
Abstract
Sodium (Na+) ions are known to regulate many signaling pathways involved in both physiological and pathological conditions. In particular, alterations in intracellular concentrations of Na+ and corresponding changes in membrane potential are known to be major actors of cancer progression to metastatic phenotype. Though the functionality of Na+ channels and the corresponding Na+ currents can be investigated using the patch-clamp technique, the latter is rather invasive and a technically difficult method to study intracellular Na+ transients compared to Na+ fluorescence imaging. Despite the fact that Na+ signaling is considered an important controller of cancer progression, only few data using Na+ imaging approaches are available so far, suggesting the persisting challenge within the scientific community. In this study, we describe in detail the approach for application of Na+ imaging technique to measure intracellular Na+ variations in human prostate cancer cells. Accordingly, we used three Na+-specific fluorescent dyes–Na+-binding benzofuran isophthalate (SBFI), CoroNa™ Green (Corona) and Asante NaTRIUM Green-2 (ANG-2). These dyes have been assessed for optimal loading conditions, dissociation constant and working range after different calibration methods, and intracellular Na+ sensitivity, in order to determine which probe can be considered as the most reliable to visualize Na+ fluctuations in vitro.
Similar content being viewed by others


Introduction
Virtually all living organisms could be unified in their feature to possess sodium (Na+) concentration gradient across a plasma membrane. It is reached due to the great difference in ionic composition between interior and exterior regions of the cell. Intracellular Na+ concentration ([Na+]i) is 10–20 times lower than extracellular Na+ concentration ([Na+]o) in every organism. Typically, [Na+]i varies around 5–15 mM depending on the cell type in mammals, whereas [Na+]o is much higher, around 120–150 mM. Such a concentration gradient is established through the activity of ion channels/transporters and pumps, and is required for maintenance of resting membrane potential. Thus, ion movement across a plasma membrane might initiate electrical excitation as well as local cytoplasmic responses. Therefore, Na+ ions have been proposed as a second messenger that could regulate many signaling pathways involved in both physiological and pathological cellular properties (Besson et al. 2015; Roger et al. 2015). In addition to being important in functioning of excitable and non-excitable tissues (e.g., nerve conduction and organogenesis), Na+ signaling has also been proposed to regulate cancer cell behaviors (Gillet et al. 2009). Indeed, Na+ signaling has been shown to play a significant role in cell polarity, motility, migration, invasiveness, and extracellular matrix remodeling. Importantly, elevated total tissue Na+ concentration has been proposed as a highly specific in vivo indicator of malignant lesions in human cancer patients (Jacobs et al. 2011; Ouwerkerk et al. 2003, 2007). Therefore, investigation of Na+ signaling dynamics at the cellular level could give us a better understanding of how a variety of carcinogenic processes are being regulated.
Various experimental approaches have been developed to study changes in [Na+]i. For example, 23Na nuclear magnetic resonance spectroscopy offers a reliable method for non-invasive quantification of [Na+]i in perfused cancer cells and tissues (Hansen et al. 1993). Flow cytometry also provides feasible [Na+]i measurements (Amorino and Fox 1995). However, due to the heterogeneity of cancer cell populations, only large differences could be accurately discriminated by these methods. Functionality of Na+ channels, and hence their active Na+ currents, can be investigated by usage of the patch-clamp technique (Armstrong 1981; Roden and George 1997). However, all of these options do not comprise the fast and easy-to-perform method to quantify [Na+]i variations as Na+ fluorescence indicator dyes do. Regarding the fact that Na+ signaling is considered as an important controller of cancer progression, it is highly surprising that only a few research groups have published their Na+ imaging results using cancer cell lines (Chen et al. 2014; Kline et al. 2000; Roger et al. 2007; Tannous et al. 2009). Indeed, in comparison to well-established calcium (Ca2+) imaging, the realization of Na+ imaging technique is much less exploited. It is more likely that the main challenge is imposed due to the relatively narrow range of the global [Na+]i change (only 10 fold), whereas for Ca2+ the differences are much larger (about 104-fold). Thus, for a similar ion flux (Na+ versus Ca2+), the latter leads to larger relative increases in ion concentration, which makes the detection of Ca2+ transients more feasible as compared to Na+.
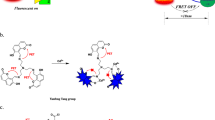
In this study, we developed an in vitro Na+ imaging approach and compared the suitability of three Na+-specific fluorescent dyes–Na+-binding benzofuran isophthalate (SBFI), CoroNa™ Green (Corona), and Asante NaTRIUM Green-2 (ANG-2) in prostate cancer cells. The dyes have been evaluated by their various parameters (e.g., loading time, working range, and sensitivity). Modulation of Na+ down the diffusion gradient, increase of intracellular Ca2+ ([Ca2+]i), inhibition of sodium/potassium ATPase (Na+/K+ ATPase), and activation of voltage-gated Na+ channels were performed to provide alterations in free [Na+]i (Fig. 1).

Schematic representation of the molecular pathways used in this study to induce alterations [Na+]i. Typical Na+ concentrations of the mammalian cells are 5–15 mM for cytosol ([Na+]int) and 120–150 mM for extracellular ([Na+]ext) medium. [Na+]o was switched from 0 to 110 mM for investigating ion diffusion down the electrochemical gradient through Na+ leak channels. Ouabain inhibits the activity of Na+/K+ ATPase and hence restrains constant expulsion of the intracellular Na+. The application of voltage-gated Na+ channels (VGSCs) opener, veratridine, should also result in [Na+]i increase. Ionomycin as an ionophore promotes Ca2+ entry through the plasma membrane and the depletion of endoplasmic reticulum (ER) Ca2+ stores and hence boosts the activity of Na+/Ca2+ exchanger (NCX)
Overall, this is the first methodological study that provides a detailed description of Na+ fluorescence imaging in three human prostate cancer cell lines: DU 145, PC-3, and PC-3M. Moreover, it addresses many technical issues related to the different Na+-specific probes and provides a better understanding on their proper application in vitro.
Materials and methods
Cell culture
In this study, human prostate cancer cell lines of epithelial origin have been used. DU 145 and PC-3 have been purchased from the American Type Culture Collection (ATCC®), whereas—PC-3M have been kindly provided by Prof. Mustafa Djamgoz (Imperial College London, UK). These cancer cell lines derive from various metastatic sites and are relatively classified by their tumorigenic potential as follows: DU 145—issue from brain, moderately metastatic (Stone et al. 1978); PC-3—issue from bone, highly metastatic (Kaighn et al. 1979); and PC-3M—issue from liver, more aggressive derivative of PC-3 (Kozlowski et al. 1984). Cells were routinely cultured at 37 °C in a humidified atmosphere with 5 % CO2 in RPMI 1640 medium (Gibco™, Thermo Fischer Scientific) containing a final concentration of 10 % fetal bovine serum (Gibco™) and 2 mM L-glutamine (Gibco™, Thermo Fischer Scientific). Twice a week, cultures were split by treatment with 0.25 % trypsin–EDTA (Gibco™, Thermo Fischer Scientific) for 5 min at 37 °C.
Solutions
The Hank’s buffer saline solution (HBSS) comprised as follows (mM): NaCl 150, KCl 5, MgCl2 1, CaCl2 2, D-glucose 10, and HEPES 10, adjusted to pH 7.4 with 1 M NaOH. For calibration purposes, two different types of media were used: Na+-free solution (Na+ was replaced by choline) and a mixture of Na+ and potassium (K+). Na+-free solution contained the following components (in mM): choline chloride 150, MgCl2 1, KCl 5, CaCl2 2, D-glucose 5.6, and HEPES 10, adjusted to pH 7.4 with 1 M KOH. Extracellular solution with high Na+ concentration consisted of (mM): NaCl 150, KCl 5, MgCl2 1, CaCl2 2, D-glucose 5.6, and HEPES 10, adjusted to pH 7.4 with 1 M NaOH. Another calibration solution consisted of an ion mixture (in mM) such as [Na+ gluconate] + [K+ gluconate] = 140, MgCl2 1, CaCl2 2, D-glucose 5.6, and HEPES 10, adjusted to pH 7.4 with 1 M KOH or NaOH. Of note, the final working concentrations of all Na+ calibration solutions ranged within 0–120 mM and contained 10 µM gramicidin and monensin. Ouabain-, ionomycin-, and veratridine-containing solutions were prepared in HBSS. All reagents have been purchased from Sigma-Aldrich, unless otherwise specified.
Sodium fluorescent imaging
All probes were purchased in a form of cell-permeant acetoxymethyl esters: Na+-binding benzofuran isophthalate (SBFI) from Interchim (Cat. No. FP-82902B), whereas CoroNa™ Green (CoroNa) and Asante NaTRIUM Green-2 (ANG-2)—from Life Technologies (Cat. No. C36676) and TEFLabs (Cat. No. 3512), accordingly. Dyes were reconstituted in dimethyl sulfoxide (DMSO) and diluted in HBSS containing 0.02 % of Pluronic® F127 to the final concentrations of 7 µM for SBFI, 10 µM for CoroNa, and 5 µM for ANG-2.
Cells were plated onto 30-mm glass coverslips and grown in standard conditions for 3 days. During this time, cells properly attached to the coverslips and had time to divide at least once. In such a way, we reassured that the cells were exhibiting healthy phenotype prior to experimental procedure. The coverslips were then washed with HBSS and loaded with 1 ml of the dye solution as follows: for SBFI–5 h in the incubator at 37 °C, for CoroNa–1 h in the incubator at 37 °C, and for ANG-2–1 h at room temperature. Afterwards, the coverslips were gently rinsed with HBSS, fixed on the imaging platform and left in the appropriate experimental solution. All recordings, including calibrations, were performed at room temperature. About 50 cells per field of view were selected for fluorescence measurements. Background fluorescence was selected as an area containing no cells and subtracted from each data point obtained during experiment.
Fluorescence was excited using an illumination DG4 system (Sutter) fitted with a xenon lamp (300 W). All recordings of Na+ fluorescence were acquired using objective 20× in the Superfluor Nikon Eclipse Ti- series inverted microscope coupled to an EMCCD camera Rolera EM-C2 (Qimaging) and processed using Metafluor 7.7.5.0 software. The excitation filters represented the following parameters in terms of wavelength and bandwidth: 340 nm/26 nm and 387 nm/11 nm for SBFI; 482 nm/35 nm for CoroNa and ANG-2. The emission filters were as follows: 510 nm/84 nm for SBFI; 536 nm/40 nm for CoroNa and ANG-2.
Data analysis and software
For the data that was plotted on a curve and fitted with logistic function, R 2 was calculated as a “least squares” method. Data were analyzed using OriginPro 2015 Beta3 software (1991–2014 OriginLab Corporation). The figures were created using CorelDRAW 11.633 software (2002 Corel Corporation).
Results
Basics of sodium imaging and ion-specific indicators
The Na+ imaging technique is based on the recordings of signals produced by fluorescent Na+ indicator dyes. These molecules are typically composed of an ion-binding site and a fluorophore unit (Fig. 2). Thus, the principle is based on dye conformational changes upon Na+ binding, which are followed by spectral changes. This can be assessed by exciting dyes’ fluorescence using specific wavelengths and measuring the emitted light, which can be registered and visualized by the software. The choice of commercially available Na+ probes is still relatively small, when comparing to the wide range of Ca2+ indicator dyes (Rose 2012). Nevertheless, there is still a significant contrast between general chemical properties of the molecules (e.g., fluorescence, ion specificity, affinity). Therefore, these differences have to be accurately considered prior to the dye employment. Importantly, a reliable Na+ indicator should be highly specific for Na+ over other ions. Hence, Na+ binding (or unbinding) should result in a significant change in the fluorescence emission. Furthermore, high dye concentrations and long-term exposure times are not advised for elimination of the possible cytotoxic effects. Thus, emission efficacy of the fluorophore should be as high as possible. To date, Na+ indicator dyes are available in the modified form of acetoxymethyl esters. In this form, the passage of the dye across the cell membrane is facilitated. Once inside, the acetoxymethyl groups are removed by cellular esterases and dyes are thus trapped in the cytoplasm.

Chemical structures of the different Na+ indicator dyes used in this study in the form of acetoxymethyl esters (AM): Na+-binding benzofuran isophthalate (SBFI), CoroNa™ Green (CoroNa), and Asante NaTRIUM Green 2 (ANG-2)
Despite new developments, the most commonly used Na+ indicator dye is still SBFI, first described in 1989 (Minta and Tsien 1989). As a ratiometric dye, it can be excited at two separate wavelengths (340 and 380 nm) and the emitted fluorescence reaches a maximum of intensity at 500 nm. The excitation spectrum is shifted upon Na+ binding onto SBFI, the Na+-bound SBFI and the Na+-free SBFI displaying excitation peaks at 340 and 380 nm, respectively. The ratio of emitted fluorescence obtained at each wavelength (F 340nm/F 380nm) thus directly correlates with [Na+]i, whereas the SBFI emission spectrum does not depend on changes in [Na+]i. Of note, the selectivity of SBFI is 18-fold higher towards Na+ than towards K+.
The properties of CoroNa have been described in detail by earlier studies (Martin et al. 2005; Meier et al. 2006). It is excited by visible light and exhibits an increase in fluorescence emission intensity upon binding of Na+ with just a little shift in wavelength. The excitation and emission maxima of CoroNa are at 492 and 516 nm, accordingly, whereas for absorbance it is near 488 nm. The selectivity of CoroNa is 4 times higher to Na+ than to K+ binding (Martin et al. 2005). Moreover, CoroNa exhibits a relatively high dissociation constant (K d) making it well suitable for measuring large [Na+]i transients (Martin et al. 2005).
Lately, a novel Na+ indicator, Asante NaTRIUM Green 1, has been described (Kim et al. 2010; Lamy and Chatton 2011). According to the manufacturer (TEFLabs Inc., Austin, TX, USA), another derivative, ANG-2, shows improved fluorescence properties, but up to now, only few data on this dye have been published (Miyazaki and Ross 2015; Roder and Hille 2014). The excitation and emission maxima of ANG-2 are around 532 and 550 nm, respectively, and Na+ binding induces an increase in fluorescence intensity. Noteworthy, ANG dyes select for Na+ over K+ about 20-fold.
Loading of the cells with sodium indicator dyes
Within the existing variety of methods for the dye delivery inside of the cell (e.g., microinjection, electroporation, patch-pipette administration, etc.), the usage of esterified compounds is the most practical. The membrane-permeable ester reaches the cytoplasm due to a hydrolytic reaction, and hence no special facilities are required. However, the disadvantage of esters is their ability to accumulate inside intracellular compartments, making the indicator insensitive to the cytosolic levels of Na+. It can be avoided by decreasing the loading temperature, which usually implies longer incubation time. Therefore, the loading conditions of each Na+ indicator have to be accurately adjusted.
In this study, we optimized loading conditions of esterified SBFI, CoroNa, and ANG-2 indicator dyes when using human prostate cancer cell lines DU 145, PC-3, and PC-3M. The loading conditions were mostly comparable to the manufacturer indications and to other publications (Diarra et al. 2001; Meier et al. 2006; Roder and Hille 2014). It has been reported that the loading time for SBFI may vary depending on the cell type. In our study, the loading times for SBFI and CoroNa were prolonged for 5 and 1 h, respectively. Furthermore, the working concentrations of CoroNa were step-wise increased from 0.5 to 10 µM. Nevertheless, even with high concentrations of CoroNa (10 µM), a gradual fluorescence signal loss was observed, most likely due to the dye leakage out of the cell. Alterations of the loading temperatures did not improve the procedure and hence were not modified. Of note, all three prostate cancer cell lines used in this study represented similar levels of loading within each of the Na+-specific dyes under conditions performed (Table 1).
Calibration of the dyes
When employing Na+ indicator dyes, [Na+]i is not measured directly. Therefore, calibration is required in order to convert fluorescence signals into defined changes in [Na+]i. Of note, constructed calibration curves are crucial for estimating multiple working parameters. For example, the dissociation constant Kd, indicative of the dye affinity for Na+, should be within the expected physiological range of [Na+]i in the biological sample. Furthermore, efficient measurements of [Na+]i changes could be recorded under conditions where the working range of the dye varies between 0.1 to 10 times of its K d value. Therefore, these parameters have to be taken into account during the experimental design. Here we describe in vitro calibration procedures of SBFI, CoroNa, and ANG-2 Na+ indicator dyes using PC-3 cells that have been sequentially exposed to solutions with different Na+ values (ranging from 0 to 120 mM) containing 10 µM monensin and gramicidin.
Calibration of ratiometric dye SBFI
Typical changes in fluorescence emitted during excitation at 340 nm (F 340nm) and 380 nm (F 380nm) have been obtained after calibration and are illustrated as F 340nm and F 380nm, respectively, in Fig. 3a, d. As expected, F 380nm inversely correlates with the changes in [Na+]i, whereas F 340nm remains essentially unaffected. Accordingly, the fluorescence ratio F 340nm/F 380nm directly corresponds to the alterations in [Na+]i (Fig. 3b, e). Nevertheless, it is still required to convert F 340nm/F 380nm recordings into [Na+] values. Typically for ratiometric dyes as SBFI, it is done by using standard equation (Grynkiewicz et al. 1985):

Calibration of SBFI in human prostate cancer PC-3 cells. The real-time [Na+]o (0–120 mM) are indicated. Calibrations performed with choline solution are represented in a–c, whereas with K+ solution are shown in d–f. a, d Changes in emitted fluorescence intensities during excitation at 340 and 380 nm. b, e Fluorescence ratio 340 nm/380 nm derived from signals shown in a and d. In c and f the data shown is derived from b and e that have been plotted versus log10 of the known Na+ concentrations and fitted using a logistic function. Data are represented as mean ± SD
where β is F 380nm(max)/F 380nm(min); K d is dissociation constant; R n is equal to F 340nm/F 380nm at each data point recorded; R n(min) and R n(max) are the fluorescence ratios F 340nm/F 380nm at the lowest and highest [Na+] concentration applied during calibration. Thus, R n, R n(min), and R n(max) are known, and β can be estimated from the values illustrated in Fig. 3a and d. In order to calculate K d, the fluorescence ratio F 340nm/F 380nm was plotted versus log10 of known [Na+] values used for the calibration (Fig. 3c, f). The derived curve was fitted using a logistic function as follows:
where p is the Hill coefficient, which represents the slope of the linear part of the curve. The EC50 was computed and attributed to β×K d value, from which K d was derived. In this study, we used different calibration solutions: in one, Na+ was substituted with choline (Fig. 3a–c), whereas in the other it was compensated by K+(Fig. 3d–f). Interestingly, the results significantly differed depending on the solutions used for calibration. In the case of choline, K d was found to be 2.4 mM, whereas the optimal working range of SBFI was proposed to vary between 0.5 and 30 mM [Na+] (Fig. 3c). In the case of the mixture of Na+ and K+ that was used for calibration, K d was found to be 20.7 mM, and the optimal working range lay between 10 and 120 mM [Na+] (Fig. 3f).
Calibration of non-ratiometric dyes CoroNa and ANG-2
The calibration of single-wavelength dyes as CoroNa and ANG-2 is relatively simple, and is done with the following equation:
where K d is dissociation constant, F equals the fluorescence signal at each data point recorded, and F min and F max are the fluorescence signals at the lowest and highest [Na+] concentration applied during calibration. The fluorescence intensities obtained after administration of various [Na+]o levels are illustrated in Figs. 4a, c and 5a, c. Afterwards, the intensity values were plotted versus log10 of [Na+] applied (Figs. 4b, d, 5b, d). The logistic function has been used for curve fitting (see Eq. 2), where the value of K d has been derived as EC50. In choline solution, for CoroNa dye K d resulted in 10.5 mM with a working range of between 1 and 60 mM (Fig. 4b), whereas for ANG-2 dye K d was 3.4 mM with a working range between 0.5 and 30 mM (Fig. 5b). When using a calibration solution that consisted of a mixture of [Na+] and [K+], parameters shifted as follows: for CoroNa, K d resulted in 43 mM, which lay in the range of 10–120 mM, whereas for ANG-2, K d resulted in 32 mM, within the range of 3.75 to 120 mM (Figs. 4d, 5d).

Calibration of CoroNa in human prostate cancer PC-3 cells. The real-time [Na+]o (0–120 mM) are indicated. Calibrations performed with choline solution are represented in a, b, whereas with K+ solution in c, d. a, c Emitted fluorescence intensities at 488 nm. In b and e the data shown is derived from a and c that have been plotted versus log10 of the known Na+ concentrations and fitted using a logistic function. Data are represented as mean ± SD

Calibration of ANG-2 in human prostate cancer PC-3 cells. The real-time [Na+]o (0–120 mM) are indicated. Calibrations performed with choline solution are represented in a, b, whereas with K+ solution in c, d. a, c Emitted fluorescence intensities at 540 nm. In b and e the data shown is derived from a and c that have been plotted versus log10 of the known Na+ concentrations and fitted using a logistic function. Data are represented as mean ± SD
Response to changes in extracellular sodium concentration
The passive transport of many ions including Na+ is carried out across the plasma membrane, down the electrochemical gradient. Thus, in order to investigate the correlation of [Na+]i variations as a response to changes in [Na+]o, we performed a so-called “Na+ switch”. First, cells were washed in Na+-free media to eliminate residual Na+ ion presence. After an initial recording carried out in Na+-free solution, [Na+]o was switched to 75 mM and then to 110 mM. As indicated in Fig. 6a, PC-3 cells responded to the Na+ switch with a 2-fold larger Na+ entry than PC-3M, whereas DU 145 exhibited relatively stable levels of [Na+]i, whatever the [Na+]o was. Although Na+ diffusion at resting membrane potential is generally accepted as a well-known phenomenon, very little is known about its precise mechanisms. From our results, it is obvious that DU 145 cells are more resistant to [Na+]o changes than PC-3 or PC-3M. Thus, it can be suggested that [Na+]i in DU 145 cells has reached some limit or saturation point, and hence no more Na+ can enter inside. Otherwise, DU145 cells may have efficient Na+ buffering mechanisms that allow its rapid extrusion upon Na+ entry. In addition, it can be hypothesized that passive Na+ transports might be mediated through some ion-specific channels, which are absent/non-functional in DU 145 cells. Nevertheless, these pathways are still scarcely described overall and have to be identified for prostate cancer cells in particular.

[Na+]i levels during Na+ switch from 0 to 75 mM and then to 110 mM concentrations. Fluorescence intensities normalized to the initial values (F/F 0). Data are represented as mean ± SD
Response to increase in cytosolic calcium concentration
Ionomycin is an ionophore that is often used as a tool for investigating Ca2+-associated mechanisms (Moskowitz and Hruska 1992; Nilsson et al. 1987). Of note, it creates pores in the lipid membrane layer that permit intracellular Ca2+ entry. Moreover, after application, ionomycin itself easily penetrates inside of the cell and reaches the endoplasmic reticulum. Consequently, the rise of [Ca2+]i is augmented due to endoplasmic reticulum depletion. In this study, we have used ionomycin in order to elevate [Ca2+]i (and hence more likely to affect) Na+/Ca2+ exchanger as the predominant cellular mechanism of Ca2+ efflux. Accordingly, in conditions where [Ca2+]i is high, the Na+/Ca2+ exchanger should allow Na+ entry, and as a result, [Na+]i increases. Here, we have observed a similar rise in [Na+]i within all prostate cancer cell lines studied (Fig. 7). This might be explained due to the nature of ionomycin, which is not a cell type-dependent reagent. Hence, its application leads to equal intracellular Ca2+ release regardless of the cell line. Consequently, [Ca2+]i rise probably affected Na+/Ca2+ exchanger and thereby provoked comparable changes in [Na+]i in all cell lines. Of note, ANG-2 was slightly less sensitive than SBFI, whereas CoroNa exhibited only partial [Na+]i fluctuations after ionomycin administration (Fig. 7).

[Na+]i levels after cytosolic Ca2+ increase induced by an application of 1 µM ionomycin. Fluorescence intensities normalized to the initial values (F/F 0). Data are represented as mean ± SD
Response to sodium/potassium pump inhibition
Na+/K+ ATPase participates in active ion transport by pumping Na+ and K+ against their concentration gradients. The enzyme is present in all higher Eukaryotes and hence its functioning has been much investigated. In particular, it has been attributed to the maintenance of the membrane resting potential, cell volume regulation, signal transduction, etc. (Dlouha et al. 1979; Quinton and Tormey 1976; Robinson 1975). Besides controlling intrinsic cellular activities, multiple studies have reported on Na+/K+ ATPase differential expression in cancer cells and its involvement in cell survival, proliferation, adhesion, migration, and invasiveness (Mijatovic et al. 2008, 2012; Weidemann 2005). Overall, Na+/K+ ATPase may be considered as one of the most important players of intracellular Na+ signaling with its prominent role in cellular (patho)-physiology. Therefore, Na+ imaging is an essential technique for visualization of Na+/K+ ATPase functioning in vitro. In the present study, the activity of Na+/K+ ATPase was examined by the application of a pump-specific inhibitor, ouabain, at a concentration of 250 µM. As expected, all cell lines tested responded with a progressive accumulation of [Na+]i (Fig. 8). Interestingly, DU 145 and PC-3 cells exhibited higher [Na+]i elevations than PC-3M cells (Fig. 8). This could imply various expression levels of this enzyme among different prostate cancer cell lines. Noteworthy, all three Na+ indicator dyes were able to detect [Na+]i increase in DU 145 and PC-3 cells (Fig. 8). However, CoroNa exhibited lower sensitivity for [Na+]i elevation in these cells and was not sufficiently effective to uncover lower response in PC-3M cells (Fig. 8).

[Na+]i levels after Na+/K+-ATPase inhibition induced by an application of 250 µM ouabain. Fluorescence intensities normalized to the initial values (F/F 0). Data are represented as mean ± SD
Response to voltage-gated sodium channel opener
Voltage-gated Na+ channels are known for their involvement in allowing action potential triggering and propagation in nerve and muscle tissues (Denac et al. 2000). However, there is growing evidence on the implementation of these channels in “non-excitable” epithelial cells (Barshack et al. 2008; Gao et al. 2009). Especially, voltage-gated Na+ channels have been found upregulated in many carcinomas, and in particular associated with their metastatic potential (Patel and Brackenbury 2015; Roger et al. 2015). Therefore, in our study we have used DU 145, PC-3, and PC-3M cells, which are considered as a highly aggressive model of human prostate cancer. Indeed, these cell lines have been reported to express voltage-gated Na+ channels (Nakajima et al. 2009; Shan et al. 2014). Thus, we have investigated their functioning using a channel-specific opener, veratridine. Surprisingly, as measured using SBFI and ANG-2, voltage-gated Na+ channel opener provoked a minor [Na+]i increase in DU 145 cells and had no effect on PC-3 and PC-3M (Fig. 9a, b). Partially, it can be explained due to the lack of the effect of veratridine itself, which has already been reported for rat prostate cancer cells (Fraser et al. 2000). In addition, though voltage-gated Na+ channels have been shown to be expressed in prostate cancer cells (Suy et al. 2012), the resting membrane potential may not be compatible with their activity. In that case, the channels may not permeate significant Na+ entry into the cells, which could also explain the lack of the effect of veratridine. We also cannot exclude that the probes used in this study are not sensitive enough to detect these variations of [Na+]i. Of note, the value of veratridine as a chemical activator of voltage-gated Na+ channels has been undermined since it was shown to have only a slight influence on the tetrodotoxin-resistant channel in non-voltage-clamped cells (Farrag et al. 2008). Overall, it has been previously suggested that veratridine may interact with other ion channels and hence its mode of action may be rather complex (Fraser et al. 2003; Romey and Lazdunski 1982; Verheugen et al. 1994).

[Na+]i levels after voltage-gated Na+ channels opening induced by an application of 50 µM veratridine. Fluorescence intensities normalized to the initial values (F/F 0). Data are represented as mean ± SD
Discussion
In vitro Na+ imaging has already been reported in various types of cells (Baquero and Gilbertson 2011; Komlosi et al. 2003; Lahn et al. 2011; Miyazaki and Ross 2015). However, only few data have been reported on its particular application in cancer cells (Chen et al. 2014; Kline et al. 2000; Roger et al. 2007; Tannous et al. 2009). Indeed, due to the relatively small differences of Na+ concentrations (as compared to the Ca2+ gradient) between intra- and extracellular compartments (around 10 times), the detection of the signal is a rather challenging procedure. Therefore, it is not surprising that only several Na+ indicator dyes, such as SBFI, CoroNa, and ANG-2, are commercially available up to date.
Since technical limitations may become decisive for a whole experimental design, it is important to take into account some major aspects of the Na+ dyes use when performing Na+ imaging. For example, the employment of such ratiometric dye as SBFI eliminates experimental variables as cell thickness or size, photobleaching, and dye concentration. However, it is sensitive to changes in intracellular pH (Diarra et al. 2001). Therefore, Na+ recordings involving significant pH alterations should be reconsidered using an alternative Na+ indicator dye. However, a ratiometric dye enables direct comparison between different experiments and quantitative measurement of signals, and, if precise [Na+]i estimation is required while using single-excitation wavelength dye, the calibration procedure has to be performed on each sample tested. The employment of the latter becomes markedly inconvenient for Na+ imaging in multiple samples, which is often required to reach statistical significance, especially when the response is transient and/or of a low magnitude.
Importantly, results of calibration were remarkably different depending on the solutions utilized. For example, when Na+ was substituted by choline, the saturation point for SBFI and ANG-2 dyes was already reached at around 20–40 mM [Na+]o with the proposed working range within 0.5 and 30 mM. In the case when Na+ concentration was compensated by K+, the saturation point as well as the working range were shifted towards higher [Na+]o. It is possible that due to electrochemical gradient, the endogenous high [K+]i creates some limitation for other cations to enter the cell. Therefore, in this case, performing calibration when increasing [K+]o would affect K+ exit out of the cell and hence impede cellular capacity for Na+ entry.
Interestingly, the calibration of all three Na+ indicator dyes has shown that CoroNa is more suitable for detecting [Na+]i variations in a wider range of concentrations. Its working range and K d are at least twice as large as SBFI or ANG-2. Indeed, CoroNa has been previously reported to discriminate substantial [Na+]i changes at high background [Na+] (Meier et al. 2006). SBFI, CoroNa, and ANG-2 are not toxic per se and there have not been any pharmacological effects of the indicators demonstrated yet. However, short exposure times and low indicator concentrations are favorable for minimizing cellular stress. In this case, the disadvantage of SBFI is its slow cell permeability, even after addition of special dispersion agent Pluronic® F127. For example, in this study, it took 5 h to charge human prostate cancer cells DU 145, PC-3, and PC-3M. Nevertheless, it has been reported that SBFI loading strongly depends on the cell type and hence it is possible that for some other in vitro models this issue will not occur (Diarra et al. 2001; Minta and Tsien 1989).
This study has been conducted to compare three different Na+-specific fluorescent dyes and their feasibility for in vitro Na+ imaging (Table 1). Once the working parameters were derived from the calibration curves, it was necessary to examine the dyes suitability within experimental conditions. Therefore, changes in free [Na+]i have been initiated via diverse pathways—ubiquitous and cell type-specific. For example, various types of human cancer cells, including prostate, have been reported to express voltage-gated Na+ channels (Fraser et al. 2014; Shan et al. 2014). Thus, application of the Na+ channel activator veratridine would lead to [Na+]i increase only in cells where functional Na+ channels are expressed on the plasma membrane. However, in this study veratridine application did not exhibit large Na+ entry, more likely due to its complex mode of action (Fraser et al. 2003). Na+/K+ ATPase, a protein that transports three Na+ out of the cell against two K+ inside, is found in the plasma membrane of all animal cells (Dlouha et al. 1979; Quinton and Tormey 1976; Robinson 1975). Accordingly, the application of ouabain, a specific inhibitor of Na+/K+ ATPase, resulted in omnipresent [Na+]i increase. Similarly, ionomycin is an ionophore that acts on any kind of cell where it enables Ca2+ entry through the plasma membrane and its release from the endoplasmic reticulum (Moskowitz and Hruska 1992; Nilsson et al. 1987). Possibly the elevated [Ca2+]i affected the activity of Na+/Ca2+ exchanger that imports Na+ and hence increased [Na+]i. In addition, Na+ entry can be provided via ion diffusion down the electrochemical gradient through Na+-specific leak channels (Pajor and Wright 1992; Parent et al. 1992; Wright et al. 1992). In this study, the function of Na+-leak channels has been investigated due to the alteration of [Na+]o. Interestingly, the changes of [Na+]o induced more rapid alterations of SBFI signal than ANG-2. Therefore, we suggest that ANG-2 is a slightly less dynamic probe than SBFI.
Of note, CoroNa has exhibited lower signal intensities than SBFI or ANG-2 throughout all experiments. This could be interpreted due to the elevated K d and working range of CoroNa, which means that the dye may work more accurately for higher [Na+] alterations. Another explanation could be attributed to the leakage of CoroNa out of the cells with time that has been observed in this study. Indeed, previous reports indicate that CoroNa may be lost quickly from intracellular medium due to its relatively small size. This can be alleviated with a constant delivery of the dye through a patch-pipette during whole-cell path clamp recordings, which enables stable intracellular CoroNa concentrations (Meier et al. 2006). In a promising novel approach, Lamy and coworkers encapsulated CoroNa Green in a PAMAM dendrimer-nanocontainer to counteract permanent dye extrusion (Lamy et al. 2012). SBFI and ANG-2 have exhibited comparable levels of signal intensities after various [Na+]i alterations. However, it has to be kept in mind that Na+ imaging conducted with the SBFI dye enables a reliable and quantitative determination of the actual [Na+]i, whereas loading conditions for ANG-2 are more practical.
In conclusion, this study presents in vitro Na+ imaging in three human prostate cancer cell lines. According to our data, CoroNa is not suitable for the detection of relatively low [Na+]i changes in the prostate cancer cell model studied here, whereas SBFI and ANG-2 can provide reliable recordings.
References
Amorino GP, Fox MH (1995) Intracellular Na+ measurements using sodium green tetraacetate with flow cytometry. Cytometry 21:248–256. doi:10.1002/cyto.990210305
Armstrong CM (1981) Sodium channels and gating currents. Physiol Rev 61:644–683
Baquero AF, Gilbertson TA (2011) Insulin activates epithelial sodium channel (ENaC) via phosphoinositide 3-kinase in mammalian taste receptor cells. Am J Physiol Cell Physiol 300:C860–C871. doi:10.1152/ajpcell.00318.2010
Barshack I, Levite M, Lang A, Fudim E, Picard O, Ben Horin S, Chowers Y (2008) Functional voltage-gated sodium channels are expressed in human intestinal epithelial cells. Digestion 77:108–117. doi:10.1159/000123840
Besson P, Driffort V, Bon E, Gradek F, Chevalier S, Roger S (2015) How do voltage-gated sodium channels enhance migration and invasiveness in cancer cells? Biochim Biophys Acta 1848:2493–2501. doi:10.1016/j.bbamem.2015.04.013
Chen D, Song M, Mohamad O, Yu SP (2014) Inhibition of Na+/K+-ATPase induces hybrid cell death and enhanced sensitivity to chemotherapy in human glioblastoma cells. BMC Cancer 14:716. doi:10.1186/1471-2407-14-716
Denac H, Mevissen M, Scholtysik G (2000) Structure, function and pharmacology of voltage-gated sodium channels. Naunyn Schmiedebergs Arch Pharmacol 362:453–479
Diarra A, Sheldon C, Church J (2001) In situ calibration and [H+] sensitivity of the fluorescent Na+ indicator SBFI. Am J Physiol Cell Physiol 280:C1623–C1633
Dlouha H, Teisinger J, Vyskocil F (1979) Activation of membrane Na+/K+-ATPase of mouse skeletal muscle by acetylcholine and its inhibition by alpha-bungarotoxin, curare and atropine. Pflugers Arch 380:101–104
Farrag KJ, Bhattacharjee A, Docherty RJ (2008) A comparison of the effects of veratridine on tetrodotoxin-sensitive and tetrodotoxin-resistant sodium channels in isolated rat dorsal root ganglion neurons Pflugers Arch 455:929–938. doi:10.1007/s00424-007-0365-5
Fraser SP, Grimes JA, Djamgoz MB (2000) Effects of voltage-gated ion channel modulators on rat prostatic cancer cell proliferation: comparison of strongly and weakly metastatic cell lines. Prostate 44:61–76. doi:10.1002/1097-0045(20000615)44:1<61
Fraser SP et al (2003) Contribution of functional voltage-gated Na+ channel expression to cell behaviors involved in the metastatic cascade in rat prostate cancer: I. Lateral motility J Cell Physiol 195:479–487. doi:10.1002/jcp.10312
Fraser SP, Ozerlat-Gunduz I, Brackenbury WJ, Fitzgerald EM, Campbell TM, Coombes RC, Djamgoz MB (2014) Regulation of voltage-gated sodium channel expression in cancer: hormones, growth factors and auto-regulation. Philos Trans R Soc Lond B Biol Sci 369:20130105. doi:10.1098/rstb.2013.0105
Gao N et al (2009) Voltage-gated sodium channels in taste bud cells. BMC Neurosci. doi:10.1186/1471-2202-10-20
Gillet L et al (2009) Voltage-gated sodium channel activity promotes cysteine cathepsin-dependent invasiveness and colony growth of human cancer cells. J Biol Chem 284(13):8680–8691
Grynkiewicz G, Poenie M, Tsien RY (1985) A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem 260:3440–3450
Hansen LL, Rasmussen J, Friche E, Jaroszewski JW (1993) Method for determination of intracellular sodium in perfused cancer cells by 23Na nuclear magnetic resonance spectroscopy. Anal Biochem 214:506–510. doi:10.1006/abio.1993.1530
Jacobs MA et al (2011) Monitoring of neoadjuvant chemotherapy using multiparametric, (2)(3)Na sodium MR, and multimodality (PET/CT/MRI) imaging in locally advanced breast cancer. Breast Cancer Res Treat 128:119–126. doi:10.1007/s10549-011-1442-1
Kaighn ME, Narayan KS, Ohnuki Y, Lechner JF, Jones LW (1979) Establishment and characterization of a human prostatic carcinoma cell line (PC-3). Invest Urol 17:16–23
Kim MK, Lim CS, Hong JT, Han JH, Jang HY, Kim HM, Cho BR (2010) Sodium-ion-selective two-photon fluorescent probe for in vivo imaging. Angew Chem Int Ed Engl 49:364–367. doi:10.1002/anie.200904835
Kline RP et al (2000) Rapid in vivo monitoring of chemotherapeutic response using weighted sodium magnetic resonance imaging. Clin Cancer Res 6:2146–2156
Komlosi P, Fuson AL, Fintha A, Peti-Peterdi J, Rosivall L, Warnock DG, Bell PD (2003) Angiotensin I conversion to angiotensin II stimulates cortical collecting duct sodium transport. Hypertension 42:195–199. doi:10.1161/01.HYP.0000081221.36703.01
Kozlowski JM, Fidler IJ, Campbell D, Xu ZL, Kaighn ME, Hart IR (1984) Metastatic behavior of human tumor cell lines grown in the nude mouse. Cancer Res 44:3522–3529
Lahn M, Dosche C, Hille C (2011) Two-photon microscopy and fluorescence lifetime imaging reveal stimulus-induced intracellular Na+ and Cl− changes in cockroach salivary acinar cells. Am J Physiol Cell Physiol 300:C1323–C1336. doi:10.1152/ajpcell.00320.2010
Lamy CM, Chatton JY (2011) Optical probing of sodium dynamics in neurons and astrocytes. Neuroimage 58:572–578. doi:10.1016/j.neuroimage.2011.06.074
Lamy CM, Sallin O, Loussert C, Chatton JY (2012) Sodium sensing in neurons with a dendrimer-based nanoprobe. ACS Nano 6:1176–1187. doi:10.1021/nn203822t
Martin VV, Rothe A, Gee KR (2005) Fluorescent metal ion indicators based on benzoannelated crown systems: a green fluorescent indicator for intracellular sodium ions. Bioorg Med Chem Lett 15:1851–1855. doi:10.1016/j.bmcl.2005.02.017
Meier SD, Kovalchuk Y, Rose CR (2006) Properties of the new fluorescent Na+ indicator CoroNa Green: comparison with SBFI and confocal Na+ imaging. J Neurosci Methods 155:251–259. doi:10.1016/j.jneumeth.2006.01.009
Mijatovic T, Ingrassia L, Facchini V, Kiss R (2008) Na+/K+-ATPase alpha subunits as new targets in anticancer therapy. Expert Opin Ther Targets 12:1403–1417. doi:10.1517/14728222.12.11.1403
Mijatovic T, Dufrasne F, Kiss R (2012) Na+/K+-ATPase and cancer. Pharm Pat Anal 1:91–106. doi:10.4155/ppa.12.3
Minta A, Tsien RY (1989) Fluorescent indicators for cytosolic sodium. J Biol Chem 264:19449–19457
Miyazaki K, Ross WN (2015) Simultaneous sodium and calcium imaging from dendrites and axons(1,2,3). eNeuro 2. doi:10.1523/ENEURO.0092-15.2015
Moskowitz DW, Hruska KA (1992) Ca2+ uptake by endoplasmic reticulum of renal cortex. I. Ionic requirements and regulation in vitro. Calcif Tissue Int 51:35–41
Nakajima T et al (2009) Eicosapentaenoic acid inhibits voltage-gated sodium channels and invasiveness in prostate cancer cells. Br J Pharmacol 156:420–431. doi:10.1111/j.1476-5381.2008.00059.x
Nilsson T, Arkhammar P, Hallberg A, Hellman B, Berggren PO (1987) Characterization of the inositol 1,4,5-trisphosphate-induced Ca2+ release in pancreatic beta-cells. Biochem J 248:329–336
Ouwerkerk R, Bleich KB, Gillen JS, Pomper MG, Bottomley PA (2003) Tissue sodium concentration in human brain tumors as measured with 23Na MR imaging. Radiology 227:529–537. doi:10.1148/radiol.2272020483
Ouwerkerk R et al (2007) Elevated tissue sodium concentration in malignant breast lesions detected with non-invasive 23Na MRI. Breast Cancer Res Treat 106:151–160. doi:10.1007/s10549-006-9485-4
Pajor AM, Wright EM (1992) Cloning and functional expression of a mammalian Na+/nucleoside cotransporter a member of the SGLT family. J Biol Chem 267:3557–3560
Parent L, Supplisson S, Loo DD, Wright EM (1992) Electrogenic properties of the cloned Na+/glucose cotransporter: I. Voltage-clamp studies. J Membr Biol 125:49–62
Patel F, Brackenbury WJ (2015) Dual roles of voltage-gated sodium channels in development and cancer. Int J Dev Biol 59:357–366. doi:10.1387/ijdb.150171wb
Quinton PM, Tormey JM (1976) Localization of Na/K-ATPase sites in the secretory and reabsorptive epithelia of perfused eccrine sweat glands: a question to the role of the enzyme in secretion. J Membr Biol 29:383–399
Robinson JD (1975) Mechanisms by which Li+ stimulates the (Na+ and K+)-dependent ATPase. Biochim Biophys Acta 413:459–471
Roden DM, George AL Jr (1997) Structure and function of cardiac sodium and potassium channels. Am J Physiol 273:H511–H525
Roder P, Hille C (2014) ANG-2 for quantitative Na(+) determination in living cells by time-resolved fluorescence microscopy. Photochem Photobiol Sci 13:1699–1710. doi:10.1039/c4,pp00061g
Roger S et al (2007) Voltage-gated sodium channels potentiate the invasive capacities of human non-small-cell lung cancer cell lines. Int J Biochem Cell Biol 39:774–786. doi:10.1016/j.biocel.2006.12.007
Roger S, Gillet L, Le Guennec JY, Besson P (2015) Voltage-gated sodium channels and cancer: is excitability their primary role? Front Pharmacol 6:152. doi:10.3389/fphar.2015.00152
Romey G, Lazdunski M (1982) Lipid-soluble toxins thought to be specific for Na+ channels block Ca2+ channels in neuronal cells. Nature 297:79–80
Rose CR (2012) Two-photon sodium imaging in dendritic spines. Cold Spring Harb Protoc 2012:1161–1165. doi:10.1101/pdb.prot072074
Shan B et al (2014) Voltage-gated sodium channels were differentially expressed in human normal prostate, benign prostatic hyperplasia and prostate cancer cells. Oncol Lett 8:345–350. doi:10.3892/ol.2014.2110
Stone KR, Mickey DD, Wunderli H, Mickey GH, Paulson DF (1978) Isolation of a human prostate carcinoma cell line (DU 145). Int J Cancer 21:274–281
Suy S et al (2012) Expression of voltage-gated sodium channel Nav1.8 in human prostate cancer is associated with high histological grade. J Clin Exp Oncol. doi:10.4172/2324-9110.1000102
Tannous BA et al (2009) Mutant sodium channel for tumor therapy. Mol Ther 17:810–819. doi:10.1038/mt.2009.33
Verheugen JA, Oortgiesen M, Vijverberg HP (1994) Veratridine blocks voltage-gated potassium current in human T lymphocytes and in mouse neuroblastoma cells. J Membr Biol 137:205–214
Weidemann H (2005) Na/K-ATPase, endogenous digitalis like compounds and cancer development—a hypothesis. Front Biosci 10:2165–2176
Wright EM, Hager KM, Turk E (1992) Sodium cotransport proteins. Curr Opin Cell Biol 4:696–702
Acknowledgments
We thank Prof. Mustafa Djamgoz for PC-3M cell line (Imperial College London, United Kingdom). The research in authors' laboratory is supported by the INSERM, SIRIC OncoLille and Laboratory of Excellence, Ion Channels Science and Therapeutics. Oksana Iamshanova was funded by la Ligue Nationale Contre le Cancer (France, GB/MA/CD 11813).
Author information
Authors and Affiliations
Corresponding author
Additional information
Special Issue: Ion Channels, Transporters and Cancer.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Iamshanova, O., Mariot, P., Lehen’kyi, V. et al. Comparison of fluorescence probes for intracellular sodium imaging in prostate cancer cell lines. Eur Biophys J 45, 765–777 (2016). https://doi.org/10.1007/s00249-016-1173-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00249-016-1173-7