
Abstract
Purpose
Overexpression of Bcl-2 family members as well as deregulated apoptosis pathways are known hallmarks of lung cancer. Non-small cell lung cancer (NSCLC) cells are typically resistant to cytotoxic chemotherapy and approaches that alter the balance between pro-survival and pro-death Bcl-2 family members have shown promise in preclinical models of NSCLC.
Methods
Here we evaluated the effects of a novel pan-Bcl-2 inhibitor GX15-070 on NSCLC survival and when combined with epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors as well as traditional cytotoxic agents. GX15-070 is a small molecule agent that binds anti-apoptotic Bcl-2 proteins and interferes with their ability to interact with pro-apoptotic proteins. We evaluated the effect of GX15-070 and correlated the effect on EGFR status as well as Bcl-2 family protein expression.
Results
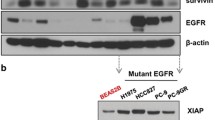
We show that GX15-070 can disrupt Mcl-1:Bak interactions in lung cancer cells. We identified differential sensitivity of a panel of lung cancer cells to GX15-070 and no clear relationship existed between EGFR status or Bcl-2 family protein expression and sensitivity to GX15-070. GX15-070 could induce apoptosis in a subset of lung cancer cell lines and this correlated with the effects on cell viability. GX15-070 combined with gefitinib was synergistic in a cell line dependent on EGFR for survival but GX15-070 could not reverse resistance to gefitinib in cell lines not dependent on EGFR for survival. Finally, we observed synergy between GX15-070 and cisplatin in NSCLC cells.
Conclusions
Based on these results, GX15-070 can trigger apoptosis in NSCLC cells and can enhance chemotherapy-induced death. These data suggest that clinical trials with GX15-070 in combination with cytotoxic chemotherapy are indicated.






Similar content being viewed by others


References
Haura EB, Cress WD, Chellappan S, Zheng Z, Bepler G (2004) Antiapoptotic signaling pathways in non-small-cell lung cancer: biology and therapeutic strategies. Clin Lung Cancer 6:113–122
Reed JC (1996) Mechanisms of Bcl-2 family protein function and dysfunction in health and disease. Behring Inst Mitt 97:72–100
Sedlak TW, Oltvai ZN, Yang E, Wang K, Boise LH, Thompson CB, Korsmeyer SJ (1995) Multiple Bcl-2 family members demonstrate selective dimerizations with Bax. Proc Natl Acad Sci U S A 92:7834–7838
Adams JM, Cory S (1998) The Bcl-2 protein family: arbiters of cell survival. Science 281:1322–1326
Cory S, Huang DC, Adams JM (2003) The Bcl-2 family: roles in cell survival and oncogenesis. Oncogene 22:8590–8607
Anton RC, Brown RW, Younes M, Gondo MM, Stephenson MA, Cagle PT (1997) Absence of prognostic significance of bcl-2 immunopositivity in non-small cell lung cancer: analysis of 427 cases. Hum Pathol 28:1079–1082
Apolinario RM, van der Valk P, de Jong JS, Deville W, van Ark-Otte J, Dingemans AM, van Mourik JC, Postmus PE, Pinedo HM, Giaccone G (1997) Prognostic value of the expression of p53, bcl-2, and bax oncoproteins, and neovascularization in patients with radically resected non-small-cell lung cancer. J Clin Oncol 15:2456–2466
Borner MM, Brousset P, Pfanner-Meyer B, Bacchi M, Vonlanthen S, Hotz MA, Altermatt HJ, Schlaifer D, Reed JC, Betticher DC (1999) Expression of apoptosis regulatory proteins of the Bcl-2 family and p53 in primary resected non-small-cell lung cancer. Br J Cancer 79:952–958
Fontanini G, Vignati S, Bigini D, Mussi A, Lucchi M, Angeletti CA, Basolo F, Bevilacqua G (1995) Bcl-2 protein: a prognostic factor inversely correlated to p53 in non-small-cell lung cancer. Br J Cancer 71:1003–1007
Gaffney EF, O’Neil AJ, Staunton MJ (1994) Bcl-2 and prognosis in non-small-cell lung carcinoma. N Engl J Med 330:1757–1758
Higashiyama M, Doi O, Kodama K, Yokouchi H, Tateishi R (1995) High prevalence of bcl-2 oncoprotein expression in small cell lung cancer. Anticancer Res 15:503–505
Higashiyama M, Doi O, Kodama K, Yokouchi H, Tateishi R (1996) Bcl-2 oncoprotein expression is increased especially in the portion of small cell carcinoma within the combined type of small cell lung cancer. Tumour Biol 17:341–344
Higashiyama M, Doi O, Kodama K, Yokouchi H, Nakamori S, Tateishi R (1997) Bcl-2 oncoprotein in surgically resected non-small cell lung cancer: possibly favorable prognostic factor in association with low incidence of distant metastasis. J Surg Oncol 64:48–54
Ikegaki N, Katsumata M, Minna J, Tsujimoto Y (1994) Expression of bcl-2 in small cell lung carcinoma cells. Cancer Res 54:6–8
Katz HR (1994) Bcl-2 protein in non-small-cell lung carcinoma. N Engl J Med 330:221
Pezzella F, Turley H, Kuzu I, Tungekar MF, Dunnill MS, Pierce CB, Harris A, Gatter KC, Mason DY (1993) Bcl-2 protein in non-small-cell lung carcinoma. N Engl J Med 329:690–694
Ritter JH, Dresler CM, Wick MR (1995) Expression of bcl-2 protein in stage T1N0M0 non-small cell lung carcinoma. Hum Pathol 26:1227–1232
Sartorius UA, Krammer PH (2002) Upregulation of Bcl-2 is involved in the mediation of chemotherapy resistance in human small cell lung cancer cell lines. Int J Cancer 97:584–592
Gautschi O, Tschopp S, Olie RA, Leech SH, Simoes-Wust AP, Ziegler A, Baumann B, Odermatt B, Hall J, Stahel RA, Zangemeister-Wittke U (2001) Activity of a novel bcl-2/bcl-xL-bispecific antisense oligonucleotide against tumors of diverse histologic origins. J Natl Cancer Inst 93:463–471
Kataoka M, Wiehle S, Spitz F, Schumacher G, Roth JA, Cristiano RJ (2000) Down-regulation of bcl-2 is associated with p16INK4-mediated apoptosis in non-small cell lung cancer cells. Oncogene 19:1589–1595
Koty PP, Zhang H, Levitt ML (1999) Antisense bcl-2 treatment increases programmed cell death in non-small cell lung cancer cell lines. Lung Cancer 23:115–127
Leech SH, Olie RA, Gautschi O, Simoes-Wust AP, Tschopp S, Haner R, Hall J, Stahel RA, Zangemeister-Wittke U (2000) Induction of apoptosis in lung-cancer cells following bcl-xL anti-sense treatment. Int J Cancer 86:570–576
Ouyang N, Ran P, Qiu Z (2000) [Bcl-2 antisense oligodeoxyribonucleotide increases apoptosis of lung carcinoma cells induced by cisplatin]. Zhonghua Jie He He Hu Xi Za Zhi 23:722–724
Ziegler A, Luedke GH, Fabbro D, Altmann KH, Stahel RA, Zangemeister-Wittke U (1997) Induction of apoptosis in small-cell lung cancer cells by an antisense oligodeoxynucleotide targeting the Bcl-2 coding sequence. J Natl Cancer Inst 89:1027–1036
Janmaat ML, Kruyt FA, Rodriguez JA, Giaccone G (2003) Response to epidermal growth factor receptor inhibitors in non-small cell lung cancer cells: limited antiproliferative effects and absence of apoptosis associated with persistent activity of extracellular signal-regulated kinase or Akt kinase pathways. Clin Cancer Res 9:2316–2326
Song L, Coppola D, Livingston S, Cress D, Haura EB (2005) Mcl-1 regulates survival and sensitivity to diverse apoptotic stimuli in human non-small cell lung cancer cells. Cancer Biol Ther, 4:267–276
Fesik SW (2005) Promoting apoptosis as a strategy for cancer drug discovery. Nat Rev Cancer 5:876–885
Banerjee D (1999) Technology evaluation: G-3139. Curr Opin Mol Ther 1:404–408
Banerjee D (2001) Genasense (Genta Inc). Curr Opin Investig Drugs 2:574–580
Morris MJ, Tong WP, Cordon-Cardo C, Drobnjak M, Kelly WK, Slovin SF, Terry KL, Siedlecki K, Swanson P, Rafi M, DiPaola RS, Rosen N, Scher HI (2002) Phase I trial of BCL-2 antisense oligonucleotide (G3139) administered by continuous intravenous infusion in patients with advanced cancer. Clin Cancer Res 8:679–683
Rudin CM, Otterson GA, Mauer AM, Villalona-Calero MA, Tomek R, Prange B, George CM, Szeto L, Vokes EE (2002) A pilot trial of G3139, a bcl-2 antisense oligonucleotide, and paclitaxel in patients with chemorefractory small-cell lung cancer. Ann Oncol 13:539–545
Rudin CM, Kozloff M, Hoffman PC, Edelman MJ, Karnauskas R, Tomek R, Szeto L, Vokes EE (2004) Phase I study of G3139, a bcl-2 antisense oligonucleotide, combined with carboplatin and etoposide in patients with small-cell lung cancer. J Clin Oncol 22:1110–1117
Reed JC, Pellecchia M (2005) Apoptosis-based therapies for hematologic malignancies. Blood 106:408–418
Tracy S, Mukohara T, Hansen M, Meyerson M, Johnson BE, Janne PA (2004) Gefitinib induces apoptosis in the EGFRL858R non-small-cell lung cancer cell line H3255. Cancer Res 64:7241–7244
Fujimoto N, Wislez M, Zhang J, Iwanaga K, Dackor J, Hanna AE, Kalyankrishna S, Cody DD, Price RE, Sato M, Shay JW, Minna JD, Peyton M, Tang X, Massarelli E, Herbst R, Threadgill DW, Wistuba II, Kurie JM (2005) High expression of ErbB family members and their ligands in lung adenocarcinomas that are sensitive to inhibition of epidermal growth factor receptor. Cancer Res 65:11478–11485
Kwak EL, Sordella R, Bell DW, Godin-Heymann N, Okimoto RA, Brannigan BW, Harris PL, Driscoll DR, Fidias P, Lynch TJ, Rabindran SK, McGinnis JP, Wissner A, Sharma SV, Isselbacher KJ, Settleman J, Haber DA (2005) Irreversible inhibitors of the EGF receptor may circumvent acquired resistance to gefitinib. Proc Natl Acad Sci USA 102(21):7665–7670
Song L, Turkson J, Karras JG, Jove R, Haura EB (2003) Activation of Stat3 by receptor tyrosine kinases and cytokines regulates survival in human non-small cell carcinoma cells. Oncogene 22:4150–4165
Chou TC, Talalay P (1984) Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul 22:27–55
Haura EB, Zheng Z, Song L, Cantor A, Bepler G (2005) Activated epidermal growth factor receptor-stat-3 signaling promotes tumor survival in vivo in non-small cell lung cancer. Clin Cancer Res 11:8288–8294
Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, Naoki K, Sasaki H, Fujii Y, Eck MJ, Sellers WR, Johnson BE, Meyerson M (2004) EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 304:1497–1500
Sordella R, Bell DW, Haber DA, Settleman J (2004) Gefitinib-sensitizing EGFR mutations in lung cancer activate anti-apoptotic pathways. Science 305:1163–1167
Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, Kris MG, Varmus H (2005) Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med 2:1–11
Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ, Joseph MK, Kitada S, Korsmeyer SJ, Kunzer AR, Letai A, Li C, Mitten MJ, Nettesheim DG, Ng S, Nimmer PM, O’Connor JM, Oleksijew A, Petros AM, Reed JC, Shen W, Tahir SK, Thompson CB, Tomaselli KJ, Wang B, Wendt MD, Zhang H, Fesik SW, Rosenberg SH (2005) An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 435:677–681
O’Brien S, Kipps TJ, Fader S, Crump M, Keating M, Anderson B, Soho C, Bole J, Turner R, Viallet J, Cheson B (2004) A phase I trial of the small molecule pan-bcl-2 family inhibitor GX15-070 administered intravenously (IV) every 3 weeks to patients with previously treated chronic lymphocytic leukemia (CLL). American Society of Hematology (ASH), Washington
Acknowledgments
We thank Dr Mark Watson (GeminX Biotechnologies Inc., Montreal, QC, Canada) for help with coimmunoprecipitation assays and Drs Pasi Janne (Dana Farber, Boston, MA, USA), Jon Kurie (MD Anderson, Houston, TX, USA), and Jeffrey Settleman (Massachusetts General Hospital Cancer Center, Boston, MA, USA) for providing cell lines. We thank Drs Warren Fiskus and Kapil Bhalla (Medical College of Georgia Cancer Center, Augusta, GA, USA) for help with synergy analysis, and Tiffany Dyn for administrative assistance. This work has been supported in part by the Molecular Imaging Core, Molecular Biology and Sequencing Core, and the Flow Cytometry Core at the H. Lee Moffitt Cancer Center & Research Institute. This work was partially funded by the H. Lee Moffitt Cancer Center & Research Institute.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Li, J., Viallet, J. & Haura, E.B. A small molecule pan-Bcl-2 family inhibitor, GX15-070, induces apoptosis and enhances cisplatin-induced apoptosis in non-small cell lung cancer cells. Cancer Chemother Pharmacol 61, 525–534 (2008). https://doi.org/10.1007/s00280-007-0499-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-007-0499-3