
Abstract
The luminescence spectra of a suite of natural sodium framework silicates including four different sodalite variants and tugtupite have been collected during X-ray irradiation as a function of temperature between 20 and 673 K. The origin of the emission bands observed in these samples is attributed to F-centres (360 nm), paramagnetic oxygen defects (400 and 450 nm), S2 − ions (620 nm) and tetrahedral Fe3+ (730 nm). Luminescence in the yellow (550 nm) is tentatively attributed to Mn2+, and red luminescence in Cr-rich pink sodalite is possibly from Cr3+ activation. Sudden reduction in luminescence intensities of emission centres was observed for all minerals in the 60–120 K range. Since it is common to all the sodalite-group minerals, we infer it is a feature of the aluminosilicate framework. Sodalite luminescence has responses from substitutions on the framework (e.g. paramagnetic oxygen defects, Fe3+) which give sodalite properties akin to other framework silicates such as feldspar and quartz. However, the presence of the sodalite cage containing anions (such as F-centres, S2 − ions) imparts additional properties akin to alkali halides. The possibility of coupling between Fe3+ and S2 − is discussed. The overall luminescence behaviour of sodalite group can be understood in terms of competition between these centre types.
Similar content being viewed by others


Introduction
The sodalite-group minerals (M8T12O24X2) are one of the most important rock-forming mineral groups (part of the feldspathoid family) in which M is a metal ion (typically Na, Ca, Mn or Zn), T is an element in tetrahedral coordination (usually Al, Si or occasionally Be) and X is an anion such as Cl− or S2−. The family includes sodalite sensu stricto (s.s.) [Na8Al6Si6O24Cl2], haüyne [(Na,Ca)4–8Al6Si6(O,S)24(SO4,Cl)1–2], lazurite [(Na,Ca)8Al6Si6O24(S,SO4,Cl)1–2] and tugtupite [Na8Be2Al2Si2Si6O24Cl2]. Some are prized as semi-precious stones (e.g. ultramarine and lapis lazuli) because the sodalite minerals show a range of colours including blue, pink, green, yellow, red and colourless. Synthetic analogues are finding applications as pigments (Schlaich et al. 2000) and efficient visible and infrared phosphors (Schipper et al. 1972; Lezhina et al. 2006). Some natural sodalites are tenebrescent, i.e. they change colour reversibly on exposure to daylight, a property which has inspired potential applications in, for example, security papers and smart coatings for blinds (Armstrong and Weller 2006). Despite being of such importance, there are relatively few published data relating to the luminescence of natural sodalites (e.g. Kirk 1955; Aierken Sidike et al. 2007; Gaft et al. 2009; Kaiheriman et al. 2014; Zahoransky 2015; Zahoransky et al. 2016). Luminescence is extremely sensitive to changes in the structure of intrinsic and extrinsic defects, and, when combined with complementary techniques, provides valuable information about the defect structure of materials. It is widely used, for example, to study the band gaps of semiconductors (e.g. Rogach et al. 2009 and refs therein) and to explore local coordination of activator ions (e.g. Jayasundera et al. 2008). In geosciences, luminescence (usually cathodoluminescence, CL) is used qualitatively to image growth zoning and quantitatively in luminescence-based dating methods such as optically stimulated luminescence (OSL).
The present study examines the light emitted by a suite of sodalite-group minerals in response to excitation by X-rays (X-ray excited optical luminescence, XEOL, sometimes referred to as radioluminescence, RL). We compare and contrast each sample to allow insights into the nature of the centres that generate luminescence. We perform XEOL as a function of temperature, the data from which are composites of XEOL with thermally stimulated luminescence (thermoluminescence, TL) (which we call XEOLTL). We compare our findings to published photoluminescence (PL) and electron spin resonance (ESR) data to understand more fully the defect structures in sodalite-group minerals that give rise to luminescence.
Previous research
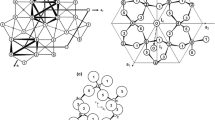
The structure of sodalite (Na8Al6Si6O24Cl2) comprises interconnected AlO4 and SiO4 tetrahedra joined at all four apices to form a three-dimensional framework (Fig. 2, Hassan and Grundy 1984). Sodalite s.s. is cubic (P \(\bar{4}\) 3n) and exhibits ordering of Al and Si such that Al atoms are never juxtaposed, i.e. ‘Löwenstein’s rule’ (Löwenstein 1954). The sodalite cages contain Na4Cl groups with Cl tetrahedrally coordinated at a special symmetry position. Sodalite s.s. has Al and Si atoms on the framework sites. Beryllium can also be accommodated on the framework by two mechanisms: the coupled substitution of Be2+ + Mn2+ = Al3+ + Na+ operates in helvite [e.g. Mn8Be6Si6O24S2] group minerals, and substitution of Be2+ + Si4+ = 2Al3+ gives rise to tugtupite [Na8Be2Al2Si8O24Cl2]. The ordering of Be, Al and Si in tugtupite results in a loss of symmetry by distortion of the cages, primarily a squashing of the cage along one axis, resulting in tugtupite being tetragonal, I \(\bar{4}\) (Danø 1966; Hassan and Grundy 1991; Antao et al. 2004; Fig. 2). The coordination of Cl in tugtupite is tetrahedral as in sodalite, but where Na is four coordinate in sodalite, it is five coordinated in tugtupite, resulting in a different electronic environment in the cage. In addition to chemical variability on the tetrahedral and metal sites, there is variability in the nature of the anion, which is Cl− in sodalite s.s. and tugtupite. Haüyne and lazurite are sodalites formed by coupled substitutions of Ca2+ + [S2−, SO4 2−] = Na+ + Cl−; hydroxysodalite has OH− > Cl− and sodalites containing carbonate (CO3 2−) ions are reported (e.g. Ballirano and Maras 2005). The literature describes synthetic sodalite analogues with, for example, Br−, I−, Se2−, Se −2 substituting for Cl− (e.g. Lushchik et al. 2001; Armstrong et al. 2003).
Materials and methods
The present study includes five minerals chosen to represent a range of composition and physical properties exhibited by sodalite-type structures. The provenance for the samples is given in Table 1, and all analytical work was carried out in the Department of Earth and Environmental Sciences at the University of St. Andrews, UK. We analyse four sodalites and the tetragonal Be–sodalite tugtupite (Na8Be2Al2Si2Si6O24Cl2), of which the most common pink form is analysed. The yellow (AF-88-IL6), turquoise (AF-12-21) and pink sodalites (AF-07-35), and the tugtupite (AF-99-173) are from the Ilímaussaq Centre, part of the Gardar Province of South Greenland (Marks and Markl 2015). Ilímaussaq is unusual in that it was a highly peralkaline, reduced magma in which sulphide dominated sulphate and methane over carbon dioxide. The fifth sample, AF-9-E, is a commercially obtained deep blue sodalite of unknown provenance, typical of the material used widely as a semi-precious stone. Note that some of the samples analysed here were also used in a detailed chemical and structural study of luminescence and tenebrescent sodalites from Ilímaussaq (Zahoransky et al. 2016).
Powder X-ray diffraction shows each sample to be single phase. Compositional data were acquired by laser ablation inductively coupled mass spectrometry (LA-ICPMS) on a Thermo Electron XSeries2 which is a quadrupole-based ICP-MS. On this instrument, sensitivity is >6 × 107 counts s−1 for 10 μg ml−1 In when used in the standard solution nebulisation mode. The instrument is coupled to a commercial New Wave UP213 frequency quintupled Nd:YAG laser. Ablation was performed by focusing the laser on the sample surface, utilising a laser repetition rate of 20 Hz and maintaining a constant energy density of ~10 J cm−2. He was utilised as carrier gas to optimise signal intensities and precision (Eggins et al. 1998). The He carrier exiting the ablation chamber was mixed with Ar prior to entering the ICP torch in order to maintain stable excitation conditions. Operating conditions were optimised before each analytical session using continuous ablation of NIST SRM 612 reference material glass, by providing maximum signal intensity and stability for Pb+, U+ and Th+ ions while suppressing oxide formation which was monitored using ThO+/Th+ (ThO+/Th+ < 0.5 %). Integration times were 20-s background, 80 s on the sample and 40-s washout between samples. Data reduction was performed using GLITTER software (Van Achterbergh et al. 2001). Raw signal counts were carefully studied as a function of time in order to exclude from calculations portions of signal deviating from a flat profile. Moreover, because the ablation time is dependent on elemental fractionation, we selected GLITTER’s option for which the software automatically uses, for each selected ablation time, segments of an unknown, the identical integrated ablation time segments of the calibrator analyses, relative to the beginning of ablation. Net background-corrected count rates for each isotope were used for calculation. On this instrument under these conditions, typical precision to 95 % confidence is ±10 % of the mean and limits of detection range between 1 and 3 ppm. However, the typical lod for sulphur, which does not readily ablate to positive ions, is estimated at ~5 ppm.
The colour of samples was quantified using a PerkinElmer UV–visible spectrometer system in reflection mode using a 150-mm integration sphere (Fig. 1). Blue, yellow and strong pink colour centres in sodalite are tentatively attributed to sulphur species (McLaughlan and Marshall 1970; Hassib et al. 1977) and pale pink colour in AF-07-35 is attributed to an F-centre (Pizani et al. 1985). Samples were examined with a commercial mineralogical UV lamp with ‘long’ (UV-C) and ‘short’ wave (UV-B) settings. The samples were analysed by XEOL and XEOLTL on a luminescence spectrometer system designed and built by Peter Townsend and co-workers at the University of Sussex, UK, but now upgraded and housed at the University of St. Andrews. The system comprises two high-sensitivity IPD detectors which are blazed in the UV-blue (200–450 nm) and red (380–800 nm) regions. A 400-nm-long pass filter is placed in front of the red detector to remove doublets of UV-blue emissions. Software written in house corrects the data from each imaging plate for system responses and merges them to generate a single spectrum covering 250–800 nm. The system has separate low-temperature (20–300 K) and high-temperature (25–400 °C, i.e. 298–673 K) stages. The measurements are observed by cooling the sample (20 K for the low-temperature stage; 25 °C for the high-temperature stage) then heating the sample at a ramp rate of 6 K min−1 with continuous X-ray irradiation. Data are collected every 5 K, and the wavelength resolution is <2 nm. The X-ray source is a Philips MCN-101 ceramic X-ray tube typically running at 20 kV and 20 mA delivering X-rays at 20 Gy min−1 (Fig. 2).

UV–visible absorption spectra of the sodalites of the present study. a Colour centres in sodalite are tentatively attributed to a variety of clustered sulphur species. The pale pink colour centre is attributed to an F-centre and b refers to sample AF-07-35 and shows both the ‘as collected’ pink sodalite and of the sample after heating to 400 °C for 10 min. The pink colour is lost on heating, leaving a colourless sodalite. The isolated spectrum of the pale pink chromophore (from subtraction of the pink and colourless forms) is given

Structures of sodalite-group minerals. Structural drawing of a sodalite and b tugtupite. The larger grey spheres are Na and the smaller lighter grey spheres are O. Note how there is a bond from Na to the bridging oxygen between Si and Be tetrahedra in tugtupite causing Na to be five coordinated in this mineral compared to tetrahedrally coordinated in sodalite. The figure is generated using data from Hassan et al. (2004) and Antao et al. (2004) with CrystalMaker®
Results
XEOL is observed from all the sodalites (Figs. 3, 4; Table 3). Some luminescence bands are consistent between samples (e.g. 360 nm is common), but nevertheless the variability between members of the same mineral group is striking.

Comparisons of XEOL as a function of temperature for the samples in Table 2. Data are normalised in intensity to allow easier comparisons of the profiles. The two temperature ranges are normalised in intensity for each range since the signals are usually much stronger at low temperature and the high-temperature spectrum would appear relatively featureless for some of the samples. Hence, a discontinuity is sometimes observed between the scales on the two runs. Three-dimensional representations of the data are also presented in supplementary materials for colour-blind readers who may not be able to appreciate the profiles from coloured maps

Wavelength profiles of the sodalite minerals at 20 and 300 K. The colours of the lines reflect the physical colours of the samples. The data are offset in the y axis to improve clarity. Data are normalised in intensity to allow easier comparison of the profiles
Pink sodalite (AF-07-35)
This sample has a pale pink colour and no obvious PL in UV-B or UV-C excitation. It has remarkably few trace elements, but contains significant Mn (11 ± 1 ppm), Fe (30 ± 3 ppm), Ga (95 ± 9 ppm) and Cr (129 ± 13 ppm) (Table 2). Its XEOL at all temperatures is dominated by strong luminescence in the UV-blue region. At low temperatures, this is centred ~400 nm, but shifts to shorter wavelength abruptly at 60 K. Thereafter, it is a broad UV-blue band covering most of the region between 300 and 450 nm, composed of overlapping broad emission. At 100 and 185 K, the luminescence profile temporarily broadens, a feature not observed for the other samples. The data show a small near IR band at ~700 nm with two prominent peaks at 60 and 100 K. The red emission fades on heating and is lost sharply at 240 K. Analysis of several pink sodalites shows that the relative proportions of UV and red luminescence are variable and some samples are dominated by red/near IR luminescence at 20 K. At room temperature, luminescence is dominated by a broad UV-blue emission centred at 365 nm, but whose flat profile hints at two peaks of similar intensity at ~300 and 365 nm. After heating to 400 °C (673 K), the pink colour is lost and the sample is colourless (Table 3).
Deep blue sodalite (AF-9-E)
This sample has the deep blue colour typical of many natural sodalites used as decorative stones and has no visible PL. Most elements are at or below the limit of detection for ICPMS, but it contains notable Mn (13 ± 1 ppm) and Fe (618 ± 62 ppm) (Table 1). Its low-temperature (20 K) XEOL lies centred predominantly in the violet (~405 nm), but on heating the intensity and wavelength change abruptly into the UV region to 370 nm at ~100 K, forming a broad, flat-topped band with a shoulder on the high-energy side, which is consistent throughout the temperature range to 400 °C (673 K).
Turquoise sodalite (AF-12-21)
The sample shows a subtle orange photoluminescence under UV-C excitation, but little under UV-B. This form has several trace element substituents including Mn (13 ± 1 ppm), Li (21 ± 2 ppm), Ga (91 ± 9 ppm) and Fe (336 ± 34 ppm). Lanthanides are above the limit of detection—the only one in our samples suite—with La (28 ± 3 ppm), Ce (14 ± 1 ppm) and Nd (9 ± 1 ppm) being the most abundant, and the remaining lanthanides ~2–5 ppm (Table 2). The XEOL at 20 K shows bands at 360–450 and ~620 nm with a characteristic vibronic structure of the latter emission (Fig. 4). Analysis of several turquoise sodalites shows that the relative proportions of the components vary considerably between samples, although all contain the same three emissions. As temperature increases, the clarity of the vibronic structure at 620 nm reduces (Fig. 3). Between 100 and 185 K, the 450-nm feature steeply fades, whereas the 350-nm emission demonstrates a commensurate increase in intensity. A kink in the intensity of the profile on warming to 240 K is observed. At high temperatures, a band at ~450 nm forms as a noticeable doublet in the luminescence profile, becoming particularly strong above 150 °C (423 K).
Yellow sodalite (AF-88-IL6)
The sample shows remarkably strong orange photoluminescence under UV-C excitation but little under UV-B. It has significant S (11 ± 1 ppm), B (47 ± 5 ppm), Fe (59 ± 6 ppm) and Ga (118 ± 12 ppm). The XEOL of yellow sodalite at 20 K shows bands at 350 and ~450 nm, but also has luminescence at ~620 nm with a characteristic vibronic structure similar to that of the turquoise sodalite, but the relative proportions are different. As with the turquoise sodalite, analysis of several samples shows variation in the relative proportions of the 350-, 450- and 620-nm components, although all samples show all three. Generally at 20 K, the 620-nm band is strong in yellow sodalite, whereas the 350-nm band is weaker than the turquoise variant. As temperature increases, the intensity of the 450-nm component falls abruptly at 100 K (and is lost by 185 K) and the vibronic structure at 620 nm becomes less distinct (Fig. 3). In the turquoise sodalite, the 620 nm dominated at low temperature, whereas the 350-nm emission dominated at RT. Despite the overall similar luminescence response of the two samples, this change was not observed in the yellow sodalite. Above room temperature, three distinct bands occur at 350, 450 and 620 nm. At and above room temperature, there is little vibronic structure visible in the 620-nm emission although its intensity becomes stronger on heating to 200 °C (473 K) and is the dominant component contrary to the turquoise where the UV emission dominates at high temperature. The intensity of all three bands decreases strongly on heating beyond 330 °C (603 K).
Tugtupite (AF-99-173)
Tugtupite shows red photoluminescence in UV-C. There are few trace substituents with Fe (21 ± 2 ppm) and Ga (147 ± 15 ppm) being the most notable. The XEOL at 20 K is dominated by a broad emission at ~550 nm with smaller emissions at ~360, 450 and 665 nm (Fig. 4). The intensity of the main 550-nm band falls steeply on heating above 130 K until it is almost absent at 150 K. Above 150 K, separate red (675 nm) and UV-blue bands (380–450 nm) dominate the luminescence up to and beyond room temperature, with two noticeable bursts of light in the 675-nm emission at 145 and 210 K. At room temperature, the luminescence comprises a broad band of at least two components (~395 and 440 nm) in the UV-blue region and one band in the red/near IR (675 nm). At 100 °C (373 K), the red/IR band intensity begins to fall and is extinct by 180 °C (453 K). The two components to the violet–blue region increase on heating to a maximum at ~160 °C (433 K) and then fall abruptly at ~200 °C (473 K).
Discussion
General features
The sodalites contain several trace elements. Be, B, Ga and Ti levels are usually above limit of detection by ICPMS (Table 2), presumably as substitutions for Al and Si on the sodalite framework. Ga (15–150 ppm) and Fe (21–618 ppm) levels are notably high, presumably either substituting tetrahedrally (as Fe3+) onto the framework, or, in the case of Fe, as Fe2+ (accompanying Mn2+) in the cages (cf. helvite group minerals). Sulphur was only detected in yellow sodalite (11 ± 1 ppm), but its limit of detection is poor (>5 ppm). This presumably exists in the cages as sulphur clusters (e.g. Aierken Sidike et al. 2007) or S2− ions (Hassan et al. 2004). Lanthanides were only detected in turquoise sodalite (total REE ≥ 65 ppm), presumably accommodated as cations in the sodalite cage. Cr (129 ± 13 ppm) is significant in pink sodalite.
The sodalite-group minerals produce luminescence from the UV through to the near infrared (Fig. 3). Despite having strong structural similarities, the responses of each sample are very varied. The behaviour is strongly temperature dependent, but some features are common. We hypothesise that common features in the luminescence relate to the aluminosilicate framework or to cage-related defects which all these minerals share. Conversely, where the behaviour is very different from sample to sample, we infer this reflects variability in the contents of the sodalite cage, or the way in which those contents interact with the lattice.
All the sodalites demonstrate abrupt changes in luminescence in the temperature range ~60–120 K. These are sudden drops in intensity for deep blue and yellow sodalites and tugtupite; in turquoise sodalite, there is a shift in emission between centres emitting at 500 nm to 360 nm and in pink sodalite, a broadening of the band with a sudden burst of light in the near IR (720 nm). In detail, the temperature changes lie at ~80 K in yellow, deep blue and turquoise sodalites; at 105 K for pink sodalite; and at ~120 K in tugtupite. These temperature contrasts are beyond the thermal lag of the sample. Despite the variations in temperature, this feature is common to all sodalites and we infer it is a property of the aluminosilicate framework. Unfortunately, we know of no data in the open literature analysing unit cell parameters as a function of temperature which would shed light on any structural causes of these discontinuities.
The sharp peaks and broadening of the profile at discreet temperatures (in pink sodalite at 60, 105 and 185 K, in turquoise sodalite at 240 K and in tugtupite at 140 K) are reminiscent of thermoluminescence, i.e. thermally stimulated emptying of electron traps emitting light which increases with temperature reaching a maximum then decreases as the temperature increased further. Therefore, these peaks are interpreted as TL peaks superimposed on the XEOL background. To explore this further, the TL of the pink sodalite was measured by X-ray irradiation of the sample at 20 K, turning off the irradiation and then ramping temperature. Such an experiment explores the stored charge in structural traps within the crystal, released by thermal stimulation. The TL of pink sodalite (Fig. 5) shows the sharp peaks and broad features seen in the XEOL data, confirming that these data are composites of XEOL and TL behaviour (XEOLTL).

Thermoluminescence (TL) of pink sodalite. TL is achieved by irradiating the sample at 20 K and then releasing stored charge by ramping temperature. The data are shown as both contour and three-dimensional formats and false coloured such that cold colours (black, blue) represent lower counts and hotter colours (orange, red) are the highest counts. The data demonstrate a complex thermoluminescence behaviour involving responses in the UV, blue and red at 60, 105, 185 and 230 K
The strong TL in pink sodalite at 105 K and in tugtupite at 145 K coincides with the temperature range of the sudden loss of luminescence intensity in all the minerals studied. Although a full discussion of the TL behaviour of sodalite-group minerals is beyond the present study, the coincidence of this TL feature with abrupt changes in luminescence quantum efficiency is consistent with a model whereby delocalisation of electrons associated with an electron traps on the sodalite framework modifies the intensity of luminescence from the sodalite host. The temperature contrast between this process in sodalite (80 K) and tugtupite (120 K) is also consistent with a feature involving the framework, since tugtupite has a different, Be-bearing framework to the other four sodalite-group minerals studied here.
UV-blue region
All the minerals of the present study, to a greater or lesser extent, show UV-blue emissions, variously observed usually at 360 or 405 nm. The most common and most intense is the 360-nm band found in pink, deep blue, turquoise and yellow sodalites from 100 K upwards. The literature on sodalite optical spectroscopy is dominated by discussions of point defects such as F-centres which occur since Na+ and Cl− are relatively weakly bound in the structure. Analogies can be drawn with luminescence centres in alkali halides, which have several UV luminescence centres resulting from interactions and clustering of vacancies and substituent ions. If we consider sodalite as an alkali halide housed by an aluminosilicate framework, then we infer that Cl− can be mobilised by X-ray irradiation as it is in alkali halides, leaving Cl− vacancies which are stabilised by several routes, including trapped electrons (F-centres), which migrate to form defect clusters. F-centres are noted in the ESR spectra of deep blue (McLaughlan and Marshall 1970; Hassib et al. 1977) and pink (Pizani et al. 1985) sodalite. To explore this, we successfully imparted a pale pink colour to transparent sodalite by electron irradiation and then returned it to a transparent form by thermal annealing above 250 °C (523 K). We observe that natural pink sodalite becomes colourless upon heating. The data are consistent with simple F-centres (v Cl) being implicated in the pink colouration in accordance with the conclusions of Hodgson et al. (1967). However, the high Cr content of this sample invites the possibility that the pink colour results from electronic transitions within Cr3+ although the loss and restoring of the colour by irradiation are more difficult to rationalise, unless it is coupled in some way to changes in Cr oxidation state. More generally, it is difficult to rule out entirely an uncharacterised species linked to redox couples such as S n−4 species. Gaft et al. (2009) attributed a 365-nm centre to Ce3+ on the basis of PL lifetime measurements. We are unable to pulse the X-ray source on our instrument, and hence, we are unable to perform lifetime experiments. However, the lanthanide concentrations of our samples and consistently low (only turquoise sodalite is above limit of detection, Table 2) and a similar centre are observed in synthetic alkali halides when Ce3+ activation is unfeasible. We note that the lifetime of a few μs measured by Gaft et al. (2009) is also consistent with F-centres in alkali halides (Akiyama and Kobashi 2009). However, from a crystal chemical point of view Ce3+ is not easily accommodated in the sodalite structure and our data (Table 2) and those of Zahoransky et al. (2016) show the content of Ce in pink sodalites from Ilímaussaq to be below the limit of detection for laser ablation ICPMS. At this stage, we do not rule out a Ce3+ signal within the UV-blue region (Gaft et al. 2009), but propose that the majority of the response at ~360 nm arises from point defects.
Blue–green region
Turquoise and pink sodalites show luminescence into the blue (i.e. ~450 nm) at low temperatures (<150 K); close inspection of the data for pink and deep blue sodalites shows that this feature is also present as a shoulder on the luminescence profiles of those samples. Blue luminescence in feldspars, another framework aluminosilicate, is attributed to oxygen paramagnetic defects (Finch and Klein 1999) or to Eu2+ activation (Gaft et al. 2005). Such centres in feldspar emit as broad bands in the region 400–460 nm. The breadth of that band results at least in part from combinations of the superposition of several overlapping emissions resulting from different centres at non-equivalent crystallographic sites (Finch and Klein 1999) and crystal anisotropy (Finch et al. 2003). In all our samples, Eu is below the limit of detection (Table 2). Pizani et al. (1985) used ESR to show that interstitial oxygen paramagnetic defects, implicated in the blue luminescence in alkali feldspar, is present in deep blue sodalite. Luminescence in this region of synthetic sodalites has also been ascribed to O2 − defects in the cage (van Doorn and Schipper 1971). By analogy with feldspars and synthetic sodalites, the blue luminescence is consistent with defects on the aluminosilicate framework rather than attributed to element activators.
Yellow region
Tugtupite has a striking yellow–orange (~550 nm) XEOL at 20 K, but on warming to 140 K, it adopts distinct red (690 nm) and UV-blue (~380 nm) emissions. Bursts of light in the red (675 nm) are thermoluminescence (our unpublished data). The transformation between the two luminescence states occurs at ~100 K. There is no fine structure in the 550-nm band to identify it as an S2 − centre (analogous to 620 nm in sodalite s.s.), and such fine structure was reported in tugtupite by Povarennykh et al. (1971) for a red (~700 nm) emission. The band appears absent from the sodalite s.s. samples in the present study. The yellow–green region is characteristic of Mn2+ emission in hosts such as apatite, feldspar and spinel (Gaft et al. 2005), and its wavelength varies across the range 540–580 nm, consistent with the broad 550 nm observed here. Furthermore, van Doorn and Schipper (1971) found that synthetic bromide sodalite doped with Mn2+ produced a PL emission at 542 nm. The Mn2+ luminescence results from forbidden d–d transitions with low transition probabilities and long (ms) lifetime, although our compositional data (Table 2) find that the Mn content is below the limit of detection. This band is problematic, and we currently tentatively attribute the low-temperature 550-nm luminescence to Mn2+ awaiting further spectroscopic characterisation. Whatever its cause, from the temperature dependence, it is hypothesised that the luminescence is only observed as long as other more favourable decay pathways are blocked and that the transition at 140 K allows access to recombination sites that emit in the UV-blue and red. Future time-resolved spectroscopy will explore this hypothesis.
Orange region
The orange (620 nm) band in yellow sodalite has a characteristic fine structure at 20 K (Fig. 3) which has also been observed in PL by Marfunin (1979) and Aierken Sidike et al. (2007). Aierken Sidike et al. (2007) did not quote the provenance of their sample—merely stating it as ‘Greenland’—but, given the similarities to our data, we suspect their sample is from the same site. They attributed the 620-nm emission to S2 − ions substituting for Cl− and the fine structure at low temperatures to symmetric vibration of sulphur in the S–S bond (Chang and Onton 1973). Analysis of S–S vapour (Surber 2005) confirms these as the spectral properties of the disulphide species. Our XEOL data comply with Surber (2005) and the PL at cryogenic temperatures of Aierken Sidike et al. (2007). Sulphur is difficult to analyse by LA-ICPMS, but we detect it in yellow sodalite (11 ± 1 ppm), which has the greatest intensity of this emission. We therefore confidently attribute the band to this species. Heating the sample causes the vibronic structure to lessen, presumably from greater interaction between the ion and the host lattice, and by room temperature cannot be confidently resolved. In detail, the reduction in vibronic structure may relate to the S2 − ion becoming increasingly free to rotate, either along its axis or perpendicular to it. The spherical Cl− ion, for which S2 − substitutes, sits at a special lattice position with tetrahedral point symmetry, whereas the S of SO4 2− in haüyne sits slightly off the special position, and an anti-phase domain structure causes superlattice reflections (Hassan et al. 2004). The dumbbell-shaped S2 − ion cannot be bound symmetrically and in alkali halides, for example, preferentially adopts particular orientations (Nishidate et al. 2003). If the S2 − ion is off-centre, the loss of vibronic structure is consistent with flipping between non-equivalent sites within the cage, or thermally assisted flips in orientation with respect to neighbouring Na ions. However, Aierken Sidike et al. (2007) and our unpublished PL data show the vibronic structure to be visible in PL at room temperature; hence, the mode of excitation influences observation of vibronic structure—more energetic excitation—may reduce the clarity of the vibronic structure. Ionising radiation (such as X-rays) may influence the populations of the S2 − centres by the interaction between disulphide and free electrons: S2 − + e− = S2 2−. Alternatively, the absence of vibronic structure may relate to how the S2 − centre is coupled to the conduction band with particularly efficient excitation through UV irradiation, in which the photon is absorbed and then emitted directly by the centre without involving the host lattice.
Red-infrared region
Red/IR luminescence in minerals of this group has also been attributed to the S2 − band (e.g. Povarennykh et al. 1971, Gorobets and Rogojine 2002). Although Povarennykh et al. (1971) reported vibronic structure in the PL red emission of tugtupite at 77 K, we see no clear evidence of vibronic structure in the red/IR RL band of our data to fingerprint it as S2 −. However, as is observed above with the S2 − centre in yellow sodalite, X-ray irradiation may not be an efficient way to observe the vibronic structure. Red/IR luminescence in framework silicates is often attributed to tetrahedrally coordinated Fe3+ substituting for Al3+ (e.g. van Doorn and Schipper 1971; Finch and Klein 1999; Brooks et al. 2002; Kaiheriman et al. 2014). However, the two samples with the strongest red/IR emissions in the present study (pink sodalite and pink tugtupite) have the lowest Fe contents (30 ± 3 and 21 ± 2 ppm, respectively). Conversely, the pink sodalite has significant Cr (129 ± 13 ppm) and Cr3+ emits with a characteristic strong wavelength in the red (Gaft et al. 2005).
The red band of sodalite-group minerals is complex and may have different sources in different samples. In pink sodalite, it may result from one or both Fe3+ and Cr3+. In tugtupite, it may be a composite of red luminescence from both S2 − and tetrahedral Fe3+ centres, with different forms of excitation (e.g. UV vs X-ray) preferentially exciting one centre more efficiently than the other. Furthermore, the quantum efficiencies of the isolated centres may be strongly influenced by coupling (and thermally assisted decoupling) of the two defect types. Interestingly, Warner and Hutzen Andersen (2012) found that orange luminescence in synthetic sodalites was dependent on the mechanism used to reduce the sulphur species and only occurred when iron was added to the reaction, hinting that interaction between the two species influences the luminescence. The detailed mechanism for this is undoubtedly complicated, but we suggest processes that contribute to such behaviour. First, the S2 − ion may be physically changing, perhaps moving in a thermally assisted way from one site to another within the aluminosilicate cage. These changes may be associated with modification of oxidation state, perhaps reducing to S2 2−, or forming or breaking S clusters by S diffusion between or within cages. It is generally assumed that the cage site contains mixes of anions and Na+ cations, but it may be cation deficient, the charge balance being offset by coupled substitutions or coupling to defect structures on the tetrahedral framework. Second, there may be thermally assisted electron hopping involving redox couples such as: Fe2+ + S2 − = Fe3+ + S2 2− or 3Fe2+ + S2 − = 3Fe3+ + 2S2−. The tenebrescence of sodalites is attributed to UV-assisted electron hopping between F-centres and S2 − (Armstrong and Weller 2006) where the F-centre is an electron trapped on a Cl− vacancy (Hodgson et al. 1967). Zahoransky et al. (2016) showed that the tenebrescence of natural sodalite is strongly influenced by the local structure and changes in the framework. Sodalites with very broad bands in the orange/red/IR may have overlapping, competing Fe3+ and S2 − centres, the relative proportions of which are dependent on redox state and the manner of excitation. At least some red/IR luminescence in sodalites from the literature cannot relate to S2 − since most natural sodalites contain predominantly sulphate (vi) and not sulphide species (Fleet et al. 2005). It is not coincidental that a strong S2 − signal is observed from rocks which formed under exceptionally reducing conditions. Greater clarification of the relative contributions of Fe3+ and S2 − to the orange—IR luminescence of sodalite will be provided by time-resolved studies. The difference in luminescence response between the sodalites and tugtupite in this region may also be influenced by the different coordination of Na, which are four coordinated in sodalites but five coordinated in tugtupite. Hence, the energy distribution in the cages is different in the two minerals.
Conclusions
Sodalite-group minerals are three-dimensional framework structures with positive (Na+) and negative (Cl− or OH−) ions in cages. Their luminescence behaviour is remarkably varied, but can be understood by analogy with alkali halides (relating to the behaviour of the cages) and tectosilicates such as feldspar (relating to features on the aluminosilicate framework). The overall behaviour results from the competition of these centre types. An abrupt change in the quantum efficiency of sodalite luminescence is observed in all sodalite-group minerals on heating above ~100 K. Deep blue and pink sodalites emit most strongly in the UV, whereas the characteristic orange UV photoluminescence of yellow and turquoise sodalite is from S2 − ions in the cage. The properties of turquoise sodalite are consistent with a mixture of yellow and deep blue variants. Tugtupite shares many features of other sodalite-group minerals, but the presence of Be on the framework allows Mn substitution (generating low-temperature Mn2+ emission) and modifies the temperature of the characteristic change in quantum efficiency.
References
Akiyama N, Kobashi K (2009) Luminescence efficiency of F centers in KI studied by lifetime measurements. Phys Rev B 80:195116
Antao SM, Hassan I, Parise JB (2004) Tugtupite: high-temperature structures obtained from in situ synchrotron diffraction and Rietveld refinements. Am Mineral 89:492–497
Armstrong JA, Weller MT (2006) Structural observation of photochromism. Chem Commun 10:1094–1096. doi:10.1039/b517715d
Armstrong JA, Dann SE, Neumann K, Marco JF (2003) Synthesis, structure and magnetic behaviour of the danalite family of minerals, Fe8[BeSiO4]6X2 (X ~ S, Se, Te). J Mater Chem 13:1229–1233
Ballirano P, Maras A (2005) Crystal chemical and structural characterization of an unusual CO3-bearing sodalite-group mineral. Eur J Mineral 17:805–812. doi:10.1127/0935-1221/2005/0017-0805
Brooks RM, Finch AA, Hole D, Townsend PD, Wu Z (2002) The red to near-infrared luminescence in alkali feldspar. Contrib Mineral Petrol 143:484–494
Chang IF, Onton A (1973) Optical properties of photochromic sulfur-doped chlorosodalite. J Electron Mater 2:17–46
Danø M (1966) The crystal structure of tugtupite—a new mineral Na8Al2Be2Si8O24(Cl, S)2. Acta Cryst 20:812–816
Eggins SM, Rudnick RL, Mcdonough WF (1998) The composition of peridotites and their minerals: a laser-ablation ICP-MS study. Earth Planet Sci Lett 154:53–71
Finch AA, Klein J (1999) The causes and petrological significance of cathodoluminescence emissions from alkali feldspar. Contrib Mineral Petrol 135:234–243
Finch AA, Hole DE, Townsend PD (2003) Orientation dependence of luminescence in plagioclase. Phys Chem Miner 30:373–381
Fleet ME, Liu XY, Harmer SL, Nesbitt HW (2005) Chemical state of sulfur in natural and synthetic lazurite by S K-edge XANES and X-ray photoelectron spectroscopy. Can Mineral 43:1589–1603
Gaft M, Reisfeld R, Panczer G (2005) Luminescence spectroscopy of minerals and materials. Springer, Berlin
Gaft M, Panczer G, Nagli L, Yeates H (2009) Laser-induced time-resolved luminescence of tugtupite, sodalite and hackmanite. Phys Chem Miner 36:127–141
Gorobets BS, Rogojine AA (2002) Luminescent spectra of minerals. RPC VIMS Publishers, Moscow
Hassan I, Grundy HD (1984) The crystal structure of sodalite-group minerals. Acta Cryst B40:6–13
Hassan I, Grundy HD (1991) The Crystal Structure and thermal expansion of tugtupite Na8[Al2Be2Si8O24]Cl2. Can Miner 29:385–390
Hassan I, Antao SM, Parise JB (2004) Haüyne: phase transition and high-temperature structures obtained from synchrotron radiation and Rietveld refinements. Mineral Mag 68:499–513
Hassib A, Beckman O, Annersten H (1977) Photochromic properties of natural sodalite. J Phys D Appl Phys 10:771–777
Hodgson WG, Brinen JS, Williams EF (1967) Electron spin resonance investigation of photochromatic sodalites. J Phys Chem 47:3719–3723
Jayasundera ACA, Finch AA, Wormald P, Lightfoot P (2008) Solvothermal synthesis and luminescent properties of two organically-templated chain-structure fluorides, [C4H14N2][MF5] (M = In, Sc). Chem Mater 20:6810–6815
Kaiheriman M, Maimaitinaisier A, Rehiman A, Sidike A (2014) Photoluminescence properties of green and red luminescence from natural and heat-treated sodalite. Phys Chem Miner 41:227–235
Kirk RD (1955) The luminescence and tenebrescence of natural and synthetic sodalite. Am Mineral 40:22–31
Lezhina M, Laeri F, Benmouhadi L, Kynast U (2006) Efficient near-infrared emission from sodalite derivatives. Adv Mater 18:280–283
Löwenstein W (1954) The distribution of aluminum in the tetrahedra of silicates and aluminates. Am Mineral 39:92–96
Lushchik C, Demidendo V, Kirm M, Kudryavtseva I, Lushchik A, Martinson I, Nagirnyi V, Vasil’chenko E (2001) Creation of F centres and multiplication of electronic excitations in Na6Al6Si6O24(NaBr)2x optical ceramics under VUV irradiation. J Phys Condens Matter 13:6133–6149
Marfunin AS (1979) Spectroscopy, luminescence and radiation centers in minerals. Springer, Berlin
Marks M, Markl G (2015) The Ilímaussaq alkaline complex, South Greenland. In: Charlier B et al (eds) Layered intrusions. Springer, Dordrecht. doi:10.1007/978-94-017-9652-1_14
McLaughlan SD, Marshall DJ (1970) Paramagnetic resonance of F-type centres in photochromatic sodalites. Phys Lett A 32:343–344
Nishidate K, Baba M, Sarjono Hasegawa M, Nishikawa K, Sokolska I, Ryba-Romanowski W (2003) First-principles study on the energetics and vibrational properties of the S2 − impurity in alkali-halide crystals. Phys Rev B 68:224307
Pizani PS, Terrile MC, Farach HA, Poole CP Jr (1985) Color centers in sodalite. Am Mineral 70:1186–1192
Povarennykh AS, Platonov AN, Tarashchan AN, Belichenko VP (1971) Tugtupite (beryllosodalite) from Ilímaussaq, South Greenland. Meddr Grønland 181(14)
Rogach AL, Klar TA, Lupton JM, Meijerink A, Feldmann J (2009) Energy transfer with semiconductor nanocrystals. J Mater Chem 19:1208–1221
Schipper DJ, van Doom CZ, Bolwijn PT (1972) Preparation of cathodochromic sodalites. J Am Ceram Soc 55:2569
Schlaich H, Lindner G-G, Fledmann J, Göbel EO, Reinen D (2000) Optical properties of Se −2 and Se2 color centres in the red Se ultramarine with the sodalite structure. Inorg Chem 39:2740–2746
Sidike Aierken, Sawuti A, Wang X-M, Zhu H-J, Kobayashi S, Kusachi I, Yamashita N (2007) Fine structure in photoluminescence spectrum of S2 − center in sodalite. Phys Chem Miner 34:477–484
Surber SE (2005) Photoelectron imaging of molecular and cluster anions. Unpublished Ph.D. thesis, University of Arizona
van Achterbergh E, Ryan CG, Jackson SE, Griffin W (2001) Data reduction software for LA–ICP–MS. In: Sylvester P (ed) Laser ablation ICPMS in the earth sciences. Mineralogical Association of Canada, Short Course Series, vol 29, pp 239–243
van Doorn CZ, Schipper DJ (1971) Luminescence of O2 −, Mn2+ and Fe3+ in sodalite. Phys Lett A 34:139–140
Warner TE, Hutzen Andersen J (2012) The effect of sulphur intercalation on the optical properties of artificial ‘hackmanite’, Na8[Al6Si6O24]Cl1.8S0.1; ‘sulfosodalite’, Na8[Al6Si6O24]S; and natural tugtupite, Na8[Be2Al2Si8O24](Cl,S)2−δ. Phys Chem Miner 39:161–168
Zahoransky T (2015) The chemical and structural control of colour, photoluminescence and tenebrescence of natural sodalites. Unpublished M.Sc. thesis, University of Tübingen, Germany
Zahoransky T, Friis H, Marks MAW (2016) Luminescence and tenebrescence of natural sodalites: a chemical and structural study. Phys Chem Minerals. doi:10.1007/s00269-016-0810-0
Acknowledgments
The refurbishment of the RLTLCL system and the establishment of it as a facility at St. Andrews were funded by NERC Grant NE/H002715/1.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

{kind=link}
Cite this article
Finch, A.A., Friis, H. & Maghrabi, M. Defects in sodalite-group minerals determined from X-ray-induced luminescence. Phys Chem Minerals 43, 481–491 (2016). https://doi.org/10.1007/s00269-016-0816-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00269-016-0816-7