of muscle development [27], and this is true for all vertebrates including amphibian and fish, pointing to an important evolutionary conservation [75, 153]. Its expression precedes all other muscle-specific structural proteins and even—with the exception of myf-5—the expression of the myogenic transcription factors myoD, myogenin, and mrf4 (myf-6) [99, 103, 112]. During early muscle development desmin and vimentin are co-expressed. Vimentin is the most abundant IF protein of immature myoblasts. Upon further differentiation into mature muscle cells, desmin is strongly upregulated, while the expression of vimentin is completely ceased [43, 152].
Post-translational modifications
Post-translational modifications do play an important role in regulating the functions of intermediate filaments. In vitro translation of chicken skeletal and smooth muscle desmin using a cell-free rabbit reticulocyte system resulted in predominantly non-phosphorylated desmin, suggesting a low amount of phosphorylated desmin under basal conditions [125]. On the other hand, desmin has been described as one of the major acceptors of 32P in differentiating chicken myotubes. Two-dimensional tryptic analysis of desmin revealed multiple sites of phosphorylation [52]. Another study showed that desmin is a substrate of the p21-activated kinase (PAK) mainly leading to phosphorylation of serine residues within the desmin “head” domain. PAK-mediated phosphorylation of desmin strongly inhibited its filament-forming ability [127]. Assembly is also inhibited by ADP ribosylation of desmin via an arginine-specific mono-ADP-ribosyltransferase of muscle, which primarily targets arginine 48 and to a lesser extent arginine 68 within the desmin “head” domain [201]. A subsequent study provided evidence that ADP-ribosylated desmin neither co-assembles with nor affects the filament formation of non-modified desmin. In contrast, the ADP-ribosylation of pre-formed desmin IFs resulted in their disassembly. Furthermore, this process is dependent on the phosphorylation of additional amino acid residues [198].
Subcellular localization and functions
Desmin immunostaining of cross-sectioned striated muscle revealed sarcoplasmic and subsarcolemmal localizations. In longitudinal sections, desmin immunostains revealed a cross-striated pattern [162]. In addition, desmin is enriched at the level of myotendinous and neuromuscular junctions of skeletal muscle as well as of intercalated discs in cardiac muscle [30, 180]. Immunogold electron microscopy demonstrated the presence of desmin at costameres, filamentous structures spanning between myofibrils and the overlaying sacolemma, filamentous inter-Z-disc structures of neighboring myofibrils and mitochondria, and at the desmosomal plaque of intercalated discs, where desmin IFs bind to desmoplakin, a member of the plakin family of crossbridging proteins [90, 111, 146, 162].
This three-dimensional filamentous extra-sarcomeric desmin cytoskeleton interlinks neighboring myofibrils (Fig. 6) and connects the myofibrillar apparatus with myonuclei, other cell organelles, and the extracellular matrix via the subsarcolemmal cytoskeleton [27, 160]. Beyond a sole mechanical integration, this complex interaction is thought to be the basis for a mechano-chemical signaling between various compartments. IFs in general and desmin in particular have also been hypothesized to directly bind to genomic DNA and to exert a role in transcription regulation and DNA organization [182]. Roles of desmin in the morphology and homeostasis of myonuclei, mitochondria, and lysosomes are discussed in the respective paragraphs on muscle pathology, desmin binding partners, and desmin mouse models. Biomechanical aspects of desmin at the level of single IFs, myoblasts, myofibers, and whole muscle are summarized in the respective paragraph on biomechanics.

Schematic of the desmin IF network in relation to the myofibrillar apparatus. In desminopathies, mutant desmin leads to structural and functional changes of the extrasarcomeric cytoskeleton, pathological protein aggregation, mitochondrial abnormalities, and signs of myofibrillar degeneration
Molecular interaction partners
Desmin exerts its multiple functions through direct and indirect binding to various other cellular molecules. In the past 4 decades of desmin research, a growing number of desmin binding partners has been described (see also a previous review on desmin interactions [38]). The following description of desmin interactions is confined to binding partners that are present in muscle cells, i.e., IF proteins, IF-associated proteins, sarcomeric proteins, membrane-associated proteins, small heat shock proteins, apoptosis-related proteins, and nucleic acids.
Interactions with other IF proteins
During muscle development and maintenance, desmin has been reported to interact with five IF proteins, i.e., vimentin, nestin, synemin (also known as desmuslin), and syncoilin, which all are expressed in a spatiotemporal pattern. In addition, the nuclear B-type lamins have been claimed to be a direct binding partner of desmin [54, 111]. With the cytoplasmic IF-proteins the situation is much clearer: Desmin and vimentin are closely related class III IF proteins, whereas nestin, synemin, and syncoilin are more distantly related class IV IF proteins. Desmin, vimentin, and nestin are co-expressed and colocalized in myoblasts. Vimentin and nestin have each been demonstrated to assemble into “heteropolymeric” IFs with desmin [74, 79, 172]. While vimentin expression is downregulated, desmin and nestin are further expressed in later stages of myogenesis [168]. In contrast to desmin, nestin is primarily retained at myotendinous and neuromuscular junctions in mature skeletal muscle [28]. Moreover, synemin was found to co-purify and colocalize with desmin and vimentin [63]. However, synemin seems not to participate in the formation of mixed filaments, but binds to pre-formed desmin or vimentin IFs [18, 81, 150]. In mature skeletal muscle, synemin is localized at the periphery of Z-discs and at the sarcolemma ([119], and own observations). Another IF protein that directly interacts and colocalizes with desmin in mature skeletal muscle is syncoilin, which is localized at neuromuscular junctions, the sarcolemma, and Z-discs. Similar to synemin, syncoilin binds to IFs but does not participate in the formation of mixed filaments and therefore should be called an IF-associated protein. Syncoilin has been postulated to play a role in the cytoskeletal anchorage of the desmin IF [142].
Interactions with IF-associated proteins
Desmin directly interacts with multiple IF-associated proteins. The proper structural and functional organization of the desmin IF system highly depends on the interaction of desmin with several plectin isoforms. In skeletal muscle, the two plectin isoforms 1d and 1f link desmin IFs to Z-discs and costameres, respectively, whereas isoform 1b links desmin to mitochondria [93]. Plectin has been reported to directly bind to the coiled-coil rod domain of desmin via its fifth plakin repeat domain and part of the following linker domain [45]. During myodifferentiation desmin has been shown to co-purify and colocalize with paranemin—the chicken ortholog of mammalian nestin [147]—at the level of Z-discs in myotubes. In contrast to plectin but similar to nestin, paranemin is downregulated upon differentiation and absent in mature muscle cells [25]. Paranemin has been described to associate with desmin IFs—but not to form heteropolymers—leading to the formation of an extended desmin IF network [163]. A further direct desmin binding protein is myospryn (or cardiomyopathy-associated protein 5), a component of the biogenesis of lysosome-related organelles complex 1 (BLOC-1). This interaction involves the desmin “head” domain and a 24-amino acid motif at the end of the SPRY domain of myospryn. Both proteins colocalize in close relation to the nucleus and the endoplasmic reticulum in neonatal cardiomyocytes as well as at intercalated discs, costameres, and lysosomes in adult cardiac muscle. It has been postulated that the desmin–myospryn interaction plays a role in lysosome biogenesis and positioning [96]. Moreover, desmin has been shown to interact with myotubularin, a protein mutated in X-linked centronuclear myopathy (XLCNM or myotubular myopathy). XLCNM-causing mutations of myotubularin were found to interfere with the desmin–myotubularin interaction resulting in an abnormal desmin IF network in skeletal muscle. Furthermore, downregulation as well as expression of mutant myotubularin both induced morphological and functional defects of mitochondria [82]. In myometrial cells, desmin has been found to directly interact with surfactant protein A (SP-A), a member of the collectin family of proteins. This interaction has been reported to inhibit the polymerization of desmin IFs [51].
Interactions with sarcomeric and membrane-associated proteins
Desmin has been reported to directly interact with components of the myofibrillar apparatus. The M-line protein myomesin-1 (synonym: skelemin) was first described as a desmin binding partner in striated muscle [143]. In addition, the coiled-coil rod domain of desmin interacts with the Z-disc section of nebulin [8]. Smooth muscle basic calponin, a major actin-, tropomyosin-, and calmodulin-binding protein, has been identified as a third sarcomeric desmin binding partner, which also interacts with the desmin rod domain [50].
The desmin IF network of muscle cells is anchored to the subsarcolemmal cytoskeleton of costameres. In this respect, desmin has been reported to bind to red blood cell spectrin and it has been postulated that non-red blood cell spectrins also may mediate the association of the desmin IF network with the plasma membrane [101]. A further link between the desmin IF network and the subsarcolemmal cytoskeleton is provided by the direct interaction of an N-terminal region of the desmin head domain with ankyrin [54]. Moreover, desmin was found to directly or indirectly interact with the nicotinic acetylcholine receptor, where it has a postulated role in the submembranous organization of the motor end plate [117].
Interactions with small heat shock proteins, apoptosis-related proteins, and nucleic acids
The two small heat shock proteins HspB1 (synonyms: Hsp25, Hsp27) and HspB5 (synonym: αB-crystallin) directly bind to desmin [19, 91]. The interaction of HspB1 with desmin depends of the phosphorylation of HspB1 at serine residue 15 [91]. Furthermore, desmin has been identified as a substrate of caspase-6 and calpains. During the process of apoptosis, caspase-6 cleaves human desmin at the conserved asparagine 264 (corresponds to mouse desmin Asp263) located in the L12 linker (Fig. 4). The N-terminal desmin cleavage product has been considered to play a role in the execution of apoptosis via a dominant-negative effect on the desmin IF network integrity [32]. Desmin also is a specific target of the proteolytic calpain (Ca2+-activated cysteine proteinase) system [60]. The limited proteolysis of desmin by calpains results in desmin “head,” “rod,” and “tail” domain cleavage products, which are no longer capable to participate in desmin IF formation, but instead heavily interfere with the proper assembly process [15, 17, 122]. The calpain system has also been attributed to exert a role in the spatiotemporal regulation of desmin protein levels during myotube formation [44]. Like other IF proteins, the head domain of desmin has been demonstrated to bind to single-stranded RNA and DNA molecules in vitro [188, 192]. This property reflects the high proportion of basic residues (12 arginines) and the absence of negatively charged residues in the non-α-helical “head” domain. The in vitro interaction of desmin and DNA is preferentially targeted to exposed single-stranded sites of repetitive and mobile DNA sequence motifs and seems to induce changes of the DNA configuration [181].
Pathophysiology
Subcellular localization and expression of wild-type and mutant desmin
Analysis of single skeletal muscle fibers from a patient with the heterozygous p.Arg350Pro desmin mutation showed the presence of pathological desmin-positive protein aggregates in conjunction with the normal cross-striated desmin staining pattern [158, 159] (Fig. 7). This finding raises the question whether mutant desmin forms mixed desmin IFs or mixed aggregates with wild-type desmin or if the mutant protein segregates from the wild-type protein. This issue cannot be solved using commercially available desmin antibodies as they do not distinguish between the wild-type and the point-mutated desmin protein species.

Indirect immunofluorescence labeling of desmin and syncoilin in an isolated muscle fiber from a desminopathy. Note the presence of multiple pathological protein aggregates in addition to the normal cross-striated staining pattern of these two proteins
Studies on desmin protein expression in desminopathies due to deletion or frame-shift mutations would allow at least the determination of the expression level of the mutant desmin species. However, only two studies reported such data yet. In one study immunoblotting revealed two desmin bands in cardiac and skeletal muscle with the lower band corresponding to a truncated desmin protein species showing an intensity of approximately 30 % of the upper wild-type band. However, no desmin mutation analysis was reported [3]. In another study on a desminopathy due to a heterozygous c.735G>C mutation, desmin immunoblotting revealed the presence of two bands at the positions of 53 kDa and 50 kDa. However, RT-PCR analysis in this particular family identified three distinct desmin mRNA species coding for wild-type and p.Glu245Asp desmin, which both are visible at 53 kDa, as well as p.Asp214_Glu245del corresponding to the truncated 50-kDa protein. Here, the amount of the truncated desmin protein varied between 5 and 43 % [35]. Two-dimensional desmin gel electrophoresis in desminopathies revealed non-uniform results. In a desminopathy due to the heterozygous p.Lys240del mutation, an aberrant and more acidic spectrum of desmin spots was visible [158, 159], whereas the p.Asp214_Glu245del desmin mutant species presented with a more alkaline pI [35].
Aberrant modifications of desmin
Desmin has been found to be oxidated and nitrated in muscle fibers containing pathological protein aggregates. These modifications may have a direct cytotoxic effect and may impair the degradation via the ubiquitin-proteasom system [87]. Beyond specific desmin modifications, skeletal muscle samples from desminopathies exhibited increased levels of glycoxidated, lipoxidated, and nitrated proteins [88].
Dysfunctions in protein quality control
A study demonstrated an increased immunoreactivity of the 20S proteasome core as well as its 19S and 11S (synonyms: PA28alpha/beta, PSME1/PSME2) regulators co-localizing with pathological protein aggregates in desminopathies. Further components of the immunoproteasome have also been reported to colocalize with the protein aggregates [46]. Another study demonstrated an enrichment and co-localization of the mutant ubiquitin UBB+1 as well as the autophagy-related p62 with desmin-positive protein aggregates in desminopathies [131]. Desmin-positive protein aggregates in desminopathies also show a positive immunoreactivity with antibodies directed against HspB1 (synonyms: Hsp25, Hsp27) and HspB5 (synonym: αB-crystallin) [160]. With regard to HspB1, two-dimensional gel electrophoresis demonstrated a shift of the main HspB1 spot to a more alkaline pI [36]. Further relationships between the expression of mutant desmin and the ubiquitin–proteasome system, autophagy, and heat shock proteins are provided in the respective paragraphs on desmin mouse models.
Mitochondrial pathology
SDH and COX stains of skeletal muscle biopsy specimens from desminopathy patients often show areas with either increased or decreased enzyme activities (Fig. 1c). With regard to a putative mitochondrial pathology, an analysis of the mitochondrial function in isolated saponin-permeablized skeletal muscle fibres from a desminopathy patient (heterozygous p.Lys240del desmin mutation) revealed an in vivo inhibition of complex I activity [158, 159].
Cytoskeletal organization
Desmin transfection studies have been performed in a variety of muscle and non-muscle cell types: BHK21 (ATCC CCL-10) hamster kidney fibroblasts express desmin and vimentin intermediate filaments [80]. HL-1 cardiac [34], C2C12 (ATCC CRL-1772), and inducible C2.7 [141] skeletal muscle-derived mouse myoblasts, human coronary artery smooth muscle (hcasmc), and neonatal rat cardiac ventricular myocytes (nrcm) [178] as well as C3H/10T1/2 (ATCC CCL-226) mouse fibroblasts ectopically expressing Myf5 or MyoD all contain desmin and vimentin IFs. 3T3 (ATCC CRL-1658) mouse fibroblasts contain vimentin. Vimentin-free mouse embryonic fibroblasts (MEFs) isolated from vimentin knockout mice and spontaneously immortalized do not express cytoplasmic IF proteins [37]. Moreover, both MCF7 (ATCC HTB-22) human epithelial cells and bovine mammary gland epithelium cells grown in the presence of hormones BMGE+H [154] are vimentin-free and express only keratin IF proteins. SW13 (ATCC CCL-105) human epithelial cells do not express any IF protein.
The overall data derived from studies using these cell types indicated that the majority of desmin mutants are incapable of forming a de novo desmin IF network, but instead form non-IF structures and desmin-positive protein aggregates. In addition, most of them induce the collapse of a pre-existing IF network such as in 3T3 cells, although several mutant desmin proteins integrate well, just like the wild-type protein, into the vimentin network [9, 11, 16, 167]. As an example, we depict here the transfection of the p.Arg406Trp desmin rod mutant into various cell types. In the vimentin-free SW13 and BMGE+H cells, it forms dot-like and short rod-like structures (Fig. 8a, b); in addition, in 3T3 cells it completely segregates from the endogenous vimentin filaments and causes the reorganization of vimentin IFs around the nucleus (Fig. 8c). Similarly, the p.Leu345Pro desmin mutant completely avoids integrating into the vimentin system (Fig. 8d–f); however, some affinity for vimentin is observed in extended vimentin IFs below the cell nucleus and outside of the area of the collapsed vimentin IFs (Fig. 8f, arrowheads denote green desmin dots aligning with the red vimentin IFs). In the collapsed area, the green and the red signals superimpose to yield a yellow signal; however, the signals differ entirely in shape, dots versus fibers, indicating that they are not contained within the same structure but are only located close to each other and thereby generate a yellow signal.

Ectopic expression of mutant desmin in cultured cells. The desmin mutant p.Arg406Trp was transfected into (a) IF-free human SW13 cells; b vimentin- and desmin-free bovine mammary gland cells (BMGE+H) containing cytokeratins; and c into mouse fibroblast 3T3 cells. Detection was by specific primary and secondary antibodies: desmin stain in green, vimentin in red. Note that the p.Arg406Trp desmin mutant does not form filaments on its own, and also in the vimentin-containing cell the desmin mutant protein stays particulate. Note furthermore that the vimentin system is re-organized (“collapsed”) around the nucleus as a consequence of the mutant desmin expression. In contrast to wild-type desmin, the mutant does not integrate into vimentin filaments [11]. Similarly, the desmin mutant p.Leu345Pro (proline is often called a “helix breaker”) segregates from the endogenous vimentin system when transfected into 3T3 cells: d desmin antibody staining; e vimentin antibody staining; f merged image highlighting green dot-like desmin structures on red vimentin filaments (arrowheads). Also in areas where both proteins are present, they clearly are in distinct structures. Images are taken from [11]
In vitro filament assembly
The first disease mutant desmin to be analyzed at the in vitro level for its assembly properties was the rather drastic p.Arg173_Glu179del desmin “rod” mutation, which misses seven amino acids in a row in coil 1B. In both cell culture after cDNA-transfection and after forced assembly of recombinant proteins, only non-IF structures were obtained [121]. Later on, several other desmin disease mutants were studied, and their ability to form IF networks in vivo was analyzed by cellular cDNA transfection [11]. In order to reveal how desmin mutations influence basic filament assembly properties, a consecutive study investigated their assembly properties at the protein level [12]. Four major classes of pathological effects were observed (Fig. 9): (1) after 10 s of assembly, filaments form by lateral association of tetramers and subsequent longitudinal annealing of ULFs (see also Fig. 5); however, upon further incubation these IF-like filaments show an abnormal lateral annealing leading to the formation of large “sheets” (Fig. 9, p.Asn342Asp); (2) a second class forms IFs similar to those generated from desmin WT, although the filaments are less regular (Fig. 9, p.Ala360Pro); (3) other mutants exhibit extended filaments at 10 s of assembly, but thereafter immediately decay into ball-like aggregates [13] (Fig. 9, p.Leu370Pro); (4) filament assembly starts with apparently normal ULFs; however, they fail to longitudinally anneal as observed with wild-type desmin and instead stay at the ULF-state or loosely associate longitudinally such that the sub-filament structure remains visible (Fig. 9, p.Arg406Trp). Notably, all desmin disease mutants that were not able to form stable filaments on their own in vitro also did not form IFs after transfection into IF-free cells. Instead, they did segregate from the endogenous IF system when transfected into vimentin- or desmin-containing cells [13]. While the finding that truncated desmin mutants did not properly assemble is not that unexpected, the observed high pathogenicity of missense mutations suggests that desmin filament assembly is a delicate process that can be easily distorted.

In vitro filament assembly of wild-type desmin compared to various mutants. Assembly was initiated at time point zero by increase of the ionic strength of the incubation buffer and stopped at 10 s and 10 min, respectively, by addition of filament buffer containing 0.2 % glutaraldehyde. The wild-type desmin (desmin WT) and the mutants, indicated within the respective panels, were analyzed after being negatively stained with uranyl acetate by transmission electron microscopy. Images are taken from [12, 13]
Biomechanics: from filaments to cells and muscle tissue
A characteristic biophysical property of IFs is their elasticity. In response to mechanical stretch, desmin filaments have been shown to extend in length in conjunction with a thinning of their diameter. Experiments demonstrated that upon a 3.4-fold extension, desmin filaments reduced their diameter from 12.6 to 3.5 nm [98]. Moreover, the phenomenon that IFs become more viscoelastic when exposed to an external mechanical force is referred to as “strain stiffening” [86, 155]. In striated muscle, the elasticity of desmin filaments has been attributed to play a cell-protective role against mechanical stress. Thus, desmin filaments may dissipate mechanical energy during muscle contraction.
In this context, mutant desmin may exert a pathogenic effect on the viscoelastic properties of the desmin filament system, which consecutively may lead to progressive muscle fiber damage and muscle weakness. The viscoelastic properties of several disease-associated mutant desmin protein species have been analyzed. Here, the focus was on desmin mutants that are able to form apparently normal filament networks in vitro and in vivo. Some of these mutants showed nanomechanical properties similar to that of wild-type desmin, while others displayed changes in their tensile properties with reduced strain stiffening. Such an increase in the resistance of desmin filaments to an external pulling force may inflict changes in the adaptability of muscle cells to mechanosensing and mechanotransduction [14, 97]. Analysis of primary cultured myoblasts derived from a patient with a heterozygous p.Arg350Pro desmin mutation revealed an aberrant response to mechanical stress. These cells displayed increased cell stiffness and a higher rate of cell death and substrate detachment [22]. A number of biomechanical studies on mice lacking desmin have been reported. One study provided evidence that the lack of desmin is associated with a reduction of the overall passive elasticity of intact isolated soleus muscle [2]. Another study focusing on the diaphragm reported a reduced stiffness and viscoelasticity of this muscle together with an increased tetanic force production [23]. A further study showed a reduced passive tensile strength after eccentric contraction associated with disrupted Z-discs in individual myofibrils. In addition, the maximal isometric force production in the extensor digitorum longus muscle was reported to be decreased, which points to a possible involvement of desmin in excitation–contraction coupling [149]. This assumption was further underlined by an additional study reporting a defective excitation–contraction coupling in intact extensor digitorum longus and soleus muscles of desmin knockout mice. Here, the increased fatigue in extensor digitorum longus muscle was assessed by continuous high frequency field stimulation and could be overcome by the addition of caffeine. This combination of results argues against a defect in the sarcoplasmic reticulum calcium storage capacity [197]. However, this interpretation was challenged by yet another study, which used a more physiological high frequency stimulation protocol with titanic trains. In this setting, a reduced fatigue in intact soleus muscle without any change in the calcium sensitivity of the contractile apparatus was observed [7]. Finally, a study described that the absence of desmin resulted in an increased number of branched myofibers in the flexor digitorum brevis muscle. This work reported neither differences in the resting sarcoplasmic calcium concentration nor in the sarcoplasmic calcium release amplitude in desmin knockout myofibers [61].
Animal models
Desmin knockout models
Des ko #1 and Des ko #2
Desmin knockout mouse models from two independent research groups were published in 1996 (Des ko #1 [104] and Des ko #2 [116]). Desmin knockout mice were viable and fertile and showed no overt defects in muscle development. However, they developed clinical and morphological signs of a progressive myopathy and cardiomyopathy [68, 104, 116]. The skeletal muscle pathology, most prominent in weight-bearing muscles, was characterized by disorganized and nonaligned fibers, Z-disc streaming, and subsarcolemmal accumulation of mitochondria. In addition, myofibrillogenesis in regenerating muscles was reported to be disturbed [104, 105, 116]. An in situ analysis of mitochondrial function revealed a reduced maximal rate of ADP-stimulated oxygen consumption [114]. Furthermore, an abnormal folding of the postsynaptic apparatus of the neuromuscular junction was noted [1]. The lack of desmin led to changes in the subcellular distribution of synemin and nestin (synonym: paranemin), but not of plectin [29].
Heart function of desmin knockout mice was investigated by a cardiac MRI study. This demonstrated significantly reduced left and right ventricular ejection fractions and cardiac output, an increased left ventricular mass, and segmental wall thinning and akinesia [171]. In vivo electrophysiological studies revealed reduced atrial but prolonged ventricular refractory periods, ventricular conduction slowing, enhanced inducibility of atrial fibrillation, and a reduced susceptibility to ventricular arrhythmias [157]. Histopathological analyses of cardiac tissue showed areas of hemorrhage, fibrosis, ischemia, and calcification [104, 116]. Isolated cardiomyocytes showed an increased cell volume [115]. In addition, changes in the morphology of intercalated discs, disruptions of the sarcolemma, as well as an abnormal shape and distribution of mitochondria were observed [179]. The mitochondrial pathology in cardiac tissue was further characterized by an aberrant conventional kinesin (synonym: kinesin-1) distribution, a lower amount of cytochrome c, and a re-localization of Bcl-2 [109]. Furthermore, ketone body and acetate metabolism, NADH shuttle proteins, amino-acid metabolism, and respiratory enzymes were affected [49].
Des ko #2/Bcl-2 tg overexpression
The issue of cardiac pathology was further addressed by crossbreeding desmin knockout mice with transgenic mice overexpressing apoptosis regulator Bcl-2 [196]. These double-mutant mice showed a reduced occurrence of fibrotic lesions in the myocardium, prevention of cardiac hypertrophy, restoration of cardiomyocyte ultrastructure, and significant improvement of cardiac function. In addition, an improved calcium handling of mitochondria was observed. Hence, it was concluded that the mitochondrial abnormalities, which are regarded as the primary cause of the cardiomyopathy in desmin knockout mice, can be ameliorated by the overexpression of Bcl-2.
Des ko #2/Des tg p.Ile451Met
In an additional study the desmin knockout strain was crossbred with another desmin mouse model with transgenic expression of the p.Ile451Met mutant [113]. Note that only mice homozygous for the desmin knockout and with presence of the p.Ile451Met desmin transgene were analyzed. In this setting, the p.Ile451Met desmin showed an aberrant subcellular distribution, was expressed at lower levels (compared to wild-type desmin in wild-type control mice), and was found to lack the N-terminal 20–30 amino acids. The latter finding has been attributed to a specific cleavage of the mutant desmin in cardiomyocytes.
Des ko #2/Des tg p.Asp263Glu/TNFtg overexpression
In the context of a mouse model for tumor necrosis factor alpha-induced cardiomyopathy, it was found that increased expression of TNF-alpha leads to degradation and changes in the subcellular distribution of desmin and to pathological aggregate formation. The latter process was attenuated in TNF-alpha overexpressing mice that additionally expressed p.Asp263Glu mutant desmin instead of the wild-type endogenous protein in the heart. This effect has been attributed to a resistance of the mutant desmin to caspase-6 cleavage [134].
Desmin transgenic mouse models
Des tg truncated/chimeric desmin
In 1996 the first transgenic desmin mouse was published [145]. In this model a hamster desmin–vimentin chimeric protein (last 129 aa of desmin replaced by last 13 aa of vimentin) was expressed under control of the hamster desmin promoter. The expression of this truncated desmin (approximately 10 % mutant compared to wild-type desmin) had a dominant-negative effect on the desmin IF network. Desmin immunostains of skeletal and cardiac muscle tissue revealed an aberrant desmin distribution characterized by a loss of the cross-striated pattern. Ultrastructural analysis showed intermyofibrillar deposits of fibrillar and membranous electron-dense material and fragmented sarcomeres as well as abnormalities of the T-tubule system.
Des tg p.Arg173_Glu179del
A second transgenic desmin mouse model expressing a desmin mutant with a small in-frame deletion of seven amino acids was reported in 2001 [194] recapitulating the human mutation reported by [121]. A three-fold overexpression of the p.Arg173_Glu179del desmin compared to the endogenous wild-type desmin led to a dominant negative effect with a disruption of the extra-sarcomeric cytoskeleton and presence of desmin-positive granular filamentous protein aggregates in cardiac tissue. These animals further showed a compromised ability of the heart to respond to β-agonist stimulation. Control animals with a three-fold overexpression of wild-type desmin showed no overt clinical or morphological effect. Skeletal muscle tissue was not addressed in this study.
Electrophysiological investigations of this mouse model showed increased P-wave duration and slowing of ventricular conduction [53]. Although no changes in the expression level of connexin-43, desmoplakin, plakoglobin, and N-cadherin were observed, immunostains showed significantly reduced signal intensities of these junctional proteins. At the ultrastructural level, intercalated discs were found to be highly convoluted and to contain fewer gap junctions and desmosomes.
Des tg p.Arg173_Glu179del/Cryabtg p.ArgR120Gly
Cardiac pathology of the p.Arg173_Glu179del desmin mouse was also analyzed after crossbreeding with transgenic mice overexpressing the p.ArgR120Gly mutant small heat shock protein αB-crystallin [193]. This αB-crystallin mutant had previously been shown to cause a cardiomyopathy characterized by desmin-positive protein aggregates in heterozygous humans and transgenic mice [187, 195]. The double-mutant mice presented with an accentuated cardiac pathology and died of congestive heart failure in the first 2 months of life. Further analyses revealed increased levels of desmin proteins and an increased abundance of pathological aggregates compared to p.Arg173_Glu179del desmin mice.
Des tg p.Arg173_Glu179del/GFPdgntg
The p.Arg173_Glu179del desmin mouse was further crossbred with a transgenic mouse model expressing the GFPdgn ubiquitin–proteasome system reporter substrate (GFP fused to an ubiquitination signal sequence) [100]. This study provided evidence that the p.Arg173_Glu179del mutant desmin impairs the proteolytic function of the ubiquitin-proteasome system by an impaired entry of ubiquitinated proteins into the 20S proteasome [110].
Des tg p.Arg173_Glu179del/GFP-LC3tg
Moreover, the p.Arg173_Glu179del desmin mouse had been crossed with a autophagy reporter mouse model expressing a GFP-LC3 fusion protein [120]. Here, expression of the desmin deletion mutant led to an increased autophagic flux associated with an increased expression of p62 [200].
Des tg p.Leu345Pro
The only transgenic mouse model expressing a desmin missense mutation in addition to the endogenous protein was reported in 2008. The animal model with low expression (approximately 10 % of total desmin) of the HA-tagged p.Leu345Pro desmin mutant was reported to show a reduced contractile function and recovery from fatigue in soleus muscle. Moreover, cardiac alterations consisting of a hypertrophic left ventricular posterior wall and decreased left ventricular chamber dimension were described. Though no MFM typical desmin-positive protein aggregates were detected, this animal presented evidence for a mitochondrial pathology characterized by swelling and vacuolization as well as increased calcium levels [94].
Conclusion and outlook
The last 2 decades have led to a number of important insights into the molecular pathogenesis of desminopathies, but the precise and sequential molecular mechanisms leading from mutant desmin to consecutive pathological protein aggregation and progressive muscle damage still remain to be elucidated. Though many desmin mutants obviously compromise the desmin filament formation, the progressive human muscle pathology cannot solely be attributed to such a simple mechanistic explanation. If desmin mutants have such a toxic effect on the desmin filament system, why does it take decades of life until clinical symptoms of muscle weakness become apparent? Our review of the currently available data on desmin and desminopathies enforces a complex and multilevel concept of disease development in which mutant desmin additionally interferes with the binding to desmin interaction partners, signaling cascades, protein quality control systems, and the function of cell organelles. Further work is needed to evaluate, broaden, and integrate these different aspects. Respective future studies shall provide essential novel insights into the atomic structure of the desmin tetramer and its assembly reactions, the biomechanics of desmin-mutant cells and tissues, the expression and subcellular localization of mutant desmin, the composition of pathological protein aggregates, the characterization of aberrant post-translational modifications and interactions of desmin with other proteins and nucleic acids, epigenetic factors, and dysfunction of various cell organelles. For studies of early disease stages, the generation and characterization of physiological cell and mouse models, i.e., with a knockin of a desmin mutation into the murine desmin gene, are necessary.
To date, no specific treatment is available for desminopathies and even basic clinical questions—e.g., is physical exercise beneficial or harmful for the course of the disease?—cannot finally be answered on the basis of the currently available literature. A regular neurological, cardiological, and pulmonological diagnostic workup is certainly mandatory for all desminopathy patients. Future studies on desminopathy-related cell and animal models will provide answers with respect to potential therapies: Do anti-aggregation drugs such as small heat shock protein inducers and autophagy and proteasome modulators have a cell-protective effect that ameliorates or even cures the progressive muscle pathology? Or should we further explore muscle-specific gene transfer approaches, for example, siRNA-based silencing of the mutant desmin allele, overexpression of the wild-type desmin, or expression of other protective proteins such as heat shock proteins and Bcl-2?
Abbreviations
- APEG1:
-
Aortic preferentially expressed protein 1 (synonym: SPEG)
- BAG-3:
-
BAG family molecular chaperone regulator 3
- BLOC-1:
-
Biogenesis of lysosome-related organelles complex 1
- CCD:
-
Cardiac conduction defects
- CHPF:
-
Chondroitin sulfate synthase 2
- CK:
-
Kreatine kinase
- COX:
-
Cytochome c oxidase
- DES:
-
Desmin gene
- DNAJB6:
-
DnaJ homolog subfamily B member 6 (synonym: heat shock protein J2)
- ECG:
-
Electrocardiogram
- EMG:
-
Electromyography
- FHL1:
-
Four and a half LIM domains protein 1
- GFAP:
-
Glial fibrillary acidic protein
- GT:
-
Gomori trichrome
- H&E:
-
Hematoxylin and eosin
- Hsp:
-
Heat shock protein
- IF:
-
Intermediate filament
- LCR:
-
5′ locus control region
- MEF:
-
Mouse embryonic fibroblast
- MFM:
-
Myofibrillar myopathy
- mrf4:
-
Muscle-specific regulatory factor 4 (synonym: myf-6)
- MRI:
-
Magnetic resonance imaging
- myf-6:
-
Myogenic factor 6 (synonym: mrf4)
- myoD:
-
Myoblast determination protein 1
- NADH-TR:
-
Reduced nicotinamide adenine dinucleotide tetrazolium reductase
- PA28:
-
Proteasome activator 28 (synonym PSME)
- PAS:
-
Periodic acid Schiff
- PSME:
-
Proteasome activator complex subunit (synonym: PA28)
- SDH:
-
Succinic dehydrogenase
- SDS-PAGE:
-
Sodium dodecyl sulfate polyacrylamide gel electrophoresis
- SHC:
-
Sequence homology class
- SPEG:
-
Striated muscle preferentially expressed protein kinase
- ULF:
-
Unit length filament
- VCP:
-
Valosin containing protein
- XLCNM:
-
X-linked centronuclear myopathy
- ZASP:
-
Z-band alternatively spliced PDZ-motif protein (synonyms: cypher, LIM domain-binding protein 3)
References
Agbulut O, Li Z, Perie S, Ludosky MA, Paulin D, Cartaud J, Butler-Browne G (2001) Lack of desmin results in abortive muscle regeneration and modifications in synaptic structure. Cell Motil Cytoskelet 49:51–66
Anderson J, Li Z, Goubel F (2001) Passive stiffness is increased in soleus muscle of desmin knockout mouse. Muscle Nerve 24:1090–1092
Arbustini E, Morbini P, Grasso M, Fasani R, Verga L, Bellini O, Dal Bello B, Campana C, Piccolo G, Febo O, Opasich C, Gavazzi A, Ferrans VJ (1998) Restrictive cardiomyopathy, atrioventricular block and mild to subclinical myopathy in patients with desmin-immunoreactive material deposits. J Am Coll Cardiol 31:645–653
Arbustini E, Pasotti M, Pilotto A, Pellegrini C, Grasso M, Previtali S, Repetto A, Bellini O, Azan G, Scaffino M, Campana C, Piccolo G, Vigano M, Tavazzi L (2006) Desmin accumulation restrictive cardiomyopathy and atrioventricular block associated with desmin gene defects. Eur J Heart Fail 8:477–483
Arias M, Pardo J, Blanco-Arias P, Sobrido MJ, Arias S, Dapena D, Carracedo A, Goldfarb LG, Navarro C (2006) Distinct phenotypic features and gender-specific disease manifestations in a Spanish family with desmin L370P mutation. Neuromuscul Disord 16:498–503
Ariza A, Coll J, Fernandez-Figueras MT, Lopez MD, Mate JL, Garcia O, Fernandez-Vasalo A, Navas-Palacios JJ (1995) Desmin myopathy: a multisystem disorder involving skeletal, cardiac, and smooth muscle. Hum Pathol 26:1032–1037
Balogh J, Li Z, Paulin D, Arner A (2003) Lower active force generation and improved fatigue resistance in skeletal muscle from desmin deficient mice. J Muscle Res Cell Motil 24:453–459
Bang ML, Gregorio C, Labeit S (2002) Molecular dissection of the interaction of desmin with the C-terminal region of nebulin. J Struct Biol 137:119–127
Bär H, Fischer D, Goudeau B, Kley RA, Clemen CS, Vicart P, Herrmann H, Vorgerd M, Schröder R (2005) Pathogenic effects of a novel heterozygous R350P desmin mutation on the assembly of desmin intermediate filaments in vivo and in vitro. Hum Mol Genet 14:1251–1260
Bär H, Goudeau B, Walde S, Casteras-Simon M, Mücke N, Shatunov A, Goldberg YP, Clarke C, Holton JL, Eymard B, Katus HA, Fardeau M, Goldfarb L, Vicart P, Herrmann H (2007) Conspicuous involvement of desmin tail mutations in diverse cardiac and skeletal myopathies. Hum Mutat 28:374–386
Bär H, Kostareva A, Sjoberg G, Sejersen T, Katus HA, Herrmann H (2006) Forced expression of desmin and desmin mutants in cultured cells: impact of myopathic missense mutations in the central coiled-coil domain on network formation. Exp Cell Res 312:1554–1565
Bär H, Mücke N, Kostareva A, Sjoberg G, Aebi U, Herrmann H (2005) Severe muscle disease-causing desmin mutations interfere with in vitro filament assembly at distinct stages. Proc Natl Acad Sci USA 102:15099–15104
Bär H, Mücke N, Ringler P, Müller SA, Kreplak L, Katus HA, Aebi U, Herrmann H (2006) Impact of disease mutations on the desmin filament assembly process. J Mol Biol 360:1031–1042
Bär H, Schopferer M, Sharma S, Hochstein B, Mücke N, Herrmann H, Willenbacher N (2010) Mutations in Desmin’s carboxy-terminal “Tail” domain severely modify filament and network mechanics. J Mol Biol 397:1188–1198
Bär H, Sharma S, Kleiner H, Mücke N, Zentgraf H, Katus HA, Aebi U, Herrmann H (2009) Interference of amino-terminal desmin fragments with desmin filament formation. Cell Motil Cytoskeleton 66:986–999
Bär H, Strelkov SV, Sjoberg G, Aebi U, Herrmann H (2004) The biology of desmin filaments: how do mutations affect their structure, assembly, and organisation? J Struct Biol 148:137–152
Baron CP, Jacobsen S, Purslow PP (2004) Cleavage of desmin by cysteine proteases: calpains and cathepsin B. Meat Sci 68:447–456
Bellin RM, Sernett SW, Becker B, Ip W, Huiatt TW, Robson RM (1999) Molecular characteristics and interactions of the intermediate filament protein synemin. Interactions with alpha-actinin may anchor synemin-containing heterofilaments. J Biol Chem 274:29493–29499
Bennardini F, Wrzosek A, Chiesi M (1992) Alpha B-crystallin in cardiac tissue. Association with actin and desmin filaments. Circ Res 71:288–294
Benvenuti LA, Aiello VD, Falcao BA, Lage SG (2012) Atrioventricular block pathology in cardiomyopathy by desmin deposition. Arq Bras Cardiol 98:e3–e6
Bergman JE, Veenstra-Knol HE, van Essen AJ, van Ravenswaaij CM, den Dunnen WF, van den Wijngaard A, van Tintelen JP (2007) Two related Dutch families with a clinically variable presentation of cardioskeletal myopathy caused by a novel S13F mutation in the desmin gene. Eur J Med Genet 50:355–366
Bonakdar N, Luczak J, Lautscham L, Czonstke M, Koch TM, Mainka A, Jungbauer T, Goldmann WH, Schröder R, Fabry B (2012) Biomechanical characterization of a desminopathy in primary human myoblasts. Biochem Biophys Res Commun 419:703–707
Boriek AM, Capetanaki Y, Hwang W, Officer T, Badshah M, Rodarte J, Tidball JG (2001) Desmin integrates the three-dimensional mechanical properties of muscles. Am J Physiol Cell Physiol 280:C46–C52
Brachvogel B, Pausch F, Farlie P, Gaipl U, Etich J, Zhou Z, Cameron T, von der Mark K, Bateman JF, Poschl E (2007) Isolated Anxa5 +/Sca-1 + perivascular cells from mouse meningeal vasculature retain their perivascular phenotype in vitro and in vivo. Exp Cell Res 313:2730–2743
Breckler J, Lazarides E (1982) Isolation of a new high molecular weight protein associated with desmin and vimentin filaments from avian embryonic skeletal muscle. J Cell Biol 92:795–806
Burkhard P, Stetefeld J, Strelkov SV (2001) Coiled coils: a highly versatile protein folding motif. Trends Cell Biol 11:82–88
Capetanaki Y, Bloch RJ, Kouloumenta A, Mavroidis M, Psarras S (2007) Muscle intermediate filaments and their links to membranes and membranous organelles. Exp Cell Res 313:2063–2076
Carlsson L, Li Z, Paulin D, Thornell LE (1999) Nestin is expressed during development and in myotendinous and neuromuscular junctions in wild type and desmin knockout mice. Exp Cell Res 251:213–223
Carlsson L, Li ZL, Paulin D, Price MG, Breckler J, Robson RM, Wiche G, Thornell LE (2000) Differences in the distribution of synemin, paranemin, and plectin in skeletal muscles of wild-type and desmin knockout mice. Histochem Cell Biol 114:39–47
Carlsson L, Thornell LE (2001) Desmin-related myopathies in mice and man. Acta Physiol Scand 171:341–348
Carmignac V, Sharma S, Arbogast S, Fischer D, Serreri C, Serria M, Stoltenburg G, Maurage CA, Herrmann H, Cuisset JM, Bär H, Ferreiro A (2009) A homozygous desmin deletion causes an Emery-Dreifuss like recessive myopathy with desmin depletion. Neuromuscul Disord 19:600
Chen F, Chang R, Trivedi M, Capetanaki Y, Cryns VL (2003) Caspase proteolysis of desmin produces a dominant-negative inhibitor of intermediate filaments and promotes apoptosis. J Biol Chem 278:6848–6853
Chernyatina AA, Nicolet S, Aebi U, Herrmann H, Strelkov SV (2012) Atomic structure of the vimentin central alpha-helical domain and its implications for intermediate filament assembly. Proc Natl Acad Sci USA 109:13620–13625
Claycomb WC, Lanson NA Jr, Stallworth BS, Egeland DB, Delcarpio JB, Bahinski A, Izzo NJ Jr (1998) HL-1 cells: a cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc Natl Acad Sci USA 95:2979–2984
Clemen CS, Fischer D, Reimann J, Eichinger L, Muller CR, Muller HD, Goebel HH, Schröder R (2009) How much mutant protein is needed to cause a protein aggregate myopathy in vivo? Lessons from an exceptional desminopathy. Hum Mutat 30:E490–E499
Clemen CS, Fischer D, Roth U, Simon S, Vicart P, Kato K, Kaminska AM, Vorgerd M, Goldfarb LG, Eymard B, Romero NB, Goudeau B, Eggermann T, Zerres K, Noegel AA, Schröder R (2005) Hsp27-2D-gel electrophoresis is a diagnostic tool to differentiate primary desminopathies from myofibrillar myopathies. FEBS Lett 579:3777–3782
Colucci-Guyon E, Portier MM, Dunia I, Paulin D, Pournin S, Babinet C (1994) Mice lacking vimentin develop and reproduce without an obvious phenotype. Cell 79:679–694
Costa ML, Escaleira R, Cataldo A, Oliveira F, Mermelstein CS (2004) Desmin: molecular interactions and putative functions of the muscle intermediate filament protein. Braz J Med Biol Res 37:1819–1830
Dagvadorj A, Goudeau B, Hilton-Jones D, Blancato JK, Shatunov A, Simon-Casteras M, Squier W, Nagle JW, Goldfarb LG, Vicart P (2003) Respiratory insufficiency in desminopathy patients caused by introduction of proline residues in desmin c-terminal alpha-helical segment. Muscle Nerve 27:669–675
Dagvadorj A, Olive M, Urtizberea JA, Halle M, Shatunov A, Bonnemann C, Park KY, Goebel HH, Ferrer I, Vicart P, Dalakas MC, Goldfarb LG (2004) A series of West European patients with severe cardiac and skeletal myopathy associated with a de novo R406 W mutation in desmin. J Neurol 251:143–149
Dalakas MC, Dagvadorj A, Goudeau B, Park KY, Takeda K, Simon-Casteras M, Vasconcelos O, Sambuughin N, Shatunov A, Nagle JW, Sivakumar K, Vicart P, Goldfarb LG (2003) Progressive skeletal myopathy, a phenotypic variant of desmin myopathy associated with desmin mutations. Neuromuscul Disord 13:252–258
Dalakas MC, Park KY, Semino-Mora C, Lee HS, Sivakumar K, Goldfarb LG (2000) Desmin myopathy, a skeletal myopathy with cardiomyopathy caused by mutations in the desmin gene. N Engl J Med 342:770–780
Duprey P, Paulin D (1995) What can be learned from intermediate filament gene regulation in the mouse embryo. Int J Dev Biol 39:443–457
Elamrani N, Brustis JJ, Dourdin N, Balcerzak D, Poussard S, Cottin P, Ducastaing A (1995) Desmin degradation and Ca(2 +)-dependent proteolysis during myoblast fusion. Biol Cell 85:177–183
Favre B, Schneider Y, Lingasamy P, Bouameur JE, Begre N, Gontier Y, Steiner-Champliaud MF, Frias MA, Borradori L, Fontao L (2011) Plectin interacts with the rod domain of type III intermediate filament proteins desmin and vimentin. Eur J Cell Biol 90:390–400
Ferrer I, Martin B, Castano JG, Lucas JJ, Moreno D, Olive M (2004) Proteasomal expression, induction of immunoproteasome subunits, and local MHC class I presentation in myofibrillar myopathy and inclusion body myositis. J Neuropathol Exp Neurol 63:484–498
Fidzianska A, Kotowicz J, Sadowska M, Goudeau B, Walczak E, Vicart P, Hausmanowa-Petrusewicz I (2005) A novel desmin R355P mutation causes cardiac and skeletal myopathy. Neuromuscul Disord 15:525–531
Fischer D, Kley RA, Strach K, Meyer C, Sommer T, Eger K, Rolfs A, Meyer W, Pou A, Pradas J, Heyer CM, Grossmann A, Huebner A, Kress W, Reimann J, Schröder R, Eymard B, Fardeau M, Udd B, Goldfarb L, Vorgerd M, Olive M (2008) Distinct muscle imaging patterns in myofibrillar myopathies. Neurology 71:758–765
Fountoulakis M, Soumaka E, Rapti K, Mavroidis M, Tsangaris G, Maris A, Weisleder N, Capetanaki Y (2005) Alterations in the heart mitochondrial proteome in a desmin null heart failure model. J Mol Cell Cardiol 38:461–474
Fujii T, Takagi H, Arimoto M, Ootani H, Ueeda T (2000) Bundle formation of smooth muscle desmin intermediate filaments by calponin and its binding site on the desmin molecule. J Biochem 127:457–465
Garcia-Verdugo I, Synguelakis M, Degrouard J, Franco CA, Valot B, Zivy M, Chaby R, Tanfin Z (2008) Interaction of surfactant protein A with the intermediate filaments desmin and vimentin. Biochemistry 47:5127–5138
Gard DL, Lazarides E (1982) Analysis of desmin and vimentin phosphopeptides in cultured avian myogenic cells and their modulation by 8-bromo-adenosine 3′,5′-cyclic monophosphate. Proc Natl Acad Sci USA 79:6912–6916
Gard JJ, Yamada K, Green KG, Eloff BC, Rosenbaum DS, Wang X, Robbins J, Schuessler RB, Yamada KA, Saffitz JE (2005) Remodeling of gap junctions and slow conduction in a mouse model of desmin-related cardiomyopathy. Cardiovasc Res 67:539–547
Georgatos SD, Weber K, Geisler N, Blobel G (1987) Binding of two desmin derivatives to the plasma membrane and the nuclear envelope of avian erythrocytes: evidence for a conserved site-specificity in intermediate filament-membrane interactions. Proc Natl Acad Sci USA 84:6780–6784
Goldfarb LG, Dalakas MC (2009) Tragedy in a heartbeat: malfunctioning desmin causes skeletal and cardiac muscle disease. J Clin Invest 119:1806–1813
Goldfarb LG, Dalakas MC (2011) Erratum for: “Tragedy in a heartbeat: malfunctioning desmin causes skeletal and cardiac muscle disease”. J Clin Invest 121:455
Goldfarb LG, Olive M, Vicart P, Goebel HH (2008) Intermediate filament diseases: desminopathy. Adv Exp Med Biol 642:131–164
Goldfarb LG, Park KY, Cervenakova L, Gorokhova S, Lee HS, Vasconcelos O, Nagle JW, Semino-Mora C, Sivakumar K, Dalakas MC (1998) Missense mutations in desmin associated with familial cardiac and skeletal myopathy. Nat Genet 19:402–403
Goldfarb LG, Vicart P, Goebel HH, Dalakas MC (2004) Desmin myopathy. Brain 127:723–734
Goll DE, Thompson VF, Li H, Wei W, Cong J (2003) The calpain system. Physiol Rev 83:731–801
Goodall MH, Ward CW, Pratt SJ, Bloch RJ, Lovering RM (2012) Structural and functional evaluation of branched myofibers lacking intermediate filaments. Am J Physiol Cell Physiol 303:C224–C232
Goudeau B, Rodrigues-Lima F, Fischer D, Casteras-Simon M, Sambuughin N, de Visser M, Laforet P, Ferrer X, Chapon F, Sjoberg G, Kostareva A, Sejersen T, Dalakas MC, Goldfarb LG, Vicart P (2006) Variable pathogenic potentials of mutations located in the desmin alpha-helical domain. Hum Mutat 27:906–913
Granger BL, Lazarides E (1980) Synemin: a new high molecular weight protein associated with desmin and vimentin filaments in muscle. Cell 22:727–738
Greenberg SA, Salajegheh M, Judge DP, Feldman MW, Kuncl RW, Waldon Z, Steen H, Wagner KR (2012) Etiology of limb girdle muscular dystrophy 1D/1E determined by laser capture microdissection proteomics. Ann Neurol 71:141–145
Gudkova A, Kostareva A, Sjoberg G, Smolina N, Turalchuk M, Kuznetsova I, Rybakova M, Edstrom L, Shlyakhto E, Sejersen T (2012) Diagnostic challenge in desmin cardiomyopathy with transformation of clinical phenotypes. Pediatr Cardiol. doi:10.1007/s00246-00012-00312-x
Hager S, Mahrholdt H, Goldfarb LG, Goebel HH, Sechtem U (2006) Images in cardiovascular medicine. Giant right atrium in the setting of desmin-related restrictive cardiomyopathy. Circulation 113:e53–e55
Halperin R, Fleminger G, Kraicer PF, Hadas E (1991) Desmin as an immunochemical marker of human decidual cells and its expression in menstrual fluid. Hum Reprod 6:186–189
Haubold KW, Allen DL, Capetanaki Y, Leinwand LA (2003) Loss of desmin leads to impaired voluntary wheel running and treadmill exercise performance. J Appl Physiol 95:1617–1622
Hedberg C, Melberg A, Kuhl A, Jenne D, Oldfors A (2012) Autosomal dominant myofibrillar myopathy with arrhythmogenic right ventricular cardiomyopathy 7 is caused by a DES mutation. Eur J Hum Genet 20:984–985
Herrmann A, Tozzo E, Funk J (2012) Semi-automated quantitative image analysis of podocyte desmin immunoreactivity as a sensitive marker for acute glomerular damage in the rat puromycin aminonucleoside nephrosis (PAN) model. Exp Toxicol Pathol 64:45–49
Herrmann H, Aebi U (1998) Structure, assembly, and dynamics of intermediate filaments. Subcell Biochem 31:319–362
Herrmann H, Aebi U (2000) Intermediate filaments and their associates: multi-talented structural elements specifying cytoarchitecture and cytodynamics. Curr Opin Cell Biol 12:79–90
Herrmann H, Aebi U (2004) Intermediate filaments: molecular structure, assembly mechanism, and integration into functionally distinct intracellular Scaffolds. Annu Rev Biochem 73:749–789
Herrmann H, Bär H, Kreplak L, Strelkov SV, Aebi U (2007) Intermediate filaments: from cell architecture to nanomechanics. Nat Rev Mol Cell Biol 8:562–573
Herrmann H, Fouquet B, Franke WW (1989) Expression of intermediate filament proteins during development of Xenopus laevis. II. Identification and molecular characterization of desmin. Development 105:299–307
Herrmann H, Häner M, Brettel M, Ku NO, Aebi U (1999) Characterization of distinct early assembly units of different intermediate filament proteins. J Mol Biol 286:1403–1420
Herrmann H, Häner M, Brettel M, Müller SA, Goldie KN, Fedtke B, Lustig A, Franke WW, Aebi U (1996) Structure and assembly properties of the intermediate filament protein vimentin: the role of its head, rod and tail domains. J Mol Biol 264:933–953
Herrmann H, Hesse M, Reichenzeller M, Aebi U, Magin TM (2003) Functional complexity of intermediate filament cytoskeletons: from structure to assembly to gene ablation. Int Rev Cytol 223:83–175
Herrmann H, Strelkov SV, Burkhard P, Aebi U (2009) Intermediate filaments: primary determinants of cell architecture and plasticity. J Clin Invest 119:1772–1783
Herrmann H, Wiche G (1987) Plectin and IFAP-300 K are homologous proteins binding to microtubule-associated proteins 1 and 2 and to the 240-kilodalton subunit of spectrin. J Biol Chem 262:1320–1325
Hirako Y, Yamakawa H, Tsujimura Y, Nishizawa Y, Okumura M, Usukura J, Matsumoto H, Jackson KW, Owaribe K, Ohara O (2003) Characterization of mammalian synemin, an intermediate filament protein present in all four classes of muscle cells and some neuroglial cells: co-localization and interaction with type III intermediate filament proteins and keratins. Cell Tissue Res 313:195–207
Hnia K, Tronchere H, Tomczak KK, Amoasii L, Schultz P, Beggs AH, Payrastre B, Mandel JL, Laporte J (2011) Myotubularin controls desmin intermediate filament architecture and mitochondrial dynamics in human and mouse skeletal muscle. J Clin Invest 121:70–85
Hong D, Wang Z, Zhang W, Xi J, Lu J, Luan X, Yuan Y (2011) A series of Chinese patients with desminopathy associated with six novel and one reported mutations in the desmin gene. Neuropathol Appl Neurobiol 37:257–270
Hübbers CU, Clemen CS, Kesper K, Boddrich A, Hofmann A, Kamarainen O, Tolksdorf K, Stumpf M, Reichelt J, Roth U, Krause S, Watts G, Kimonis V, Wattjes MP, Reimann J, Thal DR, Biermann K, Evert BO, Lochmuller H, Wanker EE, Schoser BG, Noegel AA, Schröder R (2007) Pathological consequences of VCP mutations on human striated muscle. Brain 130:381–393
Hurlimann J (1994) Desmin and neural marker expression in mesothelial cells and mesotheliomas. Hum Pathol 25:753–757
Janmey PA, Euteneuer U, Traub P, Schliwa M (1991) Viscoelastic properties of vimentin compared with other filamentous biopolymer networks. J Cell Biol 113:155–160
Janue A, Odena MA, Oliveira E, Olive M, Ferrer I (2007) Desmin is oxidized and nitrated in affected muscles in myotilinopathies and desminopathies. J Neuropathol Exp Neurol 66:711–723
Janue A, Olive M, Ferrer I (2007) Oxidative stress in desminopathies and myotilinopathies: a link between oxidative damage and abnormal protein aggregation. Brain Pathol 17:377–388
Kaminska A, Strelkov SV, Goudeau B, Olive M, Dagvadorj A, Fidzianska A, Simon-Casteras M, Shatunov A, Dalakas MC, Ferrer I, Kwiecinski H, Vicart P, Goldfarb LG (2004) Small deletions disturb desmin architecture leading to breakdown of muscle cells and development of skeletal or cardioskeletal myopathy. Hum Genet 114:306–313
Kartenbeck J, Franke WW, Moser JG, Stoffels U (1983) Specific attachment of desmin filaments to desmosomal plaques in cardiac myocytes. EMBO J 2:735–742
Kawano F, Fujita R, Nakai N, Terada M, Ohira T, Ohira Y (2012) HSP25 can modulate myofibrillar desmin cytoskeleton following the phosphorylation at Ser15 in rat soleus muscle. J Appl Physiol 112:176–186
Klauke B, Kossmann S, Gaertner A, Brand K, Stork I, Brodehl A, Dieding M, Walhorn V, Anselmetti D, Gerdes D, Bohms B, Schulz U, Zu Knyphausen E, Vorgerd M, Gummert J, Milting H (2010) De novo desmin-mutation N116S is associated with arrhythmogenic right ventricular cardiomyopathy. Hum Mol Genet 19:4595–4607
Konieczny P, Fuchs P, Reipert S, Kunz WS, Zeöld A, Fischer I, Paulin D, Schröder R, Wiche G (2008) Myofiber integrity depends on desmin network targeting to Z-disks and costameres via distinct plectin isoforms. J Cell Biol 181:667–681
Kostareva A, Sjoberg G, Bruton J, Zhang SJ, Balogh J, Gudkova A, Hedberg B, Edstrom L, Westerblad H, Sejersen T (2008) Mice expressing L345P mutant desmin exhibit morphological and functional changes of skeletal and cardiac mitochondria. J Muscle Res Cell Motil 29:25–36
Kostareva A, Sjoberg G, Gudkova A, Smolina N, Semernin E, Shlyakhto E, Sejersen T (2011) Desmin A213 V substitution represents a rare polymorphism but not a mutation and is more prevalent in patients with heart dilation of various origins. Acta Myol 30:42–45
Kouloumenta A, Mavroidis M, Capetanaki Y (2007) Proper perinuclear localization of the TRIM-like protein myospryn requires its binding partner desmin. J Biol Chem 282:35211–35221
Kreplak L, Bär H (2009) Severe myopathy mutations modify the nanomechanics of desmin intermediate filaments. J Mol Biol 385:1043–1051
Kreplak L, Herrmann H, Aebi U (2008) Tensile properties of single desmin intermediate filaments. Biophys J 94:2790–2799
Kuisk IR, Li H, Tran D, Capetanaki Y (1996) A single MEF2 site governs desmin transcription in both heart and skeletal muscle during mouse embryogenesis. Dev Biol 174:1–13
Kumarapeli AR, Horak KM, Glasford JW, Li J, Chen Q, Liu J, Zheng H, Wang X (2005) A novel transgenic mouse model reveals deregulation of the ubiquitin-proteasome system in the heart by doxorubicin. FASEB J 19:2051–2053
Langley RC Jr, Cohen CM (1986) Association of spectrin with desmin intermediate filaments. J Cell Biochem 30:101–109
Li D, Tapscoft T, Gonzalez O, Burch PE, Quinones MA, Zoghbi WA, Hill R, Bachinski LL, Mann DL, Roberts R (1999) Desmin mutation responsible for idiopathic dilated cardiomyopathy. Circulation 100:461–464
Li H, Choudhary SK, Milner DJ, Munir MI, Kuisk IR, Capetanaki Y (1994) Inhibition of desmin expression blocks myoblast fusion and interferes with the myogenic regulators MyoD and myogenin. J Cell Biol 124:827–841
Li Z, Colucci-Guyon E, Pincon-Raymond M, Mericskay M, Pournin S, Paulin D, Babinet C (1996) Cardiovascular lesions and skeletal myopathy in mice lacking desmin. Dev Biol 175:362–366
Li Z, Mericskay M, Agbulut O, Butler-Browne G, Carlsson L, Thornell LE, Babinet C, Paulin D (1997) Desmin is essential for the tensile strength and integrity of myofibrils but not for myogenic commitment, differentiation, and fusion of skeletal muscle. J Cell Biol 139:129–144
Li ZL, Lilienbaum A, Butler-Browne G, Paulin D (1989) Human desmin-coding gene: complete nucleotide sequence, characterization and regulation of expression during myogenesis and development. Gene 78:243–254
Lichtenstern T, Mücke N, Aebi U, Mauermann M, Herrmann H (2012) Complex formation and kinetics of filament assembly exhibited by the simple epithelial keratins K8 and K18. J Struct Biol 177:54–62
Lindahl Allen M, Koch CM, Clelland GK, Dunham I, Antoniou M (2009) DNA methylation-histone modification relationships across the desmin locus in human primary cells. BMC Mol Biol 10:51
Linden M, Li Z, Paulin D, Gotow T, Leterrier JF (2001) Effects of desmin gene knockout on mice heart mitochondria. J Bioenerg Biomembr 33:333–341
Liu J, Chen Q, Huang W, Horak KM, Zheng H, Mestril R, Wang X (2006) Impairment of the ubiquitin-proteasome system in desminopathy mouse hearts. FASEB J 20:362–364
Lockard VG, Bloom S (1993) Trans-cellular desmin-lamin B intermediate filament network in cardiac myocytes. J Mol Cell Cardiol 25:303–309
Lyon GE, Buckingham ME (1993) Myogenic factor gene expression in mouse somites and limb buds. In: Bernfield M (ed) Molecular basis of morphogenesis. Symposia of the society for developmental biology series, vol 13, 1st edn. John, New York, pp 155–164
Mavroidis M, Panagopoulou P, Kostavasili I, Weisleder N, Capetanaki Y (2008) A missense mutation in desmin tail domain linked to human dilated cardiomyopathy promotes cleavage of the head domain and abolishes its Z-disc localization. FASEB J 22:3318–3327
Milner DJ, Mavroidis M, Weisleder N, Capetanaki Y (2000) Desmin cytoskeleton linked to muscle mitochondrial distribution and respiratory function. J Cell Biol 150:1283–1298
Milner DJ, Taffet GE, Wang X, Pham T, Tamura T, Hartley C, Gerdes AM, Capetanaki Y (1999) The absence of desmin leads to cardiomyocyte hypertrophy and cardiac dilation with compromised systolic function. J Mol Cell Cardiol 31:2063–2076
Milner DJ, Weitzer G, Tran D, Bradley A, Capetanaki Y (1996) Disruption of muscle architecture and myocardial degeneration in mice lacking desmin. J Cell Biol 134:1255–1270
Mitsui T, Kawajiri M, Kunishige M, Endo T, Akaike M, Aki K, Matsumoto T (2000) Functional association between nicotinic acetylcholine receptor and sarcomeric proteins via actin and desmin filaments. J Cell Biochem 77:584–595
Miyamoto Y, Akita H, Shiga N, Takai E, Iwai C, Mizutani K, Kawai H, Takarada A, Yokoyama M (2001) Frequency and clinical characteristics of dilated cardiomyopathy caused by desmin gene mutation in a Japanese population. Eur Heart J 22:2284–2289
Mizuno Y, Thompson TG, Guyon JR, Lidov HG, Brosius M, Imamura M, Ozawa E, Watkins SC, Kunkel LM (2001) Desmuslin, an intermediate filament protein that interacts with alpha -dystrobrevin and desmin. Proc Natl Acad Sci USA 98:6156–6161
Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y (2004) In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell 15:1101–1111
Munoz-Marmol AM, Strasser G, Isamat M, Coulombe PA, Yang Y, Roca X, Vela E, Mate JL, Coll J, Fernandez-Figueras MT, Navas-Palacios JJ, Ariza A, Fuchs E (1998) A dysfunctional desmin mutation in a patient with severe generalized myopathy. Proc Natl Acad Sci USA 95:11312–11317
Nelson WJ, Traub P (1983) Proteolysis of vimentin and desmin by the Ca2 + -activated proteinase specific for these intermediate filament proteins. Mol Cell Biol 3:1146–1156
Nicolet S, Herrmann H, Aebi U, Strelkov SV (2010) Atomic structure of vimentin coil 2. J Struct Biol 170:369–376
Nitou M, Ishikawa K, Shiojiri N (2000) Immunohistochemical analysis of development of desmin-positive hepatic stellate cells in mouse liver. J Anat 197(Pt 4):635–646
O’Connor CM, Asai DJ, Flytzanis CN, Lazarides E (1981) In vitro translation of the intermediate filament proteins desmin and vimentin. Mol Cell Biol 1:303–309
Ohlsson M, Hedberg C, Bradvik B, Lindberg C, Tajsharghi H, Danielsson O, Melberg A, Udd B, Martinsson T, Oldfors A (2012) Hereditary myopathy with early respiratory failure associated with a mutation in A-band titin. Brain 135:1682–1694
Ohtakara K, Inada H, Goto H, Taki W, Manser E, Lim L, Izawa I, Inagaki M (2000) p21-activated kinase PAK phosphorylates desmin at sites different from those for Rho-associated kinase. Biochem Biophys Res Commun 272:712–716
Olive M, Armstrong J, Miralles F, Pou A, Fardeau M, Gonzalez L, Martinez F, Fischer D, Martinez Matos JA, Shatunov A, Goldfarb L, Ferrer I (2007) Phenotypic patterns of desminopathy associated with three novel mutations in the desmin gene. Neuromuscul Disord 17:443–450
Olive M, Goldfarb L, Moreno D, Laforet E, Dagvadorj A, Sambuughin N, Martinez-Matos JA, Martinez F, Alio J, Farrero E, Vicart P, Ferrer I (2004) Desmin-related myopathy: clinical, electrophysiological, radiological, neuropathological and genetic studies. J Neurol Sci 219:125–137
Olive M, Odgerel Z, Martinez A, Poza JJ, Bragado FG, Zabalza RJ, Jerico I, Gonzalez-Mera L, Shatunov A, Lee HS, Armstrong J, Maravi E, Arroyo MR, Pascual-Calvet J, Navarro C, Paradas C, Huerta M, Marquez F, Rivas EG, Pou A, Ferrer I, Goldfarb LG (2011) Clinical and myopathological evaluation of early- and late-onset subtypes of myofibrillar myopathy. Neuromuscul Disord 21:533–542
Olive M, van Leeuwen FW, Janue A, Moreno D, Torrejon-Escribano B, Ferrer I (2008) Expression of mutant ubiquitin (UBB + 1) and p62 in myotilinopathies and desminopathies. Neuropathol Appl Neurobiol 34:76–87
Omary MB (2009) “IF-pathies”: a broad spectrum of intermediate filament-associated diseases. J Clin Invest 119:1756–1762
Otten E, Asimaki A, Maass A, van Langen IM, van der Wal A, de Jonge N, van den Berg MP, Saffitz JE, Wilde AA, Jongbloed JD, van Tintelen JP (2010) Desmin mutations as a cause of right ventricular heart failure affect the intercalated disks. Heart Rhythm 7:1058–1064
Panagopoulou P, Davos CH, Milner DJ, Varela E, Cameron J, Mann DL, Capetanaki Y (2008) Desmin mediates TNF-alpha-induced aggregate formation and intercalated disk reorganization in heart failure. J Cell Biol 181:761–775
Park KY, Dalakas MC, Goebel HH, Ferrans VJ, Semino-Mora C, Litvak S, Takeda K, Goldfarb LG (2000) Desmin splice variants causing cardiac and skeletal myopathy. J Med Genet 37:851–857
Park KY, Dalakas MC, Semino-Mora C, Lee HS, Litvak S, Takeda K, Ferrans VJ, Goldfarb LG (2000) Sporadic cardiac and skeletal myopathy caused by a de novo desmin mutation. Clin Genet 57:423–429
Paulin D, Huet A, Khanamyrian L, Xue Z (2004) Desminopathies in muscle disease. J Pathol 204:418–427
Pfeffer G, Elliott HR, Griffin H, Barresi R, Miller J, Marsh J, Evila A, Vihola A, Hackman P, Straub V, Dick DJ, Horvath R, Santibanez-Koref M, Udd B, Chinnery PF (2012) Titin mutation segregates with hereditary myopathy with early respiratory failure. Brain 135:1695–1713
Pica EC, Kathirvel P, Pramono ZA, Lai PS, Yee WC (2008) Characterization of a novel S13F desmin mutation associated with desmin myopathy and heart block in a Chinese family. Neuromuscul Disord 18:178–182
Pinol-Ripoll G, Shatunov A, Cabello A, Larrode P, de la Puerta I, Pelegrin J, Ramos FJ, Olive M, Goldfarb LG (2009) Severe infantile-onset cardiomyopathy associated with a homozygous deletion in desmin. Neuromuscul Disord 19:418–422
Pinset C, Montarras D, Chenevert J, Minty A, Barton P, Laurent C, Gros F (1988) Control of myogenesis in the mouse myogenic C2 cell line by medium composition and by insulin: characterization of permissive and inducible C2 myoblasts. Differentiation 38:28–34
Poon E, Howman EV, Newey SE, Davies KE (2002) Association of syncoilin and desmin: linking intermediate filament proteins to the dystrophin-associated protein complex. J Biol Chem 277:3433–3439
Price MG (1987) Skelemins: cytoskeletal proteins located at the periphery of M-discs in mammalian striated muscle. J Cell Biol 104:1325–1336
Pruszczyk P, Kostera-Pruszczyk A, Shatunov A, Goudeau B, Draminska A, Takeda K, Sambuughin N, Vicart P, Strelkov SV, Goldfarb LG, Kaminska A (2007) Restrictive cardiomyopathy with atrioventricular conduction block resulting from a desmin mutation. Int J Cardiol 117:244–253
Raats JM, Schaart G, Henderik JB, van der Kemp A, Dunia I, Benedetti EL, Pieper FR, Ramaekers FC, Bloemendal H (1996) Muscle-specific expression of a dominant negative desmin mutant in transgenic mice. Eur J Cell Biol 71:221–236
Reipert S, Steinbock F, Fischer I, Bittner RE, Zeold A, Wiche G (1999) Association of mitochondria with plectin and desmin intermediate filaments in striated muscle. Exp Cell Res 252:479–491
Robson RM, Huiatt TW, Bellin RM (2004) Muscle intermediate filament proteins. Methods Cell Biol 78:519–553
Rogatsch H, Jezek D, Hittmair A, Mikuz G, Feichtinger H (1996) Expression of vimentin, cytokeratin, and desmin in Sertoli cells of human fetal, cryptorchid, and tumour-adjacent testicular tissue. Virchows Arch 427:497–502
Sam M, Shah S, Friden J, Milner DJ, Capetanaki Y, Lieber RL (2000) Desmin knockout muscles generate lower stress and are less vulnerable to injury compared with wild-type muscles. Am J Physiol Cell Physiol 279:C1116–C1122
Sandoval IV, Colaco CA, Lazarides E (1983) Purification of the intermediate filament-associated protein, synemin, from chicken smooth muscle. Studies on its physicochemical properties, interaction with desmin, and phosphorylation. J Biol Chem 258:2568–2576
Sarparanta J, Jonson PH, Golzio C, Sandell S, Luque H, Screen M, McDonald K, Stajich JM, Mahjneh I, Vihola A, Raheem O, Penttila S, Lehtinen S, Huovinen S, Palmio J, Tasca G, Ricci E, Hackman P, Hauser M, Katsanis N, Udd B (2012) Mutations affecting the cytoplasmic functions of the co-chaperone DNAJB6 cause limb-girdle muscular dystrophy. Nat Genet 44(450–455):S451–S452
Sax CM, Farrell FX, Zehner ZE (1989) Down-regulation of vimentin gene expression during myogenesis is controlled by a 5′-flanking sequence. Gene 78:235–242
Schaffeld M, Herrmann H, Schultess J, Markl J (2001) Vimentin and desmin of a cartilaginous fish, the shark Scyliorhinus stellaris: sequence, expression patterns and in vitro assembly. Eur J Cell Biol 80:692–702
Schmid E, Schiller DL, Grund C, Stadler J, Franke WW (1983) Tissue type-specific expression of intermediate filament proteins in a cultured epithelial cell line from bovine mammary gland. J Cell Biol 96:37–50
Schopferer M, Bär H, Hochstein B, Sharma S, Mücke N, Herrmann H, Willenbacher N (2009) Desmin and vimentin intermediate filament networks: their viscoelastic properties investigated by mechanical rheometry. J Mol Biol 388:133–143
Schramm N, Born C, Weckbach S, Reilich P, Walter MC, Reiser MF (2008) Involvement patterns in myotilinopathy and desminopathy detected by a novel neuromuscular whole-body MRI protocol. Eur Radiol 18:2922–2936
Schrickel JW, Stockigt F, Krzyzak W, Paulin D, Li Z, Lubkemeier I, Fleischmann B, Sasse P, Linhart M, Lewalter T, Nickenig G, Lickfett L, Schroder R, Clemen CS (2010) Cardiac conduction disturbances and differential effects on atrial and ventricular electrophysiological properties in desmin deficient mice. J Interv Card Electrophysiol 28:71–80
Schröder R, Goudeau B, Simon MC, Fischer D, Eggermann T, Clemen CS, Li Z, Reimann J, Xue Z, Rudnik-Schoneborn S, Zerres K, van der Ven PF, Fürst DO, Kunz WS, Vicart P (2003) On noxious desmin: functional effects of a novel heterozygous desmin insertion mutation on the extrasarcomeric desmin cytoskeleton and mitochondria. Hum Mol Genet 12:657–669
Schröder R, Goudeau B, Simon MC, Fischer D, Eggermann T, Clemen CS, Li Z, Reimann J, Xue Z, Rudnik-Schoneborn S, Zerres K, van der Ven PF, Fürst DO, Kunz WS, Vicart P (2007) Erratum for: “On noxious desmin: functional effects of a novel heterozygous desmin insertion mutation on the extrasarcomeric desmin cytoskeleton and mitochondria”. Hum Mol Genet 16:2989–2990
Schröder R, Schoser B (2009) Myofibrillar myopathies: a clinical and myopathological guide. Brain Pathol 19:483–492
Schröder R, Vrabie A, Goebel HH (2007) Primary desminopathies. J Cell Mol Med 11:416–426
Schröder R, Warlo I, Herrmann H, van der Ven PF, Klasen C, Blumcke I, Mundegar RR, Furst DO, Goebel HH, Magin TM (1999) Immunogold EM reveals a close association of plectin and the desmin cytoskeleton in human skeletal muscle. Eur J Cell Biol 78:288–295
Schweitzer SC, Klymkowsky MW, Bellin RM, Robson RM, Capetanaki Y, Evans RM (2001) Paranemin and the organization of desmin filament networks. J Cell Sci 114:1079–1089
Selcen D (2011) Myofibrillar myopathies. Neuromuscul Disord 21:161–171
Selcen D, Engel AG (2004) Mutations in myotilin cause myofibrillar myopathy. Neurology 62:1363–1371
Selcen D, Ohno K, Engel AG (2004) Myofibrillar myopathy: clinical, morphological and genetic studies in 63 patients. Brain 127:439–451
Sharma S, Mücke N, Katus HA, Herrmann H, Bär H (2009) Disease mutations in the “head” domain of the extra-sarcomeric protein desmin distinctly alter its assembly and network-forming properties. J Mol Med 87:1207–1219
Sjoberg G, Jiang WQ, Ringertz NR, Lendahl U, Sejersen T (1994) Colocalization of nestin and vimentin/desmin in skeletal muscle cells demonstrated by three-dimensional fluorescence digital imaging microscopy. Exp Cell Res 214:447–458
Sjoberg G, Saavedra-Matiz CA, Rosen DR, Wijsman EM, Borg K, Horowitz SH, Sejersen T (1999) A missense mutation in the desmin rod domain is associated with autosomal dominant distal myopathy, and exerts a dominant negative effect on filament formation. Hum Mol Genet 8:2191–2198
Sparn HG, Lieder-Ochs BA, Franke WW (1994) Immunohistochemical identification and characterization of a special type of desmin-producing stromal cells in human placenta and other fetal tissues. Differentiation 56:191–199
Sprinkart AM, Block W, Traber F, Meyer R, Paulin D, Clemen CS, Schröder R, Gieseke J, Schild H, Thomas D (2011) Characterization of the failing murine heart in a desmin knockout model using a clinical 3 T MRI scanner. Int J Cardiovasc Imaging 28:1699–1705
Steinert PM, Chou YH, Prahlad V, Parry DA, Marekov LN, Wu KC, Jang SI, Goldman RD (1999) A high molecular weight intermediate filament-associated protein in BHK-21 cells is nestin, a type VI intermediate filament protein. Limited co-assembly in vitro to form heteropolymers with type III vimentin and type IV alpha-internexin. J Biol Chem 274:9881–9890
Strach K, Sommer T, Grohe C, Meyer C, Fischer D, Walter MC, Vorgerd M, Reilich P, Bär H, Reimann J, Reuner U, Germing A, Goebel HH, Lochmuller H, Wintersperger B, Schröder R (2008) Clinical, genetic, and cardiac magnetic resonance imaging findings in primary desminopathies. Neuromuscul Disord 18:475–482
Sugawara M, Kato K, Komatsu M, Wada C, Kawamura K, Shindo PS, Yoshioka PN, Tanaka K, Watanabe S, Toyoshima I (2000) A novel de novo mutation in the desmin gene causes desmin myopathy with toxic aggregates. Neurology 55:986–990
Sung RK, Ursell PC, Rame JE, Bailey H, Caleshu C, Nussbaum RL, Scheinman MM (2011) QTc prolongation and family history of sudden death in a patient with desmin cardiomyopathy. Pacing Clin Electrophysiol 34:e105–e108
Szeverenyi I, Cassidy AJ, Chung CW, Lee BT, Common JE, Ogg SC, Chen H, Sim SY, Goh WL, Ng KW, Simpson JA, Chee LL, Eng GH, Li B, Lunny DP, Chuon D, Venkatesh A, Khoo KH, McLean WH, Lim YP, Lane EB (2008) The Human Intermediate Filament Database: comprehensive information on a gene family involved in many human diseases. Hum Mutat 29:351–360
Tam JL, Triantaphyllopoulos K, Todd H, Raguz S, de Wit T, Morgan JE, Partridge TA, Makrinou E, Grosveld F, Antoniou M (2006) The human desmin locus: gene organization and LCR-mediated transcriptional control. Genomics 87:733–746
Taylor MR, Slavov D, Ku L, Di Lenarda A, Sinagra G, Carniel E, Haubold K, Boucek MM, Ferguson D, Graw SL, Zhu X, Cavanaugh J, Sucharov CC, Long CS, Bristow MR, Lavori P, Mestroni L (2007) Prevalence of desmin mutations in dilated cardiomyopathy. Circulation 115:1244–1251
Thornell L, Carlsson L, Li Z, Mericskay M, Paulin D (1997) Null mutation in the desmin gene gives rise to a cardiomyopathy. J Mol Cell Cardiol 29:2107–2124
Tidball JG (1992) Desmin at myotendinous junctions. Exp Cell Res 199:206–212
Tolstonog GV, Wang X, Shoeman R, Traub P (2000) Intermediate filaments reconstituted from vimentin, desmin, and glial fibrillary acidic protein selectively bind repetitive and mobile DNA sequences from a mixture of mouse genomic DNA fragments. DNA Cell Biol 19:647–677
Traub P (1995) Intermediate filaments and gene regulation. Physiol Chem Phys Med NMR 27:377–400
van Spaendonck-Zwarts K, van Hessem L, Jongbloed JD, de Walle HE, Capetanaki Y, van der Kooi AJ, van Langen IM, van den Berg MP, van Tintelen JP (2010) Desmin-related myopathy: a review and meta-analysis. Clin Genet 80:354–366
van Spaendonck-Zwarts KY, van der Kooi AJ, van den Berg MP, Ippel EF, Boven LG, Yee WC, van den Wijngaard A, Brusse E, Hoogendijk JE, Doevendans PA, de Visser M, Jongbloed JD, van Tintelen JP (2012) Recurrent and founder mutations in the Netherlands: the cardiac phenotype of DES founder mutations p.S13F and p.N342D. Neth Heart J 20:219–228
van Tintelen JP, Van Gelder IC, Asimaki A, Suurmeijer AJ, Wiesfeld AC, Jongbloed JD, van den Wijngaard A, Kuks JB, van Spaendonck-Zwarts KY, Notermans N, Boven L, van den Heuvel F, Veenstra-Knol HE, Saffitz JE, Hofstra RM, van den Berg MP (2009) Severe cardiac phenotype with right ventricular predominance in a large cohort of patients with a single missense mutation in the DES gene. Heart Rhythm 6:1574–1583
Vernengo L, Chourbagi O, Panuncio A, Lilienbaum A, Batonnet-Pichon S, Bruston F, Rodrigues-Lima F, Mesa R, Pizzarossa C, Demay L, Richard P, Vicart P, Rodriguez MM (2010) Desmin myopathy with severe cardiomyopathy in a Uruguayan family due to a codon deletion in a new location within the desmin 1A rod domain. Neuromuscul Disord 20:178–187
Vicart P, Caron A, Guicheney P, Li Z, Prevost MC, Faure A, Chateau D, Chapon F, Tome F, Dupret JM, Paulin D, Fardeau M (1998) A missense mutation in the alphaB-crystallin chaperone gene causes a desmin-related myopathy. Nat Genet 20:92–95
Vorgias CE, Traub P (1986) Nucleic acid-binding activities of the intermediate filament subunit proteins desmin and glial fibrillary acidic protein. Z Naturforsch C 41:897–909
Vrabie A, Goldfarb LG, Shatunov A, Nagele A, Fritz P, Kaczmarek I, Goebel HH (2005) The enlarging spectrum of desminopathies: new morphological findings, eastward geographic spread, novel exon 3 desmin mutation. Acta Neuropathol 109:411–417
Wahbi K, Behin A, Charron P, Dunand M, Richard P, Meune C, Vicart P, Laforet P, Stojkovic T, Becane HM, Kuntzer T, Duboc D (2012) High cardiovascular morbidity and mortality in myofibrillar myopathies due to DES gene mutations: a 10-year longitudinal study. Neuromuscul Disord 22:211–218
Walter MC, Reilich P, Huebner A, Fischer D, Schröder R, Vorgerd M, Kress W, Born C, Schoser BG, Krause KH, Klutzny U, Bulst S, Frey JR, Lochmüller H (2007) Scapuloperoneal syndrome type Kaeser and a wide phenotypic spectrum of adult-onset, dominant myopathies are associated with the desmin mutation R350P. Brain 130:1485–1496
Wang Q, Tolstonog GV, Shoeman R, Traub P (2001) Sites of nucleic acid binding in type I-IV intermediate filament subunit proteins. Biochemistry 40:10342–10349
Wang X, Klevitsky R, Huang W, Glasford J, Li F, Robbins J (2003) αB-Crystallin Modulates Protein Aggregation of Abnormal Desmin. Circ Res 93:998–1005
Wang X, Osinska H, Dorn GW 2nd, Nieman M, Lorenz JN, Gerdes AM, Witt S, Kimball T, Gulick J, Robbins J (2001) Mouse model of desmin-related cardiomyopathy. Circulation 103:2402–2407
Wang X, Osinska H, Klevitsky R, Gerdes AM, Nieman M, Lorenz J, Hewett T, Robbins J (2001) Expression of R120G-alphaB-crystallin causes aberrant desmin and alphaB-crystallin aggregation and cardiomyopathy in mice. Circ Res 89:84–91
Weisleder N, Taffet GE, Capetanaki Y (2004) Bcl-2 overexpression corrects mitochondrial defects and ameliorates inherited desmin null cardiomyopathy. Proc Natl Acad Sci USA 101:769–774
Wieneke S, Stehle R, Li Z, Jockusch H (2000) Generation of tension by skinned fibers and intact skeletal muscles from desmin-deficient mice. Biochem Biophys Res Commun 278:419–425
Yuan J, Huiatt TW, Liao CX, Robson RM, Graves DJ (1999) The effects of mono-ADP-ribosylation on desmin assembly-disassembly. Arch Biochem Biophys 363:314–322
Yuri T, Miki K, Tsukamoto R, Shinde A, Kusaka H, Tsubura A (2007) Autopsy case of desminopathy involving skeletal and cardiac muscle. Pathol Int 57:32–36
Zheng Q, Su H, Ranek MJ, Wang X (2011) Autophagy and p62 in cardiac proteinopathy. Circ Res 109:296–308
Zhou H, Huiatt TW, Robson RM, Sernett SW, Graves DJ (1996) Characterization of ADP-ribosylation sites on desmin and restoration of desmin intermediate filament assembly by de-ADP-ribosylation. Arch Biochem Biophys 334:214–222
Acknowledgments
This work is supported by grants of the Deutsche Forschungsgemeinschaft (DFG) awarded to C.S.C, H.H., and R.S. (FOR1228: CL 381/7-1; HE 1853/9-1, 9-2; SCHR 562/13-1). For this work C.S.C. and R.S. are also supported by the Deutsche Gesellschaft für Muskelkranke e.V. (DGM). Furthermore, R.S. is supported by the Johannes & Frieda Marohn Stiftung. S.V.S. was supported by the Katholieke Universiteit Leuven Research Supplement (OT) grant 07/071 and by the Research Foundation Flanders (FWO) grant G.0709.12. We are grateful to Prof. Oliver Friedrich (University of Erlangen-Nuremberg) for his input on biomechanical aspects, to Mr. cand. med. Gerrit Haaker (University Hospital Erlangen) for assistance with data acquisition for Table 1, and Mr. Matthias Bähre for his assistance in preparation of Figs. 5 and 6.
Open Access
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Clemen, C.S., Herrmann, H., Strelkov, S.V. et al. Desminopathies: pathology and mechanisms. Acta Neuropathol 125, 47–75 (2013). https://doi.org/10.1007/s00401-012-1057-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-012-1057-6