
Abstract
Natural killer (NK) cells have been used in several clinical trials as adaptive immunotherapy. The low numbers of these cells in peripheral blood mononuclear cells (PBMC) have resulted in various approaches to preferentially expand primary NK cells from PBMC. While some clinical trials have used the addition of interleukin 2 (IL-2) to co-stimulate the expansion of purified NK cells from allogeneic donors, recent studies have shown promising results in achieving in vitro expansion of NK cells to large numbers for adoptive immunotherapy. NK cell expansion requires multiple cell signals for survival, proliferation and activation. Thus, expansion strategies have been focused either to substitute these factors using autologous feeder cells or to use genetically modified allogeneic feeder cells. Recent developments in the clinical use of genetically modified NK cell lines with chimeric antigen receptors, the development of expansion protocols for the clinical use of NK cell from human embryonic stem cells and induced pluripotent stem cells are challenging improvements for NK cell-based immunotherapy. Transfer of several of these protocols to clinical-grade production of NK cells necessitates adaptation of good manufacturing practice conditions, and the development of freezing conditions to establish NK cell stocks will require some effort and, however, should enhance the therapeutic options of NK cells in clinical medicine.
Similar content being viewed by others


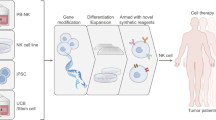
Introduction
Natural killer (NK) cells are potent effectors of the innate immune system and form the first line of defense against diseases, including malignancies. They are members of the innate lymphoid cell family and characterized in humans by expression of the phenotypic marker CD56 (neural cell adhesion molecule) in the absence of CD3 (T-cell co-receptor) [1]. The NK cell cytotoxic attack is immediate, does not require prior antigen-priming and is instead orchestrated in a unique way by an array of receptors with activating or inhibitory functions. Important activating receptors include the C-type lectin-like receptors CD94/NKG2C and NKG2D and the natural cytotoxicity receptors (NCR) NKp30, NKp44 and NKp46, which recognize ligands on tumor cells or virally infected cells. NK cell inhibition is essentially mediated by interactions of the polymorphic inhibitory killer cell immunoglobulin-like receptors (KIRs) with their cognate human–leukocyte–antigen (HLA) ligands. These HLA-ligands are bound by KIR using simplified amino acid (AA) structures in the alpha-1 helix of the HLA molecule. HLA-ligands are subdivided into three major motifs, HLA-Group 1 or C1 (Ser77/Asn80), HLA-Group 2 or C2 (Asn77/Lys80) and HLA-Bw4 (AA position 77–83) which bind predominantly to inhibitory KIR characterized by a long extracellular immunoglobulin domain (Table 1). Other NK cells inhibitory receptors specific for HLA class I molecules are CD94/NKG2A with the non-classical class I molecule HLA-E as ligand and the leukocyte Ig-like receptor-1 binding to HLA-G1 [2, 3]. NK cell inhibition is essential to prevent their attacks on healthy ‘self’-tissue; on the other hand, it needs to be overcome in therapeutic application of NK cells against transformed cells. The discovery of donor-derived alloreactive NK cells present in T-cell-depleted HLA haplo-identical grafts for hematopoietic stem cell transplantation (HSCT) was a milestone in the field of NK cell therapy [4, 5]. Such haplo-mismatched NK cells exerted potent anti-leukemia effects in the absence of graft-versus-host disease (GvHD) [6]. Haplo-identical HSCT provides a valuable treatment option for high-risk leukemia patients and, however, remains afflicted with transplant-related morbidity and mortality [7, 8]. Severe infection risks after the intensive transplant conditioning regimens and GvHD caused by alloreactive T cells in the graft are major complications. Novel developments to optimize this type of therapy are under way. An important approach is the focus on the generation of grafts with defined cell compositions to achieve potent graft-versus-leukemia (GvL) effects with low incidences of severe side effects.
Several studies showed that NK cell alloreactivity has a clinical benefit in overall survival and reduced relapse rates for patients after HSCT in HLA-identical settings [8–10]. Patients with a ‘mismatch’ of KIR and HLA-ligands (‘receptor–ligand’ concept) in graft-versus-recipient direction showed reduced relapse rates underlining the GvL effect of allogeneic NK cells [8]. In contrast to the earlier clinical studies of NK cell mismatches based on solely HLA (KIR–ligand) typing of donor and recipient (‘ligand–ligand’ concept), these studies included the typing of KIR receptors beside HLA genes of the donor (Fig. 1). This strategy allowed for the differential analysis of the ‘mismatch’ effect comparing the ‘ligand–ligand’ versus the ‘receptor–ligand’ concept. While the ‘ligand–ligand’ concept is based on the differences of the HLA-pattern (e.g., HLA-C locus antigens) between recipients and donors, the ‘receptor–ligand’ concept requires the typing of KIR genes in order to adjust the KIR expression in the donor and hence alloreactive NK cells in the stem cell graft. A comparative analysis based on identical clinical study cohorts of adult patients transplanted with matched unrelated donors has been performed by Leung, Handgretinger and co-workers showing that the selection of donors (grafts) should be based on the ‘receptor–ligand’ model [10]. This study and other underlined the importance to perform KIR gene typing of preferential donors in order to adjust the clinical risk [8–11]. Most of these studies have underlined the clinical relevance of the inhibitory NK cell receptors KIR2DL1, KIR2DL2/3 and KIR3DL1 interacting with the three HLA ligand motifs C1, C2 and Bw4, respectively.

Schematic view of NK cell KIR receptor inhibition by C1, C2 and Bw4 ligands expressed by recipient tumor cells. a The patient expresses C1, C2 and Bw4 KIR ligands and KIR2DL1-, KIR2DL2/3- and KIR3DL1-positive donor NK cells are inhibited (KIR–HLA match, no KIR–ligand missing). b The patient expresses C1, and Bw4 KIR ligands (C2 is missing), single-KIR2DL1+ donor NK cells are not inhibited and can take part in the graft-versus-tumor effect (KIR–HLA mismatch, KIR–ligand missing)
Functions of activating KIR receptors on the outcome of HSCT are only recently being unveiled. Graft recipients from HLA-identical siblings had a reduced relapse rate, if donor genotypes contained the activating KIR receptor genes KIR2DS1 and KIR2DS2 [12, 13]. Recipients of grafts from donors homozygous or heterozygous for the KIR group B haplotype, containing more than one activating KIR gene in comparison to KIR haplotype A, displayed better overall survival [14, 15]. This effect was even more pronounced if donors were homozygous for centromeric B gene motifs containing KIR2DS2 when compared to haplotype A donors missing KIR2DS2 [14]. In addition, recent studies could demonstrate protective effects for telomeric activating KIR3DS1 and KIR2DS1 on transplant outcome. While patients receiving unrelated grafts from KIR2DS1 donors had a reduced relapse rate, patients receiving grafts for KIR3DS1 donors were characterized by a lower risk of grade II–IV GvHD and mortality [16]. In the study of Venstrom et al, the effect of KIR2DS1 was mediated by grafts from donors with KIR2DS1 genes on a C1-positive HLA background (C1/C1 or C1/C2), while patients receiving grafts from donors with KIR2DS1 genes on a C2/C2 HLA background had significantly higher relapse rates [17].
In addition to donor KIR gene content, KIR allelic polymorphism has been shown to have an impact on recipient outcome after HSCT. KIRs that regulate NK cells are highly polymorphic and KIR alleles encode receptors that have stronger signaling function than others [18]. Bari and colleagues showed that patients who received a KIR2DL1-R(245)-positive graft with HLA-C receptor–ligand mismatch had the best survival and lowest risk of leukemia progression compared with those patients who received a KIR2DL1-C(245) homozygous graft [19]. Thus, KIR allelic subtyping of allogeneic NK could be beneficial for donor selection in the transplant setting as well as for immunotherapies optimizing GvL effects.
Recent publications point toward rapid immune reconstitution and sustained persistence of NK cells with high-level surface expression of CD94/NKG2C in patients with cytomegalovirus (CMV)-reactivation post-HSCT [20]. A development of ‘memory’ NK cells in such patients has been postulated. A reduction in the risk of leukemia relapse within the first-year post-HSCT was found, however, no increase in overall survival [21]. Further investigations are necessary to identify the underlying mechanisms and to better estimate the extent of the therapeutic potential of NK cells in transplant settings involving CMV reactivation [22].
The therapeutic potential of NK cells has led to several approaches in immunotherapy that can be summarized under the following principles.
-
(1)
Autologous NK cells can be activated and potentiated through systemic administration of cytokines like interleukin (IL)-2, IL-12, IL-15, IL-18, IL-21 and type I IFNs. Despite safe administration of ex vivo activated and expanded autologous NK cells using cytokines and the generation of PBMCs with enhanced cytotoxicity against NK-resistant targets, no clinical responses in cancer patients were seen [23, 24].
-
(2)
Allogeneic NK cells in adoptive cell transfer have shown beneficial cytotoxic effects killing malignant cells/tumors based on the ‘KIR mismatch’ principle [25, 26]. This approach is highly effective in HLA haplo-identical transplantation settings, but requires a more detailed analysis of HLA and NK KIR gene pattern if used in HSCT using HLA matched related or unrelated donors. Donor lymphocyte infusion (DLI) takes advantage of NK cell alloreactivity of cells that are expanded and activated in vitro prior to adoptive transfer using various cytokines (IL-2, IL-15 or IL-21) and growth factors [27–29]. In addition, monoclonal antibodies blocking inhibitory KIRs can be used to stimulate NK cell function [30, 31].
-
(3)
Antibody-dependent cytotoxic cell lysis (ADCC) NK cells express the activating receptor type IIIA Fc receptor (CD16). This receptor enables NK cells to recognize antibodies on target cells, which triggers subsequently the destruction of the cells via ADCC. This effect can be augmented using monoclonal antibodies that stimulate endogenous or adoptive NK cells. Evidence for NK cell-mediated ADCC has been given in clinical studies using antibody treatment of non-Hodgkin lymphoma with rituximab (anti-CD20) [32, 33], multiple myeloma with daratumumab in combination with all-trans retinoic acid [34] or human anti-KIR antibody IPH2102 and lenalido [31], metastatic breast cancer with herceptin (anti-trastuzumab) [35] and metastatic colorectal cancer or squamous cell carcinoma of the head and neck by the epidermal growth factor receptor (EGFR) inhibitor cetuximab [36].
-
(4)
NK cell lines/chimeric antigen receptor modification There are seven established NK cells lines: NK-92, YT, NKL, HANK-1, KHYG-1, NK-YS and NKG [37, 38]. These cell lines are ideal candidates for the expansion under GMP conditions. However, only the human NK-92 cell line has shown to be safe and efficient in clinical trials [39–41]. Recently gene transfer of CARs into primary NK cells or NK-92 has brought new therapeutic options [42, 43].
Stimulation of NK cell activity to enhance immunotherapy
It was discovered early on that exposure to stimulatory factors such as the cytokine IL-2 enhanced NK cell potency significantly. This property was already exploited clinically in the 1980s by investigators from the National Cancer Institute (NCI, USA) [44, 45]. However, clinical outcomes of these original studies did not match expectations. Early clinical trials aimed to ‘in vivo’ expand NK cells and to improve their antitumor activity by administrating systemic cytokines, such as IL-2, into the patients with poor clinical outcome due to high toxicity of IL-2. Similarly, low-dose IL-2 administration after autologous stem cell transplantation with lower side effects showed reduced cytotoxic functionality.
In another approach, leukapheresis products were IL-2-stimulated in vitro for a short term (overnight or a few days), to generate lymphokine-activated killer (LAK) cells for re-application to patients. However, such LAK cells were essential T cells with the effector NK cells substituting only a minor fraction. Short-term stimulation of leukapheresis products was insufficient to achieve notable expansion and activation of the NK cells that represent only 10–20 % of peripheral blood lymphocytes. Alternatively, high doses of IL-2 were directly administered to patients to activate NK cells in vivo. However, this clinical treatment modality was afflicted with serious side effects [46]. In addition, IL-2 leads to the stimulation of regulatory T cells; thus, NK cell ex vivo stimulation with other cytokines would be favorable [47, 48]. Recently, there are indications that brief pre-activation of NK cells with novel cytokines such as IL-12, IL-15 and IL-18 induces CD25 (low-affinity IL-2 receptor alpha chain) expression on NK cells [49]. Thus, immunotherapy with cytokine-cocktail pre-activated NK cells may pose a novel treatment option in the near future. Significant technological advances, a better understanding of NK cell biology and the discovery of novel stimulatory factors paved the way for entirely new clinical study designs. Recently the field of NK cell therapy is rapidly internationally emerging and a variety of pioneering approaches is under development or in clinical testing extensively reviewed elsewhere [50, 51].
Selection and expansion of primary NK cells for clinical application
Several protocols have been established aiming to generate sufficient numbers and purity of NK cells by keeping their functional capabilities [27–29, 50]. However, only a few strategies have been developed which follow the stringent requirements of GMP. Besides cellular efficacy of NK cell components, GMP-based manufacturing is essential for clinical application [28, 52, 53]. In general, NK cells can be expanded from PBMC, umbilical cord blood (CB) or bone marrow (BM) and hESC or iPS.
NK cells from peripheral blood mononuclear cells
NK cell is in PBMC with a range of 5–20 %. Therefore, various protocols have been used to isolate and preferentially expand primary NK cells from PBMC [50]. The common principle is a combination of cell selection and depletion using immunomagnetic beads [52]. These protocols use leukapheresis products for the clinical-grade purification of NK cells by depleting CD3 cells followed by selection of CD56 cells [28] or in combination with subsequent short-term (14-day) expansion with IL-2 [52]. Clinical-grade expansion of NK cells in lymphokine-activated killer (LAK) cell cultures for 28 days with IL-15 has been reported [27]. NK cell expansion requires multiple signals for survival, proliferation and activation. Thus, expansion strategies have been focused either to substitute these factors using autologous feeder cells and/or to use genetically modified allogeneic feeder cells Functional activity is defined by cytotoxicity against various malignant cell lines and expression pattern of NK cell receptor (cluster of differentiation (CD)-16, natural killer group-2 member D (NKG2D), CD69, NKp30, NKp44, NKp46 and CD158b. Expansion of NK with autologous PBMC as feeder cells has been shown to generate functional active NK cells with a therapeutic cells dosage [54]. Using GMP-compliant components and autologous feeder cells, purified NK cells were effectively expanded (2500-fold at day 17) [54]. Similarly, large-scale expansion of GMP-compliant NK cells with cytolytic activity against tumor cells has been reported using autologous PBMCs in the presence of OKT3 and IL-2 at 14 day [55]. Other feeder cells such as Jurkat T-lymphoblast subline KL-1 have been used which achieved expansion of NK cells accompanied by reciprocal inhibition of T-cell growth [56]. Promising results were also obtained by the leukemia cell line K562, genetically altered to express membrane-bound form of IL-15 and the 4-1BB (CD137L), which has led to an 277-fold expansion after 3 weeks (21 days) in culture [57]. More recently, K562 cell lines have been engineered to express membrane-bound IL-21 along with CD137L [58]. Expansion was highly selectively for NK cells and reached 100-fold by 3 weeks while CD3+ T cells went from initially 60–1 %. [59]. Genetically modified K562 feeder cells have also the benefit to be used as frozen stock (vials) [60]. In addition to the expansion of total NK cells, strategies for selective expansion of individual NK cell subpopulations have been developed. These strategies are based on the observation that despite a fixed number of inherited KIR genes, each individual NK cell expresses a different number of KIR genes on the cell surface [61]. NK cells with particular sets of inhibitory receptors have been named ‘single-KIR’ cells in the context of developing KIR mismatched cell components for therapeutic use. ‘Single-KIR’ NK cells have been shown to lysis human acute myeloid leukemia (AML) cells in vitro and in vivo [62, 63]. Selection and expansion of clinical-grade single-KIR+ NK cell subsets have been established using a GMP-based approach [63].
NK cells from umbilical cord blood or bone marrow-derived CD34+ cells
CD34+ hematopoietic progenitors from umbilical CB or BM are considered as an excellent source for cell therapeutic applications [64]. Earlier studies have been challenged to reach efficient numbers of NK cells considering the low number of NK cells in CB units. Therefore, different protocols have been developed for the generation of NK cells from CD34+ cells from BM and later from CB using co-culturing systems with stromal cell lines and a combination of cytokines that promote the development of NK cells [65–68]. Many culture systems contain in general components of animal origin (e.g., bovine serum), and this will require changes to adapt them for clinical applications. More recently, a cell culture platform for the ex vivo expansion and NK differentiation from CB-derived CD34+ cells has been established. This method uses clinical-grade serum-free culture medium, no feeder cells and a mixture of heparin and cytokines as a substitute for the extracellular microenvironment of BM in static cell culture bags and automated bioreactor [69]. Expansion of up to 10*10 CD34+-derived NK cells NK cells was feasible with high levels of activating receptors (e.g., NKG2D and NCR) and the ability to efficiently lyse myeloid leukemia and melanoma cell lines, as well as primary leukemia blasts [70]. A phase I trial in elderly AML patients using NK cells based upon this expansion technique is in progress (CCMO no. NL31699 and Dutch Trial Register nr. 2818) [70].
NK cells from embryonic stem cells or induced pluripotent stem cells
The potential to use NK cell from hESC or iPS for immunotherapeutic applications has been reviewed recently by Francisco Borrego [71]. In contrast to research and clinical studies with NK cell from PBMC and or CD34+CB cells, the generation of NK cells form hESC or iPS is a relative new area with an sophisticated approach [72]. Currently, experimental design is focused to optimize the culture conditions to generate these cells. Efficient generation of functional NK cells from hESC has been described CD34+ with acquired functional receptors and cytolytic activity [73, 74]. In addition, these cells could lyse malignant cells by both direct cell-mediated cytotoxicity and ADCC. Clinical-scale generation of NK cells has been published by Kaufmann and co-workers [75]. This method allows the production of mature and functional NK cells from hESC and iPS after expansion for at least 2 month using IL-21 expressing antigen-presenting cells. Harvested NK cell numbers are sufficient to be used for treatment of single patients.
Future perspectives
Current insights into the cellular mechanism geared by receptor–ligand interactions and structural analysis of receptor binding affinities will lead to develop more specific targeting strategies against malignant hematopoietic or solid tumor cells using NK cell-based therapies. Future studies will lead to a better understanding of the complex interactions between inhibitory and activating signaling mechanisms including memory and education of NK cells. This should open a broad therapeutic field in using NK cells for either individualized or ‘off-the-shelf’ cell therapy. The promising results in overcoming the limited number of NK cells by expansion and the feasibility to use GMP-compliant techniques for selection and depletion should enable to tailor NK cell immunotherapy for various malignant diseases. Recent development in the clinical use of gene modified NK cell lines (e.g., CAR) and the development of expansion protocols for the clinical use of NK cell from hESC or iPS are challenging improvements for NK cell-based immunotherapy. Transfer of these protocols to clinical-grade production of NK cells necessitates adaptation of GMP conditions, and the development of freezing conditions to establish NK cell stocks will require some effort and, however, should enhance the therapeutic options of NK cells in clinical.
Abbreviations
- AA:
-
Amino acid
- ADCC:
-
Antibody-dependent cytotoxic cell lysis
- AML:
-
Acute myeloid leukemia
- BM:
-
Bone marrow
- CAR:
-
Chimeric antigen receptors
- CB:
-
Cord blood
- CMV:
-
Cytomegalovirus
- DLI:
-
Donor lymphocyte infusions
- EGFR:
-
Epidermal growth factor receptor
- GMP:
-
Good manufacturing practice
- GvHD:
-
Graft-versus-host disease
- GvL:
-
Graft-versus-leukemia
- hESC:
-
Human embryonic stem cells
- HLA:
-
Human leukocyte antigen
- HSCT:
-
Hematopoietic stem cell transplantation
- iPS:
-
Induced pluripotent stem cells
- KIR:
-
Killer cell immunoglobulin-like receptor
- LAK:
-
Lymphokine-activated killer
- MHC:
-
Major histocompatibility complex
- NCR:
-
Natural cytotoxicity receptors
- NK:
-
Natural killer
- PBMC:
-
Peripheral blood mononuclear cells
References
Luetke-Eversloh M, Killig M, Romagnani C (2013) Signatures of human NK cell development and terminal differentiation. Front Immunol 4:499
Navarro F, Llano M, Bellon T, Colonna M, Geraghty DE, Lopez-Botet M (1999) The ILT2(LIR1) and CD94/NKG2A NK cell receptors respectively recognize HLA-G1 and HLA-E molecules co-expressed on target cells. Eur J Immunol 29:277–283
Moretta L, Pietra G, Montaldo E, Vacca P, Pende D, Falco M, Del Zotto G, Locatelli F, Moretta A, Mingari MC (2014) Human NK cells: from surface receptors to the therapy of leukemias and solid tumors. Front Immunol 5:87
Ruggeri L, Capanni M, Urbani E, Perruccio K, Shlomchik WD, Tosti A, Posati S, Rogaia D, Frassoni F, Aversa F, Martelli MF, Velardi A (2002) Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science 295:2097–2100
Locatelli F, Pende D, Mingari MC, Bertaina A, Falco M, Moretta A, Moretta L (2013) Cellular and molecular basis of haploidentical hematopoietic stem cell transplantation in the successful treatment of high-risk leukemias: role of alloreactive NK cells. Front Immunol 4:15
Aversa F, Martelli MF, Velardi A (2012) Haploidentical hematopoietic stem cell transplantation with a megadose T-cell-depleted graft: harnessing natural and adaptive immunity. Semin Oncol 39:643–652
Martelli MF, Di Ianni M, Ruggeri L, Pierini A, Falzetti F, Carotti A, Terenzi A, Reisner Y, Aversa F, Falini B, Velardi A (2014) “Designed” grafts for HLA-haploidentical stem cell transplantation. Blood 123:967–973
Hsu KC, Keever-Taylor CA, Wilton A, Pinto C, Heller G, Arkun K, O’Reilly RJ, Horowitz MM, Dupont B (2005) Improved outcome in HLA-identical sibling hematopoietic stem-cell transplantation for acute myelogenous leukemia predicted by KIR and HLA genotypes. Blood 105(12):4878–4884
Park S, Kim K, Jang JH, Kim SJ, Kim WS, Kang ES, Jung CW (2015) KIR alloreactivity based on the receptor-ligand model is associated with improved clinical outcomes of allogeneic hematopoietic stem cell transplantation: result of single center prospective study. Hum Immunol 76(9):636–643
Leung W, Iyengar R, Triplett B, Turner V, Behm FG, Holladay MS, Houston J, Handgretinger R (2005) Comparison of killer Ig-like receptor genotyping and phenotyping for selection of allogeneic blood stem cell donors. J Immunol 174(10):6540–6545
Cooley S, Weisdorf DJ, Guethlein LA, Klein JP, Wang T et al (2010) Donor selection for natural killer cell receptor genes leads to superior survival after unrelated transplantation for acute myelogenous leukemia. Blood 116:2411–2419
Venstrom JM, Dupont B, Hsu KC, Pittari G, Gooley TA, Chewning JH, Spellman S, Haagenson M, Gallagher MM, Malkki M, Petersdorf E (2014) Donor activating KIR2DS1 in leukemia. N Engl J Med 371(21):2042
Pittari G, Liu XR, Selvakumar A, Zhao Z, Merino E, Huse M, Chewning JH, Hsu KC, Dupont B (2013) NK cell tolerance of self-specific activating receptor KIR2DS1 in individuals with cognate HLA-C2 ligand. J Immunol 190(9):4650–4660
Cooley S, Trachtenberg E, Bergemann TL, Saeteurn K, Klein J, Le CT et al (2009) Donors with group B KIR haplotypes improve relapse-free survival after unrelated hematopoietic cell transplantation for acute myelogenous leukemia. Blood 113:726–732
Oevermann L, Michaelis SU, Mezger M, Lang P, Toporski J, Bertaina A, Zecca M, Moretta L, Locatelli F, Handgretinger R (2014) KIR B haplotype donors confer a reduced risk for relapse after haploidentical transplantation in children with ALL. Blood 124(17):2744–2747
Venstrom JM, Gooley TA, Spellman S, Pring J, Malkki M, Dupont B et al (2010) Donor activating KIR3DS1 is associated with decreased acute GvHD in unrelated allogeneic hematopoietic cell transplantation. Blood 115:3162–3165
Venstrom JM, Pittari G, Gooley TA, Chewning JH, Spellman S, Haagenson M, Gallagher MM, Malkki M, Petersdorf E, Dupont B, Hsu KC (2012) HLA-C-dependent prevention of leukemia relapse by donor activating KIR2DL1. N Engl J Med 367:805–816
Yawata M, Yawata N, Draghi M, Little AM, Partheniou F, Parham P (2006) Roles for HLA and KIR polymorphisms in natural killer cell repertoire selection and modulation of effector function. J Exp Med 203:633–645
Bari R, Rujkijyanont P, Sullivan E, Kang G, Turner V, Gan K, Leung W (2013) Effect of donor KIR2DL1 allelic polymorphism on the outcome of pediatric allogeneic hematopoietic stem-cell transplantation. J Clin Oncol 31(30):3782–3790
Foley B, Cooley S, Verneris MR, Pitt M, Curtsinger J, Luo X, Lopez-Verges S, Lanier LL, Weisdorf D, Miller JS (2012) Cytomegalovirus reactivation after allogeneic transplantation promotes a lasting increase in educated NKG2C+ natural killer cells with potent function. Blood 119:2665–2674
Green ML, Leisenring WM, Xie H, Walter RB, Mielcarek M, Sandmaier BM, Riddell SR, Boeckh M (2013) CMV reactivation after allogeneic HCT and relapse risk: evidence for early protection in acute myeloid leukemia. Blood 122:1316–1324
Zambello R, Teramo A, Barila G, Gattazzo C, Semenzato G (2014) Activating KIRs in chronic lymphoproliferative disorder of NK cells: protection from viruses and disease induction? Front Immunol 5:72
Burns LJ, Weisdorf DJ, DeFor TE, Vesole DH, Repka TL, Blazar BR et al (2003) IL-2-based immunotherapy after autologous transplantation for lymphoma and breast cancer induces immune activation and cytokine release: a phase I/II trial. Bone Marrow Transplant 32:177–186
Sakamoto N, Ishikawa T, Kokura S, Okayama T, Oka K, Ideno M, Sakai F, Kato A, Tanabe M, Enoki T, Mineno J, Naito Y, Itoh Y, Yoshikawa T (2015) Phase I clinical trial of autologous NK cell therapy using novel expansion method in patients with advanced digestive cancer. J Transl Med 13(1):277
Miller JS, Soignier Y, Panoskaltsis-Mortari A et al (2005) Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood 105(8):3051–3057
Ruggeri L, Mancusi A, Capanni M, Martelli MF, Velardi A (2011) Exploitation of alloreactive NK cells in adoptive immunotherapy of cancer. Curr Opin Immunol 17(2):211–217
Suck G, Oei VY, Linn YC, Ho SH, Chu S, Choong A, Niam M, Koh MB (2011) Interleukin-15 supports generation of highly potent clinical-grade natural killer cells in long-term cultures for targeting hematological malignancies. Exp Hematol 39:904–914
Brehm C, Huenecke S, Quaiser A, Esser R, Bremm M, Kloess S, Soerensen J, Kreyenberg H, Seidl C, Becker PS, Mühl H, Klingebiel T, Bader P, Passweg JR, Schwabe D, Koehl U (2011) IL-2 stimulated but not unstimulated NK cells induce selective disappearance of peripheral blood cells: concomitant results to a phase I/II study. PLoS One 6(11):e27351
Suck G, Koh MB (2010) Emerging natural killer cell immunotherapies: large-scale ex vivo production of highly potent anticancer effectors. Hematol Oncol Stem Cell Ther 3:135–142
Binyamin L, Alpaugh RK, Hughes TL, Lutz CT, Campbell KS, Weiner LM (2008) Blocking NK cell inhibitory self-recognition promotes antibody-dependent cellular cytotoxicity in a model of anti-lymphoma therapy. J Immunol 180:6392–6401
Nijhof IS, Lammerts van Bueren JJ, van Kessel B, Andre P, Morel Y, Lokhorst HM, van de Donk NW, Parren PW, Mutis T (2015) Daratumumab-mediated lysis of primary multiple myeloma cells is enhanced in combination with the human anti-KIR antibody IPH2102 and lenalidomide. Haematologica 100(2):263–268
Alderson KL, Sondel PM (2011) Clinical cancer therapy by NK cells via antibody-dependent cell-mediated cytotoxicity. J Biomed Biotechnol 2011:379123
Du J, Lopez-Verges S, Pitcher BN, Johnson J, Jung SH, Zhou L, Hsu K, Czuczman MS, Cheson B, Kaplan L, Lanier LL, Venstrom JM (2014) CALGB 150905 (Alliance): rituximab broadens the antilymphoma response by activating unlicensed NK cells. Cancer Immunol Res 2(9):878–889
Nijhof IS, Groen RW, Lokhorst HM, van Kessel B, Bloem AC, van Velzen J, de Jong-Korlaar R, Yuan H, Noort WA, Klein SK, Martens AC, Doshi P, Sasser K, Mutis T, van de Donk NW (2015) Upregulation of CD38 expression on multiple myeloma cells by all-trans retinoic acid improves the efficacy of daratumumab. Leukemia 29(10):2039–2049
Daldrup-Link HE, Meier R, Rudelius M, Piontek G, Piert M, Metz S, Settles M, Uherek C, Wels W, Schlegel J, Rummeny EJ (2005) In vivo tracking of genetically engineered, anti-HER2/neu directed natural killer cells to HER2/neu positive mammary tumors with magnetic resonance imaging. Eur Radiol 15(1):4–13
Monteverde M, Milano G, Strola G, Maffi M, Lattanzio L, Vivenza D, Tonissi F, Merlano M, Lo Nigro C (2015) The relevance of ADCC for EGFR targeting: a review of the literature and a clinically-applicable method of assessment in patients. Crit Rev Oncol Hematol 95(2):179–190
Suck G, Branch DR, Keating A (2006) Irradiated KHYG-1 retains cytotoxicity: potential for adoptive immunotherapy with a natural killer cell line. Int J Radiat Biol 82(5):355–361
Gong JH, Maki G, Klingemann HG (1994) Characterization of a human cell line (NK-92) with phenotypical and functional characteristics of activated natural killer cells. Leukemia 8(4):652–658
Tonn T, Becker S, Esser R, Schwabe D, Seifried E (2001) Cellular immunotherapy of malignancies using the clonal natural killer cell line NK-92. J Hematother Stem Cell Res 10(4):535–544
Tonn T, Schwabe D, Klingemann HG, Becker S, Esser R, Koehl U, Suttorp M, Seifried E, Ottmann OG, Bug G (2013) Treatment of patients with advanced cancer with the natural killer cell line NK-92. Cytotherapy 15(12):1563–1570
Arai S, Meagher R, Swearingen M et al (2008) Infusion of the allogeneic cell line NK-92 in patients with advanced renal cell cancer or melanoma: a phase I trial. Cytotherapy 10(6):625–632
Boissel L, Betancur-Boissel M, Lu W, Krause DS, Van Etten RA, Wels WS, Klingemann H (2013) Retargeting NK-92 cells by means of CD19- and CD20-specific chimeric antigen receptors compares favorably with antibody-dependent cellular cytotoxicity. Oncoimmunology 2(10):e26527
Hermanson DL, Kaufman DS (2015) Utilizing chimeric antigen receptors to direct natural killer cell activity. Front Immunol 6:195
Rosenberg SA, Lotze MT, Muul LM, Leitman S, Chang AE, Ettinghausen SE, Matory YL, Skibber JM, Shiloni E, Vetto JT et al (1985) Observations on the systemic administration of autologous lymphokine-activated killer cells and recombinant interleukin-2 to patients with metastatic cancer. N Engl J Med 313:1485–1492
Rosenberg SA (1988) The development of new immunotherapies for the treatment of cancer using interleukin-2. A review. Ann Surg 208:121–135
Rosenberg SA, Lotze MT, Yang JC, Aebersold PM, Linehan WM, Seipp CA, White DE (1989) Experience with the use of high-dose interleukin-2 in the treatment of 652 cancer patients. Ann Surg 210:474–484
Knorr DA, Bachanova V, Verneris MR, Miller JS (2014) Clinical utility of natural killer cells in cancer therapy and transplantation. Semin Immunol 26(2):161–172
Skrombolas D, Frelinger JG (2014) Challenges and developing solutions for increasing the benefits of IL-2 treatment in tumor therapy. Expert Rev Clin Immunol 10(2):207–217
Leong JW, Chase JM, Romee R, Schneider SE, Sullivan RP, Cooper MA, Fehniger TA (2014) Preactivation with IL-12, IL-15, and IL-18 induces CD25 and a functional high-affinity IL-2 receptor on human cytokine-induced memory-like natural killer cells. Biol Blood Marrow Transplant 20:463–473
Klingemann H (2015) Challenges of cancer therapy with natural killer cells. Cytotherapy 17(3):245–249
Childs RW, Berg M (2013) Bringing natural killer cells to the clinic: ex vivo manipulation. Hematology/the Education Program of the American Society of Hematology American Society of Hematology Education Program, pp 234–246
Koehl U, Brehm C, Huenecke S, Zimmermann SY, Kloess S, Bremm M, Ullrich E, Soerensen J, Quaiser A, Erben S, Wunram C, Gardlowski T, Auth E, Tonn T, Seidl C, Meyer-Monard S, Stern M, Passweg J, Klingebiel T, Bader P, Schwabe D, Esser R (2013) Clinical grade purification and expansion of NK cell products for an optimized manufacturing protocol. Front Oncol 3:118
Linn YC, Yong HX, Niam M, Lim TJ, Chu S, Choong A, Chuah C, Goh YT, Hwang W, Loh Y, Ng HJ, Suck G, Chan M, Koh M (2012) A phase I/II clinical trial of autologous cytokine-induced killer cells as adjuvant immunotherapy for acute and chronic myeloid leukemia in clinical remission. Cytotherapy 14(7):851–859
Ahn Yong-Oon, Kim Saerom, Kim Tae Min, Song Eun Young, Park Myoung Hee, Heo Dae Seog (2013) Irradiated and activated autologous PBMCs induce expansion of highly cytotoxic human NK cells in vitro. J Immunother 36:373–381
Lim O, Lee Y, Chung H, Her JH, Kang SM, Jung MY, Min B, Shin H, Kim TM, Heo DS, Hwang YK, Shin EC (2013) GMP-compliant, large-scale expanded allogeneic natural killer cells have potent cytolytic activity against cancer cells in vitro and in vivo. PLoS One 8(1):e53611
Lim SA, Kim TJ, Lee JE, Sonn CH, Kim K, Kim J, Choi JG, Choi IK, Yun CO, Kim JH, Yee C, Kumar V, Lee KM (2013) Ex vivo expansion of highly cytotoxic human NK cells by cocultivation with irradiated tumor cells for adoptive immunotherapy. Cancer Res 73(8):2598–25607
Fujisaki H, Kakuda H, Shimasaki N, Imai C, Ma J, Lockey T, Eldridge P, Leung WH, Campana D (2009) Expansion of highly cytotoxic human natural killer cells for cancer cell therapy. Cancer Res 69(9):4010–4017
Denman CJ, Senyukov VV, Somanchi SS, Phatarpekar PV, Kopp LM et al (2012) Membrane-bound IL-21 promotes sustained ex vivo proliferation of human natural killer cells. PLoS One 7(1):e30264
Wang X, Lee DA, Wang Y, Wang L, Yao Y, Lin Z, Cheng J, Zhu S (2013) Membrane-bound interleukin-21 and CD137 ligand induce functional human natural killer cells from peripheral blood mononuclear cells through STAT-3 activation. Clin Exp Immunol 172(1):104–112
Baek HJ, Kim JS, Yoon M, Lee JJ, Shin MG, Ryang DW, Kook H, Kim SK, Cho D (2013) Ex vivo expansion of natural killer cells using cryopreserved irradiated feeder cells. Anticancer Res 33(5):2011–2019
Uhrberg M, Valiante NM, Shum BP, Shilling HG, Lienert-Weidenbach K, Corliss B, Tyan D, Lanier LL, Parham P (1997) Human diversity in killer cell inhibitory receptor genes. Immunity 7:753–763
Langenkamp U, Siegler U, Jörger S, Diermayr S, Gratwohl A, Kalberer CP, Wodnar-Filipowicz A (2009) Human acute myeloid leukemia CD34+CD38- stem cells are susceptible to allorecognition and lysis by single KIR-expressing natural killer cells. Haematologica 94(11):1590–1594
Siegler U, Meyer-Monard S, Jörger S, Stern M, Tichelli A, Gratwohl A, Wodnar-Filipowicz A, Kalberer CP (2010) Good manufacturing practice-compliant cell sorting and large-scale expansion of single KIR-positive alloreactive human natural killer cells for multiple infusions to leukemia patients. Cytotherapy 12(6):750–763
Schönberg K, Fischer JC, Kögler G, Uhrberg M (2011) Neonatal NK-cell repertoires are functionally, but not structurally, biased toward recognition of self HLA class I. Blood 117(19):5152–5156
Fluevano M, Domogala A, Blundell M, Jackson N, Pedroza-Pacheco I, Derniame S et al (2014) Frozen cord blood hematopoietic stem cells differentiate into higher numbers of functional natural killer cells in vitro that mobilized hematopoietic stem cells or freshly isolated cord blood hematopoietic stem cells. PLoS One 9:e87086
Pinho MJ, Punze M, Sousa M, Barros A (2011) Ex vivo differentiation of natural killer cells from human umbilical cord blood CD34+ progenitor cells. Cell Commun Adhes 18:45–55
Frias AM, Porada CD, Crapnell KB, Cabral JM, Zanjani ED, Almeida-Porada G (2008) Generation of functional natural killer and dendritic cells in a human stromal- based serum-free culture system designed for cord blood expansion. Exp Hematol 36:61–68
Spanholtz J, Preijers F, Tordoir M, Trilsbeek C, Paardekooper J, deWitte T et al (2011) Clinical-grade generation of active NK cells from cord blood hematopoietic progenitor cells for immunotherapy using a closed-system culture process. PLoS One 6:e20740
Spanholtz J, Tordoir M, Eissens D, Preijers F, van der Meer A, Joosten I et al (2010) High log-scale expansion of functional human natural killer cells from umbilical cord blood CD34-positive cells for adoptive cancer immunotherapy. PLoS One 5:e9221
Chouaib S, Pittari G, Nanbakhsh A, El Ayoubi H, Amsellem S, Bourhis JH, Spanholtz J (2014) Improving the outcome of leukemia by natural killer cell-based immunotherapeutic strategies. Front Immunol 5:95
Eguizabal C, Zenarruzabeitia O, Monge J, Santos S, Angel Vesga Maruri N, Arrieta A, Rinon M, Tamayo-Orbegozo E, Amo L, Larrucea S, Borrego F (2014) Natural killer cells for cancer immunotherapy: pluripotent stem cells-derived NK cells as an immunotherapeutic perspective. Front, Immunol Article 439
Woll PS, Grzywacz B, Tian X, Marcus RK, Knorr DA, Verneris MR et al (2009) Human embryonic stem cells differentiate into a homogeneous population of natural killer cells with potent in vivo antitumor activity. Blood 113:6094–6101
Woll PS, Martin CH, Miller JS, Kaufman DS (2005) Human embryonic stem cell-derived NK cells acquire functional receptors and cytolytic activity. J Immunol 175:5095–5103
Knorr DA, Bock A, Brentjens RJ, Kaufman DS (2013) Engineered human embryonic stem cell-derived lymphocytes to study in vivo trafficking and immunotherapy. Stem Cells Dev 22:1861–1869
Knorr DA, Ni Z, Hermanson D, Hexum MK, Bendzick L, Cooper LJ et al (2013) Clinical-scale derivation of natural killer cells from human pluripotent stem cells for cancer therapy. Stem Cells Transl Med 2:274–283
Acknowledgments
This work was supported by the LOEWE Center for Cell and Gene Therapy Frankfurt funded by the Hessisches Ministerium für Wissenschaft und Kunst funding reference number: III L 4- 518/17.004.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
None of the authors has a conflict of interest to declare.
Additional information
This article is part of the Symposium-in-Writing “Natural killer cells, ageing and cancer”, a series of papers published in Cancer Immunology, Immunotherapy.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Becker, P.S.A., Suck, G., Nowakowska, P. et al. Selection and expansion of natural killer cells for NK cell-based immunotherapy. Cancer Immunol Immunother 65, 477–484 (2016). https://doi.org/10.1007/s00262-016-1792-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00262-016-1792-y