
Abstract
Temperature field measurements in liquids are demonstrated using zinc oxide (ZnO) thermographic phosphor particles. The particles are added to the liquid as a tracer. Following laser excitation, the temperature-dependent luminescence emission of the particles is imaged and the temperature is determined using a two-colour intensity ratio method. The particle size requirements for accurate temperature tracing in turbulent flows are calculated using a numerical heat transfer model. Particle–water mixtures were prepared using ultrasonic dispersion and characterised using scanning electron microscope imaging and laser diffraction particle-sizing, indicating that the particle size is 1–2 \(\upmu\)m. The particle luminescence properties were characterised using spectroscopic and particle luminescence imaging techniques. Using 355 nm laser excitation, the luminescence signal is the same in water and in air. However, 266 nm excitation is used to avoid spectral overlap between Raman scattering from water and the detected ZnO luminescence emission. It is shown that 266 nm excitation can be used for temperature measurements in water using mass loads as low as 1–5 mg L\(^{-1}\), corresponding to measured particle number densities 0.5–2.5 \(\times \,10^{12}\) particles \(\hbox {m}^{-3}\). In this range, the measured intensity ratio is independent of the mass load. The dependence of the intensity ratio on the laser fluence is less pronounced using excitation at 266 nm compared to 355 nm. A single-shot, single-pixel temperature precision of ±2–3 \(^{\circ }\hbox {C}\,(1\sigma )\) can be achieved over a temperature range spanning \(50\,^{\circ }\hbox {C}\). The technique was applied to a convection experiment to measure the temperature fields in a buoyant thermal plume, demonstrating the suitability of these imaging diagnostics for the investigation of thermal convection and heat transfer.
Similar content being viewed by others
1 Introduction
To investigate turbulent flows encountered in engineering applications and in nature it is necessary to measure various quantities such as the fluid temperature, flow velocity, and concentrations of mixtures or specific chemical species. For example, turbulent natural convection occurs in systems of geophysical and astrophysical importance like the atmosphere, oceans, the Earths mantle and in stars (Ahlers et al. 2009), as well as in, e.g. the ventilation of buildings and the solidification of metals. These systems are therefore of fundamental interest and are often studied numerically and experimentally. In the laboratory such flows are frequently investigated in scaled, simpler experiments with well-defined boundary conditions, for example the Rayleigh–Bénard configuration, i.e. a horizontal layer of fluid bounded by a heated bottom surface and a cooled top surface. In such thermally driven flows the two parameters of primary interest are temperature and velocity. To understand coherent flow structures in the boundary layer (e.g. Robinson 1991; Haramina and Tilgner 2004; Li et al. 2012; Puits et al. 2014) and the large-scale flow dynamics (e.g. Hartlep et al. 2003; Resagk et al. 2006; Schumacher and Emran 2015), it would be especially interesting to measure both temperature and velocity at the same time in order to study correlations and the interaction between buoyancy forces and the turbulent flow. The same is true for the investigation of heat transfer between a fluid and components actively cooled by forced convection, where measurements of temperature and velocity are needed to determine the heat flux. For electronic devices including computer chips, the measurements must also necessarily be at a small scale of 100’s \(\upmu \hbox {m}\).
Optical measurement techniques have the benefit that they do not rely on physical probes, e.g. thermistors at fixed locations that may perturb the flow. However, the primary advantage of optical methods is that there is the possibility to acquire instantaneous snapshots of whole regions of the flow field with a high spatial resolution. When the fluid in question is a liquid, in some circumstances temperature imaging techniques based on thermochromic liquid crystals (TLCs) (Smith et al. 2001) or laser-induced fluorescence (LIF) of temperature-sensitive dyes (such as rhodamine, e.g. Sakakibara and Adrian 1999) or particles can be used.
Established approaches based on TLCs have a very impressive temperature precision (0.01–0.1 \(^\circ \hbox {C}\), e.g. Schmeling et al. 2014) and can be combined with particle image velocimetry (PIV) to obtain the velocity field (Dabiri 2009). However, the temperature range is limited to a few \(^\circ \hbox {C}\). The temporal resolution is restricted by the long response times of TLCs (\(\sim\)10 ms for 10 \(\upmu \hbox {m}\) crystals in Schmeling et al. 2015) and the spatial resolution in the out-of-plane dimension is limited to several mm by the white light illumination sources employed (Dabiri 2009). Additionally, global vs local calibration strategies, the dependence of the colour scatter on particle size, and hysteresis or damage due to shear forces need to be considered to achieve the optimum precision and accuracy.
A temperature precision in the range 0.1–1 \(^\circ \hbox {C}\) (e.g. Sakakibara and Adrian 2004) can be achieved using two-colour rhodamine LIF thermometry, covering a wider temperature range (e.g. 20 \(^\circ \hbox {C}\), with a precision \({\pm }1.4\,^\circ \hbox {C}\) in Robinson et al. 2008). The technique has been applied simultaneously with PIV using additional tracer particles, and even in 3D throughout a whole liquid volume using a rapid scanning technique (Hishida and Sakakibara 2000).
Temperature-sensitive particles can also be synthesised by incorporating a temperature-dependent luminophore (e.g. EuTTA dye) into ion-exchange particles and used as a tracer for thermometry (Someya et al. 2010). The luminescence lifetime-based measurement requires a long integration time (\({\sim }\hbox {ms}\)) since camera exposures must be distributed across the decay waveform. The same particle luminescence images can be used for simultaneous velocimetry by evaluating the particle motion, either between laser pulses (Someya et al. 2009) or between camera exposures (Someya et al. 2010). The latter approach presumably degrades the temperature precision since the particles are not stationary during the integration period for the temperature measurement. While experimentally simple, this method necessitates a tradeoff between temporal and spatial resolution and measurement precision.
These tracers and the array of techniques based upon them have several attractive characteristics for different measurement situations. However, considering all the requirements of a suitable measurement technique—adequate spatial and temporal resolution, temperature range and precision, and compatibility with simultaneous velocity field measurements—they also have various and sometimes significant limitations. Of the methods listed above, the large particle size for TLCs and ion-exchange particles makes their tracing ability questionable in gas flows, a medium in which rhodamine LIF thermometry could also not be applied. From a practical standpoint, a flexible technique appropriate for measurements in gases and liquids would be very useful.
Thermographic Particle Image Velocimetry (thermographic PIV) is a laser-based technique for simultaneous two-dimensional temperature and velocity measurements. The method is based on thermographic phosphors, which are solid materials with temperature-dependent luminescence properties. Phosphor particles are seeded into the flow under investigation. A laser is used to excite the particles in the measurement plane, and their temperature-sensitive luminescence emission is imaged with cameras to determine the particle temperature using a two-colour intensity ratio method. Simultaneously, visible laser light scattered by particles in the same plane is recorded to determine the velocity field using a conventional particle image velocimetry (PIV) approach. Appropriately sized particles must be used, so that they closely follow the turbulent flow motion, and the particle temperature matches that of the surrounding fluid (Fond et al. 2012). So far primarily applied to gas flows (e.g. Omrane et al. 2008; Rothamer and Jordan 2012; Jordan and Rothamer 2012; Lawrence et al. 2013; Lipzig et al. 2013), this technique permits simultaneous, single-shot temperature-velocity imaging (Fond et al. 2012; Neal et al. 2013) at fast (multi-kHz) sampling rates (Abram et al. 2013), with a high (2o C) precision (Abram et al. 2015), using simple instrumentation and a single tracer.
In liquids, thermographic phosphor particles have previously been used as a tracer for average temperature measurements of single droplets using the lifetime (Omrane et al. 2004a, b) and spectral ratio (Omrane et al. 2004a) methods, as well as for lifetime-based planar measurements of droplets and sprays, using a fast framing camera for detection (Omrane et al. 2004c). The previously described two-colour intensity ratio technique has the advantage of providing 2D temperature measurements with a short integration time (\({<}\upmu \hbox {s}\)), using a simple two-camera or single-camera\(+\)stereoscope system. There are so far only two demonstrations of this two-colour temperature imaging in liquids. These determined the average temperature distribution in an n-dodecane spray using the thermographic phosphor \(\hbox {Mg}_4\hbox {GeO}_{5.5}\hbox {F:Mn}^{4+}\) (Brübach et al. 2006), and the temperature fields in burning methanol droplets (Särner et al. 2008). Neither paper addressed the temperature tracing response of the particles or investigated the particle morphology and luminescence characteristics in detail. The latter study used the phosphor ZnO:Ga. Our research group has recently shown that it is the intrinsic edge emission of zinc oxide (ZnO) itself that is redshifted with temperature, and have used ZnO particles for sensitive temperature measurements in gases (Abram et al. 2015).
In this paper we build on this work and the aforementioned liquid studies by extending the application of ZnO particles to temperature measurements in liquids. An experimental setup for characterising phosphor particles in liquids is developed (Sect. 2). The particle size appropriate for temperature and velocity tracing in liquids is determined and the size distribution and morphology of the particles is investigated (Sect. 3). The luminescence properties of ZnO particles dispersed in water are characterised and compared to gas-phase measurements using the same particles (Sects. 4, 5). The technique is also demonstrated in a convection experiment in water to image the development of a buoyant thermal plume (Sect. 6).
2 Experimental setup
2.1 Test object

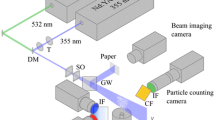
Setup diagram for characterisation of ZnO particles. The same setup was used for the temperature imaging demonstration
A mixture of deionised water and ZnO particles (96479, Sigma-Aldrich) was contained in a 28 mL (\(40\times 20\times 35\) mm) fused silica cuvette placed on a heated magnetic stirring plate. Prior to the characterisation and calibration measurements, using a calibrated type-K thermocouple and meter (maximum error \({\pm }0.5\,^{\circ }\hbox {C}\)) it was verified that when heating the dispersion with the stirrer switched on, the temperature was uniform throughout the liquid to within \(1\,^{\circ }\hbox {C}\). For demonstration purposes, to generate a strongly nonuniform-temperature field a \(10\times 10\) mm resistance heating block was fixed flat on the bottom of the inside of the cuvette and powered using a direct-current power supply.
2.2 Temperature imaging
Either the third (355 nm) or fourth (266 nm) harmonics of an Nd:YAG laser (GCR-150, Spectra-Physics) were used to excite the particles. Using +500 and −40 mm cylindrical lenses, the beam was formed into a light sheet intersecting the centre of the cuvette as shown in Fig. 1. To determine the laser fluence, the light sheet thickness was measured by reflecting the beam using a fused silica window and imaging the fluorescence of a paper target for 266 nm (this method can actually be used for a range of wavelengths, see Fond et al. 2015a), or directly illuminating the sensor of a webcam for 355 nm (e.g. Pfadler et al. 2009) (see also Fig. 1). A photodiode-based energy-monitoring unit (Energy monitor V9, LaVision), calibrated using a pyroelectric energy detector (ES245C, Thorlabs), was used to measure the energy of the laser on a shot-to-shot basis.
The particle luminescence emission was detected using two \(2\times 2\) hardware-binned interline transfer CCD cameras (Imager ProX 2M, LaVision) fitted with 50 mm f/1.4 objectives (Nikon). The exposure time was 5 \(\upmu \hbox {s}\). However, the luminescence lifetime, which determines the effective measurement duration, is below 1 ns. A 50:50 plate beamsplitter (46642) and two band-pass filters at 387-11 (84094) and 425-50 nm (86961, all from Edmund Optics) were used to separate and filter the two detection channels. To achieve spatial overlap between images prior to division, the reflection camera was mounted on micrometre translation stages and a laboratory jack. By adjusting these and by changing the vertical angle of the beamsplitter to account for rotation, the field-of-view of each camera (\(40\times 32\) mm) can be precisely matched. The mean residual displacement between particle images was \({<}0.24\; 2\times 2\)-binned pixels, as determined by a cross-correlation algorithm, and so no software mapping was used.
Luminescence images were acquired at a rate of 10 Hz. The background (camera offset) was subtracted before applying a cutoff filter \({>}20\) counts to remove low signals, and smoothing using a \(9\times 9\) moving average filter. The in-plane resolution was 450 \(\upmu \hbox {m}\) at 90 % contrast, as measured using an equivalently processed image of a resolution target. For the purpose of characterising the particle luminescence emission, average luminescence intensities and intensity ratios were extracted from a small region in the centre of the processed images as described in Sects. 4.3–4.5.
2.3 Particle counting
The particle number density was measured using a particle counting system (see Fond et al. 2015b). This consisted of an additional Nd:YAG laser at 532 nm and a third CCD camera. The laser beam was formed into a sheet overlapping the measurement plane. Mie scattering images were processed using MATLAB to determine the number of particles, and together with the probe volume size as defined by the measured green light sheet thickness (full-width half-maximum of the Gaussian beam profile) and camera field-of-view, the particle number density (particles \(\hbox {m}^{-3}\)) was determined. To ensure fair comparison between results, care was taken that the fluence of the green laser, which could affect the lower limit of particle detection using this method, was the same as that employed in previous gas-phase experiments (Abram et al. 2015).
The loss in resolution due to broadening of the light sheet by multiple scattering is considered to have a negligible effect on the measured number density, since the majority of the Mie scattering is in the forward direction along the direction of propagation of the laser beam. However, in this context it should be noted that, for thermometry, multiple scattering of luminescence signal between particles may reduce the measurement accuracy, especially in very large seeded volumes or where a large dynamic range of luminescence signal exists (e.g. in chemically reacting gas flows). Testing this in simple, generic configurations is an ongoing area of investigation.
2.4 Spectroscopy
Two setups were used for spectroscopic investigations of ZnO particles. For measuring the excitation spectrum, a fluorescence spectrometer (RF-5301PC, Shimadzu) was used. The device houses a 150 W xenon lamp and monochromator for excitation, monochromator and photomultiplier tube (PMT) for detection, and an additional PMT to automatically compensate for the excitation light power. The ZnO particle dispersion could be placed inside the sample holder in a cuvette which was continuously stirred using a magnetic stirrer. An additional filter (387-11 nm, see above) was placed on the detection line to additionally block scattered excitation light. The background was recorded using deionised water as a reference and subtracted from the results. For the measurements presented here, the signal-to-background ratio was greater than 10 across the investigated wavelength range (250–370 nm).
To obtain luminescence spectra, ZnO powder was contained in a ceramic crucible and excited at either 355 or 266 nm using the pulsed Nd:YAG laser described above. The laser beam diameter was adjusted using an iris and reflected onto the sample using a dichroic mirror appropriate for the excitation wavelength. Luminescence was collected using an f/4 lens; spectrally dispersed using a 300-mm focal length f/4 spectrometer (Acton SP-2300i, Princeton Instruments) with a grating groove density of 300 g mm\(^{-1}\) and entrance slit width of 100 \(\upmu \hbox {m}\); and detected using an interline transfer CCD camera (Imager ProX 2M, LaVision) with an exposure time of 5 \(\upmu\)s. The transmittance of the complete detection system was calibrated using the reference spectrum of a tungsten halogen lamp (LS-1, Ocean Optics).
3 Particle size and particle mixture preparation
3.1 Particle tracing properties
The inherent accuracy of the measurement technique depends on the time it takes for the particles to respond to changes in the fluid temperature and velocity. The matter has previously been addressed for gas flows, normally using properties for air (Fond et al. 2012). Here the analysis is extended to liquids.
The finite difference numerical model previously developed by our group (see Fond et al. 2012) was used to solve the heat conduction equation to evaluate the particle response time to a step change in the liquid temperature. This approach is able to account for local temperature-dependent fluid properties, a finite fluid volume, i.e. insulated system boundary conditions, and radiative heat transfer to the surroundings. The temperature-dependent properties of water, which is the liquid used in these experiments, were included in the model to account for changes in the thermal conductivity over the working temperature range (0–100 \(^{\circ }\hbox {C}\)). However, a semi-infinite medium, i.e. with the temperature of the liquid fixed far from the particle, was considered because of the low particle number densities of \(10^{11}-10^{12}\) particles \(\hbox {m}^{-3}\) used in these experiments and the high specific heat capacity of water. Radiative heat transfer was also not included owing to the near-ambient temperatures under consideration.
The calculated response times are adequately fast for many turbulent liquid flows of interest: \(\tau _{T,95\%} <35\,\upmu\)s for spherical ZnO particles with a diameter of \(5\,\upmu \hbox {m}\) in water between 0–100 \(^{\circ }\hbox {C}\). The particles can be significantly larger in a liquid than in a gas, where in air for example, particles with a diameter of 1 \(\upmu \hbox {m}\) have to be used to achieve a similar response time at \(20\,^{\circ }\hbox {C}\). The numerical model captures the thermal inertia of the fluid via the term including the thermal diffusivity, which is preserved in the time-dependent heat conduction equation for the fluid. We note that a simple lumped capacitance approach (Wadewitz and Specht 2001), as employed by Lipzig et al. (2013), may not be sufficiently accurate because the volumetric heat capacity, much larger for liquids like water, is not included in the time-independent heat conduction equation. This point will be explored in more detail in a future article.
For the velocity response time, the equation of motion for a spherical particle in a fluid has to be solved:
U is the velocity, \(U_{\mathrm{s}} =U_{\mathrm{p}}-U_{\mathrm{f}}\) is the slip velocity between the particles and the fluid, \(\mu\) the fluid dynamic viscosity, \(\rho\) is the density, \(d_{\mathrm{p}}\) the particle diameter, and g the acceleration due to gravity; subscripts p and f refer to the particle and fluid respectively. Since in water the particle Reynolds number is below unity for slip velocities up to 0.1 m s\(^{-1}\) for particles with a 5 \(\upmu \hbox {m}\) diameter, Stokes’ drag law has been assumed in the second term (note that at higher slip velocities the drag force would be larger than that predicted by Stokes’ drag law, so these calculated response times are a conservative estimate). The last three terms on the right-hand side describe the positive fluid pressure gradient, the added mass, and the buoyancy and gravity forces acting on the particle. The density of ZnO is \(5.6\,\hbox {g\,cm}^{-3}\), the same order of magnitude as water, so we then assume \(\rho _{\mathrm{p}} \approx \rho _{\mathrm{f}}\) and solve for velocity difference to yield the particle velocity relaxation time:
The retained added mass and pressure gradient terms have the effect of increasing the factor to \(\frac{1}{4}\), rather than the \(\frac{1}{6}\) normally present in the \(\tau _{U,95\%}\) expression for gases where usually \(\rho _{\mathrm{p}} \gg \rho _{\mathrm{f}}\) is assumed and so \(\rho _{\mathrm{f}} \approx 0\). The absolute particle density appears in the expression, indicating that neutrally buoyant particles in air respond faster than denser neutrally buoyant particles in, for example, water. The dynamic viscosity of water is nearly two orders of magnitude higher than for air, so as for temperature the particles can be considerably larger in liquids and still offer adequate tracing capability. For spherical ZnO particles 5 \(\upmu \hbox {m}\) in diameter, \(\tau _{U,95\%} = 35\;\upmu \hbox {s}\) in water at 20 \(^{\circ }\hbox {C}\). Typically, the temperature dependence of the dynamic viscosity is more pronounced in liquids and this should be considered if using larger particles in highly turbulent liquid flows.
The particle diameter is normally a critical parameter due to its quadratic dependence, especially when seeding solid particles into gas flows. We conclude that due to the differing properties of liquids and gases the constraints on the particle diameter are relaxed considerably for liquids. Larger particles can be used while still obtaining fast response times.
3.2 SEM imaging and particle size measurements
When adding ZnO particles to the liquid, agglomerates so large as to be visible to the eye were clearly present and these could not be removed by simply stirring the sample. Therefore, the particle–water mixtures were dispersed using an ultrasonic homogeniser (Sonopuls UW2070, Bandelin). The particle size distributions with and without this treatment were characterised using laser diffraction measurements (Mastersizer 2000, Malvern Instruments), as shown in Table 1. The median particle diameter based on volume does not change significantly (\(d_{(v,50)}\sim\)1–2 \(\upmu \hbox {m}\)) and is suitable for flow tracing purposes, but Fig. 2 shows that the distribution without ultrasound is bimodal, containing a number of very large particles in the range 10–100 \(\upmu \hbox {m}\). Therefore, ultrasonic dispersion was routinely used for the following experiments to prepare the particle–water mixtures, using successive dilutions of an initial mixture down to mass loads in the range 1–5 mg L\(^{-1}\) used for the actual measurements (Sect. 3.3).

Particle size distribution of ZnO powder, with and without ultrasonic dispersion
A qualitative overview of scanning electron microscope (SEM) images of the particles shows that the primary particles are submicron, on the scale of 100 nm. The image in Fig. 3 indicates that after ultrasonic dispersion the primary particles remain agglomerated, and that the majority of particles have projected sizes in the range 1–2 \(\upmu \hbox {m}\). Although there are a few particles slightly larger than this, these are of a size still adequate for accurate flow tracing (\({\sim }5\,\upmu \hbox {m}\)), and there are no very large agglomerates on the order \(100\,\upmu \hbox {m}\). The particle-sizing measurements (Fig. 2) can be expected to contain some inaccuracy since the particles are not homogeneous spheres, an assumption underlying the Mie theory that the device utilises. However, the SEM image also supports the picture that ultrasonic dispersion produces a particle size distribution favourable for flow tracing: in the range 1–2 \(\,\upmu \hbox {m}\), and free of large agglomerates. Due to improved drag characteristics and an increased ratio of particle surface area to particle volume, the respective time responses for both velocity and temperature are therefore expected to be even faster than the calculations above.

SEM image of ZnO powder following ultrasound treatment
3.3 Particle mixture preparation
The particle-sizing measurements indicate that ultrasound dispersion is necessary to remove large agglomerates, and so the following procedure was used to generate the liquid-particle dispersion. First, 20 mg of ZnO particles was added to 40 mL deionised water. In this dense initial dispersion containing large agglomerates the ultrasonic homogeniser (see above) was used for 2 min, after which the mixture appeared homogeneous and turbid. This was then diluted in two subsequent steps to the required mass load (1–5 mg L\(^{-1}\)), which appears completely clear to the eye in an ordinary one litre laboratory beaker.
3.4 Particle counting measurements
Using the previously developed particle counting system (Fond et al. 2015b) it is possible to directly measure the particle number density and therefore know how many particles contribute to the measured luminescence signal. This system was used to measure the number of particles for a fixed mass load of 1 mg L\(^{-1}\) in a dilution prepared using the procedure described above. For this measured mass load, the particle number density is \(4.8 \times 10^{11}\) particles \(\hbox {m}^{-3}\). From repeated measurements using new dilutions, the particle number density varied by only ±12 % of this mean value. Measured mass loads were used to obtain the required particle number densities and unless stated otherwise the particle number densities for all subsequent experiments described below are subject to this small error.
It was noted from the particle counting measurements that the particle number density was not temporally stable on the scale of several tens of minutes, where both the recorded luminescence intensity and number of counted particles gradually decreased proportionally over time. The temporal course of this behaviour was not consistent during repeat tests with the magnetic stirrer switched on. This stirring action should prevent the settling of particles, and the settling velocity assuming \(d_{(v,50)}=1.2\,\upmu \hbox {m}\) spherical particles is calculated to be 1 cm h\(^{-1}\) in quiescent water. Simple settling of particles was therefore not considered to be a likely reason for the observed behaviour. Switching the stirrer off, using deionised water from a different source, or using a borosilicate cuvette did not improve the situation. If a solution was left for 24 h, it was not possible to obtain the same particle number density, even if the particles were redispersed by ultrasound. In all cases, after 30 min had elapsed the presence of a weak luminescence signal emanating from the walls of the cuvette indicated that the particles are presumably attracted to them.
In this case, the ratio of the container surface area to total liquid volume is not favourable and we consider that the use of a larger cuvette or a recirculation system would allow the experimenter to establish a stable number density of particles over longer (hours) periods of time. This will increase the repeatability of some characterisation tests performed using liquid-particle mixtures requiring a fixed number of particles for an extended duration.
4 Results
4.1 Interference from Raman scattering
The previous gas-phase temperature measurements with ZnO particles used 355 nm excitation (Abram et al. 2015). Using this wavelength and a laser energy of 3 mJ/pulse, a series of initial test measurements were taken, first with an empty cuvette, and a second with deionised water only. The first of these tests revealed no signal in either channel, but in the second test, a weak signal could be detected in the 425-50 nm channel. The 425-50 interference filter has an optical density (OD) 5 at 355 nm. To ensure this signal was not laser light scattered at 355 nm an additional notch filter blocking 355 nm (also OD 5) was temporarily installed, but this did not reduce the signal intensity. An integrated fibre-optic-coupled CCD spectrometer (BRC641E, BW-Tek) was used to measure the spectrum of the signal, revealing a peak at 404 nm as shown in Fig. 4. The spectral location of the interfering signal suggests it is Raman-scattered 355 nm laser light corresponding to the water O–H stretch at 3400 \(\hbox {cm}^{-1}\). This was confirmed by installing a polarisation filter (Hoya) on the 425-50 nm detection channel and measuring the ratio between the polarised and depolarised components of the signal. Our value of 0.3 is similar to the Raman scattering depolarisation ratio in liquid water measured by Bray et al. (2013) (0.2 at 3400 \(\hbox {cm}^{-1}\)).

Spectrum recorded from a water-filled cuvette containing no phosphor particles illuminated with 355 nm laser light. The left peak is the laser line, and the right peak is the interfering Raman signal at \({\sim }404\) nm
Instead of the third harmonic of an Nd:YAG laser, using the fourth harmonic at 266 nm would shift this Raman line to \(\sim\)293 nm, which is well-separated from the ZnO edge luminescence and outside the passbands of both filters (see Fig. 6). This is a simple means to avoid generating the Raman signal at a wavelength directly overlapping the ZnO edge emission, and was the approach adopted for all subsequent measurements. Other possible solutions are discussed in Sect. 5.
4.2 Excitation and emission spectra
Measurements were taken to identify differences between excitation at the UV Nd:YAG harmonics at 355 and 266 nm. A dispersion of ZnO particles in a 3 mL cuvette was placed inside the fluorescence spectrometer. A continuous scan monitoring the ZnO edge emission peak at 387 nm is shown in Fig. 5. The luminescence signal is normalised to 355 nm. Using the continuous output of the xenon lamp, the relative signal using 266 nm excitation is about 55 % of the 355 nm value.

Excitation spectrum of ZnO particles dispersed in water, normalised to 355 nm
Using the setup described in Sect. 2.4, the luminescence emission was also measured using pulsed (10 ns) excitation at 355 and 266 nm with the Nd:YAG laser, at a fluence of 1.2 mJ \(\hbox {cm}^{-2}\). The results shown in Fig. 6 are for aggregated powder placed in a crucible. The spectra are normalised to indicate that there is little difference in the lineshape or emission peak position using different excitation wavelengths. The Raman scattering peak and the temperature-shifted ZnO emission spectrum at 95 \(^\circ \hbox {C}\) are also shown, to illustrate the spectral overlap. It should be noted that the relative signal level using 355 or 266 nm excitation determined by such a test is not a reliable indicator of the signal from an individual ZnO particle due to multiple scattering effects in the bulk powder, and this evaluation is left for measurements in water (Sect. 4.3).

Emission spectrum of ZnO particles at 23 \(^\circ \hbox {C}\) using 266 and 355 nm excitation (this study), and 355 nm excitation at 95 \(^\circ \hbox {C}\) (Abram et al. 2015). The measured spectrum (not to scale) of the Raman scattering from water at \(\sim\)404 nm and the transmission curves of the interference filters used in this study are also marked on the plot
4.3 Luminescence signal
The luminescence signal per particle is an important parameter which directly affects the precision of the temperature measurement. Since in these water experiments the particles are dispersed in a very different manner to that previously used for gas flows, the particle size might be quite different. Therefore initially the luminescence signal was measured, with the first aim to confirm that the signal per particle was the same in air as in water. For air, these data are already available from the previous study using the same ZnO particles (Abram et al. 2015). To obtain similar data in water, a dispersion of ZnO particles was prepared and illuminated using the laser at 355 nm. Simultaneously, the laser energy was measured on a shot-to-shot basis. The average signal in the 387-11 nm detection channel was calculated for each laser shot and is plotted as a function of laser fluence in Fig. 7.
For comparison, the same detection channel was considered for both air and water experiments (387-11 nm filter). The data were compared at a fluence of 40 mJ \(\hbox {cm}^{-2}\) and \(1\times 10^{11}\) particles \(\hbox {m}^{-3}\). The laser sheet thickness chosen in each experiment, which changes the number of particles contained in the probe volume and therefore the luminescence signal, was also accounted for. Though the same f/1.4 camera objectives were used in each study, different CCD cameras were used and so the ADC conversion factors, quantum efficiency and hardware-binned pixel size, as well as the magnification, were all accounted for according to the equation presented in Fond et al. (2015a). The calculation shows that for ZnO using 355 nm excitation, the measured signal per particle for air and water is the same (numerical values are within 6 %). A single ZnO particle emits \({\sim }3\times 10^{6}\) photons using a fluence of 40 mJ \(\hbox {cm}^{-2}\), using a 10 ns laser pulse at 355 nm. For both sets of measurements, the main sources of uncertainty are the absolute laser fluence, the particle number density measurement, and the camera ADC, which can differ from that stated by the manufacturer. Considering a 10 % error for each of these factors in each experiment, the overall uncertainty is estimated to be 25 %.

Filtered luminescence signal against laser fluence for 355 and 266 nm excitation, for ZnO particles dispersed in water. The data have been corrected for small differences in the mass load using scattering from the particles at 532 nm, and for the laser sheet thickness
Since the intention was to use 266 nm excitation to eliminate Raman interference, the second aim of these measurements was to check the luminescence signal per particle using this excitation wavelength. For this, the mass load was kept same as for the 355 nm measurements described above. As previously mentioned, the number density for a given measured mass load is subject to a ±12 % error. Therefore to improve the accuracy of the excitation wavelength comparison, the scattering signal from the particles at 532 nm was monitored using an additional CCD and the average signal from each dataset was used to correct for small variations in the particle number density. The measurements were also corrected for the UV laser sheet thickness. Since the same setup was used for both excitation wavelengths, the main source of error in the signal per particle comparison stems from the UV laser fluence measurement, for an overall uncertainty of 15 %.
The results are displayed in Fig. 7, showing that the signal is approximately a factor two lower for 266 nm excitation. This corresponds to the measured excitation spectrum (Fig. 5). The saturation behaviour is also prominent, where the rate of increase of the luminescence emission decreases with laser fluence. This finding is consistent with studies of ZnO (Abram et al. 2015) and BAM:Eu\(^{2+}\) (Fond et al. 2015a) particles in air.
4.4 Seeding density effects
The temperature is inferred from the ratio of the luminescence signal intensities recorded by each camera, in specific spectral regions determined by the interference filters (see Fig. 6). Therefore, possible codependencies of the intensity ratio on other experimental parameters should be investigated. The band gap of ZnO particles is at 3.37 eV (Rodnyi and Khodyuk 2011), corresponding to \(\sim\)368 nm, and the room temperature luminescence emission peak is shifted only slightly to \(\sim\)385 nm. Therefore there is some overlap between absorption and emission and so luminescence could be reabsorbed by particles on the detection path. For example, with the filters used here this effect would dominate in the 387-11 nm channel, so the intensity ratio might be expected to increase with the particle number density.
This was checked for a range of mass loads in the range 1–5 mg L\(^{-1}\), corresponding to particle number densities between \(5\times 10^{11}\) and \(2.5\times 10^{12}\) particles \(\hbox {m}^{-3}\), by measuring the average intensity ratio using the two-colour thermometry system. The results shown in Fig. 8 indicate that within the measurement uncertainty there is no obvious trend in the intensity ratio. Any changes in the particle number density during experiments are therefore supposed to have no effect on the measurement accuracy.

Intensity ratio measured for different mass loads of ZnO particles dispersed in water. The uncertainty in the intensity ratio was determined from a repeat measurement with a different 2 mg L\(^{-1}\) dilution. In addition, for these experiments the uncertainty in the measured mass load was ±10 %
4.5 Excitation laser fluence
The previous study using ZnO particles in air found that there was a dependence of the intensity ratio on the excitation laser fluence at 355 nm (Abram et al. 2015). This was checked for the 266 nm excitation adopted for the measurements in water. Single-shot intensity ratios were evaluated from each instantaneous image and the laser energy was measured on a shot-to-shot basis. These results are shown in Fig. 9. Also, in this plot the results using 355 nm excitation in air are reproduced for comparison.

Intensity ratio against laser fluence. The 266 nm results were measured in water as part of this study, and the 355 nm results are from Abram et al. (2015). The results are normalised to 5 mJ \(\hbox {cm}^{-2}\) to show the relative trend
The results indicate that there is also some dependence of the intensity ratio on the laser fluence when using excitation at 266 nm. However, the dependence is less prominent. Between 5 and 20 mJ \(\hbox {cm}^{-2}\), the ratio increases by 55 % for 266 nm excitation as oppose to a 75 % increase using 355 nm. As discussed in Sect. 5, this is a benefit of 266 nm excitation, since in temperature imaging experiments this dependence must be corrected for.
Although the emission spectrum of the bulk ZnO powder is very similar using each excitation wavelength (Fig. 6), in comparing the liquid and gas-phase measurements the absolute value of the intensity ratio is different for each excitation wavelength (hence the normalisation in Fig. 9). However, different cameras and fields of view were employed in each case. Variations in camera gain or the effective filter transmission curves in different imaging configurations might be responsible this difference in the absolute intensity ratio values.
4.6 Temperature calibration
To calibrate the intensity ratio against temperature, a particle–water dispersion (5 mg L\(^{-1}\)) was continuously stirred and heated using the heating plate. At various intermediate temperatures, sets comprising of 100 images were acquired with the 266 nm laser energy set at 10 mJ \(\hbox {cm}^{-2}\) (800 \(\upmu \hbox {m}\) light sheet thickness). Though the fluence-dependence of the intensity ratio is reduced at higher fluence, this fluence was chosen because in practice the available laser energy may be a limiting factor in experiments with significantly larger probe volumes. Intensity ratio images were divided by an average intensity ratio image at room temperature as a correction for spatial nonuniformity in light collection efficiency and spatial variation in the laser fluence. The laser energy was measured at each calibration temperature so that slight variations in laser fluence could be corrected for. The temperature calibration points and quadratic fit are shown in Fig. 10. The mean deviation of the calibration points from the fit is 0.3 \(^\circ \hbox {C}\), and the maximum deviation is 0.6 \(^\circ \hbox {C}\). The displayed errorbar is based on the error of the thermocouple and meter used to reference the absolute temperature (\({\pm }0.5\,^\circ \hbox {C}\)). The sensitivity, which at 24 \(^\circ \hbox {C}\) is \({\sim }0.7\,\%/^\circ \hbox {C}\), is across the range 20–70 \(^\circ \hbox {C}\) very similar to that obtained using 355 nm excitation in air using the same filter combination (Abram et al. 2015).

Temperature calibration curve using 266 nm excitation at a fluence of 10 \(\hbox {mJ\,cm}^{-2}\). The uncertainty in the absolute calibration temperature is ±0.5 °C
Since the temperature fields are uniform the same images used for the calibration were also used to assess the temperature precision. Pulse-to-pulse fluctuations in the laser energy were continuously measured using the energy monitor, allowing correction of each single-shot image using the laser fluence-intensity ratio data of Fig. 9. The calibration curve was used to convert the corrected intensity ratio fields into temperature. The single-shot precision (the average standard deviation (\(1\sigma\)) of the processed pixels in each instantaneous temperature image) was \({\pm }2.9\,^\circ \hbox {C}\) at \(25\,^\circ \hbox {C}\) (1.0 % of the absolute temperature) and \({\pm }2.1\,^\circ \hbox {C}\) (0.6 %) at \(72\,^\circ \hbox {C}\), for an in-plane spatial resolution of \(450\,\upmu \hbox {m}\). The reason for the improved temperature precision is the improved sensitivity with increasing temperature using this filter combination. The shot-to-shot standard deviation (1\(\sigma\)) of the average temperature was \({\pm }1.4\,^\circ \hbox {C}\) (0.5 %) at \(25\,^\circ \hbox {C}\).
5 Discussion
In this study we use 266 nm excitation to avoid the interfering Raman scattering from water, thereby making use of the broad absorption band of ZnO. In general, broad absorption bands are an attractive feature of thermographic phosphors. Excitation flexibility is beneficial in this case because spectral discrimination of the luminescence signal and Raman interference on the detection side would be difficult to achieve. Inspection of Fig. 6 shows that choosing a different filter combination to avoid integrating the Raman signal at 404 nm would severely compromise the signal level for measurements in the temperature range 0–100 \(^\circ \hbox {C}\). The short-lived edge luminescence emission has a lifetime of 100's ps, which also precludes temporal discrimination using ns laser pulses. Even using a polarisation filter on the appropriate channel, the depolarised component of the Raman scattering would still be transmitted. Water was used in these experiments because it is easy to handle and used in numerous practical applications, but different liquids will have different Raman spectra and might permit 355 nm excitation for measurements in this temperature range.
The comparison of the two excitation wavelengths indicates the signal is a factor two lower for 266 nm. For the same laser fluence, there are less excitation photons at 266 nm, partly explaining the difference. The room temperature absorption spectrum in Klingshirn (2007) shows continuous absorption above the band gap energy, and aside from a small minima at around 325 nm (which corresponds well to our excitation measurements) the absorption coefficient gradually increases farther into the UV spectral region. However, the absorption coefficient is very similar at 266 and 355 nm (\({\sim }2.7\times 10^5\; \hbox {cm}^{-1}\)). Together with our excitation measurements, this may suggest that the quantum efficiency at 266 nm is lower than at 355 nm. However, it should be noted that the luminescence signal per particle and the excitation spectrum were obtained with a very different excitation irradiance. This complicates the interpretation of these results due to saturation effects or nonlinear emission behaviour due to the high exciton density in the material when using pulsed laser excitation of the order MW \(\hbox {cm}^{-2}\). Nevertheless, if necessary the lower signal using 266 nm excitation can be regained by using twice the mass load of particles in the liquid.
It was not possible to compare the trend of intensity ratio with laser fluence in water with both excitation wavelengths due to the interfering Raman signal when using 355 nm. However, it is likely that the previously identified effects of excitation irradiance and laser-induced heating (Abram et al. 2015) are similar irrespective of whether the particle is immersed in a liquid or gas (noting that, in a liquid or gas, the timescale of particle-fluid heat transfer is several orders of magnitude longer than the near-instantaneous heating effect produced by the 10 ns laser pulse, and the <1 ns emission lifetime of ZnO). This paper is devoted to characterisation of the particles for use in liquids, but this trend will be confirmed in future experiments in air flows. The results of this work show that the relative change in the intensity ratio with laser fluence using 266 nm excitation in water is less pronounced than when using 355 nm excitation in air. For a fixed variation in the laser energy, the induced variation in the measured intensity ratio is lower. This is beneficial for the temperature measurement because there is less demand on the correction employed for spatial and temporal variations in the fluence, as discussed in Abram et al. (2015).
The average spatial laser fluence distribution is addressed by dividing by a time-average uniform-temperature intensity ratio image. Additionally though, the laser fluence across the sheet thickness is not uniform, so a particle will be illuminated by a different fluence depending on its depth position within the light sheet. However, at particle number densities \(10^{11}-10^{12}\) particles \(\hbox {m}^{-3}\) there are so many particles in each probe volume that this has a negligible effect on the measurement precision (Abram et al. 2015). Also, it was shown in that work that, for ZnO particles in the temperature range of these experiments, the change in the normalised intensity ratio with temperature is the same irrespective of laser fluence, so a single calibration curve can be used to convert the intensity ratio images to temperature (Abram et al. 2015). Yet we still emphasise the need to reduce the variation in laser fluence to minimise residual error. For example, because the relative sensitivity of the intensity ratio to laser fluence varies with the fluence, even for a temporally stable laser beam profile, overall laser energy fluctuations will cause a variation in the measured temperature that depends on the average fluence distribution within the measurement volume, i.e. a fixed pattern error. For this experimental setup, the spatial fluence variation in the measurement volume was at maximum 50 %, which, using data from Fig. 9, leads to a calculated \({\sim }1\,^\circ \hbox {C}\) error due to temporal laser energy fluctuations. For these measurements, this is on the order of the random noise and is included in the temperature precision estimation. Larger variations must be avoided, by using for example a beam homogeniser (see, e.g. Pfadler et al. 2006). In this regard, we also note that the spatial variation in laser fluence caused by refractive index gradients is difficult to correct for. The intensity ratio dependence on the laser fluence for ZnO may cause difficulties in strongly nonuniform-temperature flows.
We consider two possible explanations for the different dependencies of the intensity ratio on the laser fluence with 355 and 266 nm excitation wavelengths. On the one hand, if laser-induced heating is primarily responsible for the behaviour, since the difference between excitation and emission wavelengths is larger using 266 nm excitation more energy would be dissipated in the particles as heat, leading to a stronger dependence of intensity ratio on laser fluence. In comparing the trends, these results are therefore somewhat surprising. On the other hand, Abram et al. (2015) showed that the increase in the intensity ratio with laser fluence was in part due to the high excitation irradiance. For the same laser fluence, there are less photons at 266 nm, and so the photon irradiance is lower. This may affect the density of excited centres in each particle, causing a spectral shift of the luminescence emission (Klingshirn 2007) and thereby altering the relative trend in the intensity ratio with laser fluence.
The SEM images show that after ultrasound treatment the particles remain irregularly shaped agglomerates, and the particle-sizing measurements indicate that larger agglomerates are broken up. The measurements of luminescence signal per particle confirmed that the signal per particle in air and in water is very similar using 355 nm excitation. Since we do not anticipate any influence of the environment on the particle luminescence emission, and that there is likely to be no significant difference in the absorption of laser light by the particles stemming from the different refractive indices of air and water, these measurements do suggest that the size of the ZnO agglomerates is similar, whether using ultrasound in water or seeding in a gas flow. In our experience, we have found that seeders for gas flows that produce a high degree of centrifugation, high velocities leading to high shear forces, and agitation of the particle bed produces an aerosol free of large agglomerates, with improved spatial homogeneity and temporal consistency. It would seem that larger agglomerates are effectively broken up by these processes and the resulting dispersion is similar to that produced using ultrasound treatment in a liquid.
6 Temperature imaging demonstration

Evolving temperature fields above a \(10\times 10\) mm resistance heating block. Only every fifth image of the 10 Hz recording is displayed. In the second and third images, a little bubble can be seen rising with the buoyant plume
The resistance heating block was fixed inside the cuvette, which was filled with a dispersion of particles. The 266 nm laser light sheet intersected the centre of the cuvette. Starting from a uniform room temperature, the heating block was switched on, while luminescence images were continuously acquired at a rate of 10 Hz. These were processed to produce temperature images using the previously recorded calibration curve.
A time sequence of the evolving temperature field is shown in Fig. 11. A plume forms above the block and rises upward due to buoyancy. The plume increases in temperature and grows, forming a convective current within the cuvette. This 10 Hz sampling rate is sufficient to resolve the flow behaviour in time, but only every fifth image of the later part of the recording is shown in Fig. 11. The full temperature difference is \({\sim }50\,^\circ \hbox {C}\) and the plume is clearly visible against the background noise (\({\pm }5\,^\circ \hbox {C}\) at the 95 % (\(2\sigma\)) confidence level). A video of the plume development is available online as supplementary material to this article.
7 Conclusions
Calculations were performed to establish a suitable particle size for flow tracing in liquids, showing that spherical ZnO particles 5 \(\upmu \hbox {m}\) in diameter have response times for temperature and velocity below 35 \(\upmu \hbox {s}\) in water at 20 \(^{\circ }\hbox {C}\), fast enough for most turbulent liquid flows of interest. SEM images of particle–water mixtures following ultrasonic dispersion indicated that the particles remain in an agglomerated form with a projected size on the scale of 1–2 \(\upmu \hbox {m}\). This supported the results of the laser diffraction particle-sizing measurements which showed that ultrasonic dispersion is essential to remove large agglomerates. The number of particles was directly measured for a given mass load finding that, using the developed dispersion method, 1 mg L\(^{-1}\) corresponds to a particle number density \(4.8 \times 10^{11}\) particles \(\hbox {m}^{-3}\). The particle luminescence properties were characterised using spectroscopic and particle luminescence imaging techniques, determining that:
-
Using 355 nm laser excitation, the luminescence signal in water and in air is the same (to within 25 %). A single ZnO particle emits \({\sim }3\times 10^{6}\) photons using a fluence of 40 mJ \(\hbox {cm}^{-2}\), using a 10 ns laser pulse at 355 nm.
-
In water 355 nm excitation generates Raman scattering at \(\sim\)404 nm which spectrally overlaps the ZnO luminescence emission.
-
The interfering Raman scattering can be avoided by using 266 nm radiation (the fourth harmonic of an Nd:YAG laser) to excite ZnO.
-
Compared with 355 nm excitation, the luminescence signal per particle is a factor two (15 % uncertainty) lower using 266 nm.
-
Compared with 355 nm excitation, the dependence of the intensity ratio on laser fluence is lower using 266 nm.
-
The intensity ratio is independent of the mass load in the range 1–5 mg L\(^{-1}\), corresponding to particle number densities between \(5\times 10^{11}\) and \(2.5\times 10^{12}\) particles \(\hbox {m}^{-3}\).
The technique was successfully used to measure the temperature field in a thermal plume developing above a resistance heating block. It is straightforward to use the same tracer particles for PIV, enabling simultaneous velocity measurements. The diagnostics have a short integration time (\(\sim\)ns), can be applied at kHz repetition rates, have a good spatial resolution of the order 100's \(\upmu \hbox {m}\), and could be used to measure over a broad temperature range (\({>}100\,^\circ \hbox {C}\)) while maintaining a good precision (\({\pm }2{-}3\,^{\circ }\hbox {C}, 1\sigma\)), serving as a useful addition to laser diagnostics for temperature-velocity imaging in liquids.
Future work in this area should focus on the following:
-
To further investigate the dependence of the luminescence signal and intensity ratio on the laser fluence using 266 nm excitation in air, and acquire spectrally resolved measurements of water-particle mixtures using 355 and 266 nm excitation.
-
A well-known difficulty of measuring in liquids is the laser striping effects caused by refractive index gradients in strongly nonuniform-temperature flows. For ZnO, the identified dependence of the intensity ratio on the laser fluence is a potential issue. Sensitive phosphors that exhibit a reduced spectral dependence on laser fluence may be required.
-
An additional helpful outcome of this study is related to the fact that characterising phosphors on a single particle basis is essential to find new, useful phosphors and design thermographic PIV experiments. While spectroscopic studies covering the appropriate temperature range and tests suited to the specific application are both often necessary, this work shows that liquids are a good medium in which to measure the signal per particle at room temperature, the quantity of interest. Only a small amount (<0.1 mg for the 28 mL cuvette used in this study) of powder is needed for a simpler experiment, compared with the requirement for much larger (100 g) quantities of particles for the seeders typically used for gas flows. Experiments like these can be used to perform carefully controlled investigations of single phosphor particles to systematically investigate the effect of, e.g. particle size, host compound, dopant/sensitiser concentration and other important parameters related to particle morphology and phosphor composition.
References
Abram C, Fond B, Heyes A, Beyrau F (2013) High-speed planar thermometry and velocimetry using thermographic phosphor particles. Appl Phys B 111:155–160
Abram C, Fond B, Beyrau F (2015) High-precision flow temperature imaging using ZnO thermographic phosphor tracer particles. Opt Express 23:19,453–19,468
Ahlers G, Grossmann S, Lohse D (2009) Heat transfer and large-scale dynamics in turbulent Rayleigh–Bénard convection. Rev Mod Phys 81:503–537
Bray A, Chapman R, Plakhotnik T (2013) Accurate measurements of the Raman scattering coefficient and the depolarization ratio in liquid water. Appl Opt 52:2503–2510
Brübach J, Patt A, Dreizler A (2006) Spray thermometry using thermographic phosphors. Appl Phys B 83:499–502
Dabiri D (2009) Digital particle image thermometry/velocimetry: a review. Exp Fluids 46:191–241
du Puits R, Li L, Resagk C, Thess A, Willert C (2014) Turbulent boundary layer in high Rayleigh number convection in air. Phys Rev Lett 112:1–5
Fond B, Abram C, Heyes A, Kempf A, Beyrau F (2012) Simultaneous temperature, mixture fraction and velocity imaging in turbulent flows using thermographic phosphor tracer particles. Opt Express 20:22,118–22,133
Fond B, Abram C, Beyrau F (2015a) Characterisation of the luminescence properties of \(\text{ BAM:Eu }^{2+}\) particles as a tracer for thermographic particle image velocimetry. Appl Phys B 121:495–509
Fond B, Abram C, Beyrau F (2015b) On the characterisation of tracer particles for thermographic particle image velocimetry. Appl Phys B 118:393–399
Haramina T, Tilgner A (2004) Coherent structures in boundary layers of Rayleigh–Bénard convection. Phys Rev E 69(056):306
Hartlep T, Tilgner A, Busse F (2003) Large-scale structures in Rayleigh–Bénard convection at high Rayleigh numbers. Phys Rev Lett 91(064):501
Hishida K, Sakakibara J (2000) Combined planar laser-induced fluorescence-particle image velocimetry for velocity and temperature fields. Exp Fluids 29:129–140
Jordan J, Rothamer D (2012) Pr:YAG temperature imaging in gas-phase flows. Appl Phys B 110:285–291. doi:10.1007/s00340-012-5274-4
Klingshirn C (2007) ZnO: material, physics and applications. Chem Phys Chem 8:782–803
Lawrence M, Zhao H, Ganippa L (2013) Gas phase thermometry of hot turbulent jets using laser induced phosphorescence. Opt Express 21:12,260–12,281. doi:10.1364/OE.21.012260
Li L, Shi N, du Puits R, Resagk C, Schumacher J, Thess A (2012) Boundary layer analysis in turbulent Rayleigh-Bénard convection in air: experiment versus simulation. Phys Rev E 86:12
Neal N, Jordan J, Rothamer D (2013) Simultaneous measurements of in-cylinder temperature and velocity distribution in a small-bore diesel engine using thermographic phosphors. SAE Int J Eng 6:19
Omrane A, Juhlin G, Ossler F, Aldén M (2004a) Temperature measurements of single droplets by use of laser-induced phosphorescence. Appl Opt 43:3523–3529
Omrane A, Santesson S, Aldén M, Nilsson S (2004b) Laser techniques in acoustically levitated micro droplets. Lab Chip 4:287–291
Omrane A, Särner G, Aldén M (2004c) 2D-temperature imaging of single droplets and sprays using thermographic phosphors. Appl Phys B 79:431–434
Omrane A, Petersson P, Aldén M, Linne M (2008) Simultaneous 2D flow velocity and gas temperature measurements using thermographic phosphors. Appl Phys B 92:99–102
Pfadler S, Beyrau F, Löffler M, Leipertz A (2006) Application of a beam homogenizer to planar laser diagnostics. Opt Express 14:10,171–10,180
Pfadler S, Dinkelacker F, Beyrau F, Leipertz A (2009) High resolution dual-plane stereo-PIV for validation of subgrid scale models in large-eddy simulations of turbulent premixed flames. Combust Flame 56:1552–1564
Resagk C, du Puits R, Thess A, Dolzhansky F, Grossmann S, Araujo F, Lohse D (2006) Oscillations of the large scale wind in turbulent thermal convection. Phys Fluids 18(095):105
Robinson S (1991) Coherent motions in the turbulent boundary layer. Annu Rev Fluid Mech 23:601–639
Robinson G, Lucht R, Laurendau N (2008) Two-color planar laser-induced fluorescence thermometry in aqueous solutions. Appl Opt 47:2852–2858
Rodnyi P, Khodyuk I (2011) Optical and luminescence properties of zinc oxide: a review. Opt Spectrosc 111:776–785
Rothamer D, Jordan J (2012) Planar imaging thermometry in gaseous flows using upconversion excitation of thermographic phosphors. Appl Phys B 106:435–444. doi:10.1007/s00340-011-4707-9
Sakakibara J, Adrian R (1999) Whole field measurement of temperature in water using two-color laser induced fluorescence. Exp Fluids 26:7–15
Sakakibara J, Adrian R (2004) Measurement of temperature field of a Rayleigh–Bénard convection using two-color laser-induced fluorescence. Exp Fluids 37:331–340
Särner G, Richter M, Aldén M (2008) Two-dimensional thermometry using temperature-induced line-shifts of ZnO:Zn and ZnO:Ga fluorescence. Opt Lett 33:1327–1329
Schmeling D, Bosbach J, Wagner C (2014) Simultaneous measurement of temperature and velocity fields in convective air flows. Meas Sci Technol 25:16
Schmeling D, Bosbach J, Wagner C (2015) Measurements of the dynamics of thermal plumes in turbulent mixed convection based on combined PIT and PIV. Exp Fluids 56:134
Schumacher J, Emran M (2015) Large-scale mean patterns in turbulent convection. J Fluid Mech 776:96–108
Smith C, Sabatino D, Praisner T (2001) Temperature sensing with thermochromic liquid crystals. Exp Fluids 30:190–201
Someya S, Yoshida S, Li Y, Okamoto K (2009) Combined measurement of velocity and temperature distributions in oil based on the luminescent lifetimes of seeded particles. Meas Sci Technol 20:9
Someya S, Ochi D, Li Y, Tominaga K, Ishii K, Okamoto K (2010) Combined two-dimensional velocity and temperature measurements using a high-speed camera and luminescent particles. Appl Phys B 99:325–332
van Lipzig J, Yu M, Dam N, Luijten CCM, de Goey LPH (2013) Gas-phase thermometry in a high-pressure cell using \(\text{ BaMgAl }_{10}\text{ O }_{17}\): Eu as a thermographic phosphor. Appl Phys B 111:469–481. doi:10.1007/s00340-013-5360-2
Wadewitz A, Specht E (2001) Limit value of the Nusselt number for particles of different shape. Int J Heat Mass Transf 44:967–975
Acknowledgments
Miriam Pougin gratefully acknowledges the support of Studienstiftung des deutschen Volkes. The authors would like to thank the following researchers at Otto-von-Guericke-Universität Magdeburg for their time, expertise and/or equipment: Ulf Betke and Stefan Rannabauer (SEM imaging); Benoit Fond (helpful discussions); Katja Guttmann (fluorescence spectrometer); Werner Hintz and Andreas Schlinkert (ultrasound and particle-sizing measurements); Volker Lorenz (deionised water); and Helmut Weiß (Nd:YAG laser).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Abram, C., Pougin, M. & Beyrau, F. Temperature field measurements in liquids using ZnO thermographic phosphor tracer particles. Exp Fluids 57, 115 (2016). https://doi.org/10.1007/s00348-016-2200-2
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00348-016-2200-2