
Abstract
We examined the intraindividual variation present in the first ribosomal internal transcribed spacer (ITS1) of Anopheles farauti to determine the level of divergence among populations for this important malarial vector. We isolated 187 clones from 70 individuals and found regional variation among four internal tandem repeats. The data were partitioned prior to analysis given the presence of a paralogous ITS2 sequence, called the 5′-subrepeat, inserted in the ITS1 of most clones. A high level of homogenization and population differentiation was observed for this repeat, which indicates a higher rate of turnover relative to the adjacent ‘core’ region. Bayesian analysis was performed using several substitutional models on both a combined and a partitioned data set. On the whole, the ITS1 phylogeny and geographic origin of the samples appear to be congruent. Some interesting exceptions indicate the spread of variant repeats between populations and the retention of ancestral polymorphism. Our data clearly demonstrate concerted evolution at the intraspecific level despite intraindividual variation and a complex internal repeat structure from a species that occupies a continuous coastal distribution. A high rate of genomic turnover in combination with a high level of sequence divergence appears to be a major factor leading to its concerted evolution within these populations.
Similar content being viewed by others

Introduction
The concerted evolution of repeated sequence families is facilitated by a process of homogenization that involves reciprocal and nonreciprocal recombination of repeated DNA within and between chromosomes. This results in a pattern of accumulated interspecific divergence and intraspecific similarity among the products of gene duplication (Elder and Turner 1995; Graur and Li 2000). Recombinant processes such as unequal crossing-over, gene conversion, and retroposition directly affect the copy number of variant repeats in the genome and, as a consequence, generate non-Mendelian patterns of segregation (Brosius 2003; Graur and Li 2000). These exchanges cause a gain or loss of repeated DNA with a ‘turnover’ frequency that can occur several orders of magnitude faster than rates of point mutation (Dover 2000). When this DNA turnover is coupled with the spread of chromosomes by sexual reproduction, then these genomic changes have an even greater potential to change the genetic variation present within populations and therefore within a species.
The relative importance of the genomic-level versus population-level processes that generate intraspecific divergence within repeated sequence families remains largely unknown and is often misunderstood (Williams 1990; Liao 1999; Dover 2002). Interactions between these levels are expected to occur, though one can dominate, depending on the rates of recombinant exchanges within and between chromosomes (Dover 2000). For example, restrictions to the interchromosomal exchange of repeats can lead to the formation of divergent chromosomal lineages (Schindelhauer and Schwarz 2002). These differences may then accumulate between individuals, enabling natural selection and genetic drift to become indirectly involved in the fixation dynamics of a variant repeat, i.e., how it is spread or removed from a population. By comparison, interchromosomal exchanges reduce interindividual variation and provide the opportunity for a population of sexually reproducing organisms to undergo cohesive genetic changes (Luchetti et al. 2003). Such exchanges can be rapid when present (e.g., Wendel et al. 1995) and may also be restricted to particular regions within a gene family, due to localized recombinant activity (Gonzalez and Sylvester 2001).
The fixation of gene family variation within a population is not expected to occur instantaneously and so partial homogenization may be observed with differences in copy number, repeat length, and point mutational changes. The first study of intraspecific concerted evolution to use DNA sequence data found an absence of intraindividual and intrapopulation variation, plus a large degree of between-population divergence, in the HindIII satellite DNA family of pupfish (Elder and Turner 1994). Such high rates of homogenization and the fixation of variation within local breeding populations have obvious practical applications in identifying population subdivisions. Few studies have since focused at the intraspecific level to identify the pattern of DNA sequence polymorphism in repeated sequence families. However, their utility as genetic markers depends on a high rate of turnover of variant repeats within the genome for concerted evolution to occur at the population level. The rapid turnovers of R1 and R2 retrotransposable elements present in the ribosomal DNA (rDNA) of Drosophila melanogaster are a prime example of this (Averbeck and Eickbush 2005).
The ribosomal RNA multigene family is a frequently used genetic marker and the classic example of concerted evolution in a tandemly repeated sequence family (Graur and Li 2000). The rRNA genes are evolutionary conserved and variation within the gene family is usually homogenized within species. The rDNA internal transcribed spacers (ITS1 and ITS2) are regions of divergent noncoding DNA separated by a small rRNA-encoding subunit (5.8S rDNA) and flanked by the 18S and 28S rDNA. These spacers are usually divergent between species and homogenized within species. The rDNA ITS can be sequenced directly from PCR products if length variation is absent and homogenization is complete. However, previous examples that demonstrated spacer conservation (e.g., Fritz et al. 1994; Schön et al. 1998) have subsequently been refuted by intraindividual and intrapopulation sampling, coupled to the isolation of individual repeats by cloning (Onyabe and Conn 1999; Gandolfi et al. 2001). Whether expected or not, the presence of intraindividual variation in the ITS1 can confound phylogenetic relationships (Famà et al. 2000; Harris and Crandall 2000). There are also examples of length variation within individuals (Fenton et al. 1998) and between closely related species (von der Schulenburg et al. 2001). Poor phylogenetic resolution among populations can result from low levels of divergence (e.g., Schizas et al. 1999; Presa et al. 2002), but direct sequencing may also be a confounding factor in which the presence of polymorphisms within individuals is underestimated.
We present the results of an intraspecific study demonstrating concerted evolution of the ITS1 in Anopheles (Cellia) farauti. This is the most coastally restricted and widely dispersed mosquito in the An. punctulatus group and an important malarial vector in the Australian biogeographical region (Beebe and Cooper 2002). A previous study of this species found that the ITS1 contains a degree of population divergence, whereby individuals contain one of five geographically distinct PCR-RFLP genotypes (Beebe et al. 2000a). In this study we analyzed the complex distribution pattern of variant ITS1 repeats within and among populations of An. farauti. This spacer is characterized by the presence of long internal tandem repeats which contain regional variation for both copy number and variant repeat types. Bayesian analysis was used to estimate a phylogeny of the ITS1 from a series of substitutional models and data partitions. The congruence of the phylogeny with the sample origin and the level of resolution present demonstrates that the homogenization of variant repeats occurs fast enough to reduce some variation within regional populations. Remarkably, the divergence of the ITS1 reflects recent patterns of population structure in a species that occupies a continuous coastal habitat.
Materials and Methods
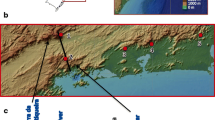
Mosquito specimens used here came from the Australian Army Malarial Institute field collections of An. farauti (Fig. 1). Additional samples came from an inbred colony originally from the island of Rabaul, sampled between 1996 and 2001. Three other species of An. farauti were included in a phylogenetic analysis: An. farauti 8 (a putatively new taxon), An. irenicus, and An. farauti 6 as an outgroup species.

Map of the southwest Pacific showing An. farauti collection sites and the abbreviated location names referred to in the text. Abbreviations are detailed in Table 1
DNA Extraction, PCR Amplification, Cloning, and Sequencing
Genomic DNA was extracted using a CTAB-based method (Reineke et al. 1998). Each sample of DNA was resuspended in 30 μl of TE buffer (10 mM Tris-HCl, pH 8, 0.1 mM EDTA) containing 250 ng of Rnase (Roche). The whole ITS was isolated by PCR amplification, using primers F-ITS/18S (5′-GATTCCACCGCGAGTTGCAGTTGG-3′), which amplifies 112 bp of the 18S gene, and R-ITS/28S (5′-CCGTTTCGCTCGCAGCTACTCAGG-3′), which amplifies 108 bp of the 28S gene. PCRs were performed with ~10 ng of DNA, each primer at 500 nM, 5% DMSO, each dNTP at 50 μM, 1 unit of Pfu DNA polymerase (MBI Fermentas), 1 × PCR buffer supplied (pH 8.8), and 2 mM MgSO4. Thermocycling conditions were one cycle at 95°C for 3 min, followed by 20-25 cycles at 95°C for 30 s, 69°C for 30 s, and 75°C for 3.5 min, then one cycle at 75°C for 3 min. PCR products were electrophoresed on 1% agarose gel and the bands excised, then purified with a gel extraction kit (QIAGEN). An internal ITS1 repeat, called the 5′-subrepeat, and flanking regions were isolated using ~10 ng of DNA in a 10-μl-volume PCR reaction containing primers F-ITS/18S and R-ITS1precore (5′-CCTGTCGGATTGTGACATGTTC-3′) at 400 nM each, 10 mM Tris-HCl, pH 8.9, 40 mM KCl, 1.5 mM MgCl2, 6% DMSO, each dNTP at 75 μM, 0.25 unit of Taq DNA polymerase, and 0.125 unit of Pfu DNA polymerase. Thermocycling conditions were one cycle at 95°C for 3 min, followed by 30 cycles at 95°C for 30 s, 65°C for 30 s, and 72°C for 1 min, then one cycle at 72°C for 3 min.
Cloned copies of the ITS were isolated from the PCR gel extractions. An aliquot of each extract was phosphorylated in a new tube containing 1 unit of T4 polynucleotide kinase, 1 × ligase buffer, and 5% PEG 4000 (all MBI Fermentas). These were incubated at 37°C for 20 min, then denatured at 65°C for 20 min. A 50-ng aliquot of dephosphorylated SmaI-cut pGEM-3Zf(+) (Promega) was added during the last 5 min, before cooling to 4°C. The samples were incubated for 16 h after adding 2 units of T4 DNA ligase (MBI Fermentas), 1 × ligase buffer, and 5% PEG 4000. Ligation reactions were denatured at 70°C for 15 min. Standard techniques for transformation and identification of recombinant cells (e.g., Sambrook and Russell 2001) were carried out using CaCl2-competent Eschericha coli cells of the DH5α strain. Cells were recovered for no longer than 40 min after transformation. Insert-containing plasmids were purified using the GeneElute miniprep kit (Sigma-Aldrich). The clones chosen for sequencing were size-selected by PCR amplification and electrophoresis of the ITS1 in order to sample a range of length variants from each individual.
Both forward and reverse DNA strands were sequenced using the BigDye terminator cycle sequencing kit v2.0 and an ABI 377 sequencer (Applied Biosystems). The PCR primers F-ITS/18S, R-ITS/28S, and R-ITS1precore were used for sequencing plus six other primers: F-Anf-central (5′-TCCTACCACCAACAGTCGTAG-3′), R-Anf-central (5′-GCGTGCTACGACTGTTGGTG-3′), R-HinfI/ITS2 (5′-CGGGAGCGGGCAACGGATTG-3′), F5′-ITS1core (5′-GAGTTGAACATGTCACAATCCGACAGG-3′), R3′-ITS1core (5′-GCCGGTCGGCTTTTTAATATACGCTAC-3′), and F-ITS1postcore (5′-AAGCAGCAAGTAGAGCATCCC-3′).
Sequence Analysis
Multiple sequences were manually aligned because of internal subrepetition. Substitutional models were assessed for the ITS1 and the same data divided into partitions that form adjacent regions within the spacer; the 5′-subrepeat and the ‘core.’ These were modeled using the Bayesian Information Criterion (BIC) employed in DT-ModSel (Minin et al. 2003) and the Akaike Information Criterion (AIC) employed in Modeltest v3.7 (Posada and Crandall 1998). DT-ModSel parameter estimates were used to determine average pairwise distances with maximum likelihood (ML) correction and pairwise deletion of gaps, using PAUP* v4.0 (Swofford 2002). An a priori assessment of the suitability of the data for phylogenetic analysis was performed using the likelihood-mapping method (Strimmer and von Haeseler 1997) employed in TREE-PUZZLE v5.2 (Schmidt et al. 2002) with parameter estimates taken from DT-ModSel. Recombination detection methods for noncoding data were also employed using the Recombination Detection Program, RDP v2.0 (Martin and Rybicki 2000). Homogeneity of base composition among the aligned sequences was determined using the chi-square test in TREE-PUZZLE.
Bayesian analysis was used for phylogenetic reconstruction and substitutional model parameter estimation, using MrBayes v3.1 (Ronquist and Huelsenbeck 2003). The ITS1 data were partitioned for Bayesian analysis given an a priori expectation that characteristic differences may be present between the 5′-subrepeat and the core regions as a result of their separate origins and potentially different rates of recombination within the rDNA locus. Another data partition was formed using all the potentially informative gaps in the alignment. Adjacent gaps were treated as single insertion events if they were part of a tandem repeat. The best-performing model for the ITS1 and core, according to the BIC, was a constrained version of the general time-reversible (GTR) model with a single transitional class and the two transversional classes (the TVM model). In addition, a proportion of sites is treated as potentially variable according to a gamma distribution (Γ), and the remainder considered invariant (I). The BIC model for the 5′-subrepeat was the Hasegawa, Kishino, and Yano (HKY) model + Γ + I. The model with the highest AIC weight (Posada and Buckley 2004) for the ITS1 was TVM + Γ + I, while the second highest was GTR + Γ + I. The model with the highest AIC weight for the core was the GTR + Γ + I, and for the 5′-subrepeat the highest was TVM + Γ + I; the second was GTR + Γ + I. The TVM model is not available in MrBayes v3.1 and was generalized to the GTR model. The gamma distribution shape parameter (α) was approximated to four rate categories, with α < 1 indicating mutational ‘hot spots.’
Bayesian analysis consisted of four independent MCMC runs for each of the following models: a single GTR + Γ + I model, the mixed model HKY + Γ + I (5′-subrepeat) and GTR + Γ + I (core), and the mixed model GTR + Γ + I for both partitions. The base frequency estimates, proportion of invariants (p-inv), substitution rate matrix, and α were treated as either linked or unlinked parameters when partitions were enforced. Default settings in MrBayes v3.1 for the priors were used for each parameter, except the ‘temperature’ parameter was reduced to 0.10 to improve the MCMC sampling among four chains. Analyses were run for 10 million generations, sampled every 500, to yield 20,000 trees for each of the MCMC runs. The mean, 95% credibility intervals (CI), and effective sample size of each parameter were determined after stationarity from four independent analyses, using Tracer v1.3 (Rambaut and Drummond 2004). A burn-in period of 1,500,000 MCMC generations was used and convergence of MCMC chains to a stationary distribution was checked visually by inspection of the log-likelihood scores for each parameter plotted against generation time. An estimate of the ITS1 phylogeny was made using the maximum posterior probability (MAP) tree (Rannala and Yang 1996). Topological uncertainty was determined from a 70% majority-rule consensus of 70,000 trees pooled from four analyses, using PAUP* v4.0.
Results
Identical and Duplicate Sequences
A total of 187 ITS1 clones were sequenced from 70 An. farauti individuals (GenBank accession numbers EF530745–EF530918, EF042693–EF042694, EF042699, EF042702, EF042704, and EF042710–EF042717), ranging from 1–7 clones each and sampled from 26 locations (Table 1). An additional 16 sequences came from three other species: 9 from An. irenicus (EF042722–EF042730), 3 from An. farauti 6 (EF042741–EF042743), and 4 from a putative new species, An. farauti 8 (EF042718–Ef042721). The homogenized rDNA gene regions and identical ITS1 sequences were removed prior to analysis (Table 1). Duplicate sequences, i.e., those differing by short tandem repeats (mainly a dinucleotide GTn), were also excluded given the redundancy of indels under a likelihood model. In all, 11 identical pairs of sequences (all An. farauti) and 20 duplicate pairs (3 An. irenicus and 17 An. farauti) were present. Five of these became duplicates after an alignment edit of a 21-bp repeat, and two by the removal of a 67-bp repeat (see ITS1 alignments section). Twenty-four of the identical and duplicate pairs came from the same individual or population. The remaining seven pairs were from an adjacent population or nearby region. Note that, for brevity, the population origin of each clone is abbreviated using a letter, followed by a numeral to denote an individual and its respective clone (Table 1).
Internal Repetition
Four types of tandem repeat are present within the ITS1 of An. farauti: the 5′-subrepeat (287–291 bp) located at the 5′-end of the spacer, the ‘core repeat’ (360–362 bp), a 21-bp repeat (including one dispersed copy), and a 67-bp repeat at the 5′-end (Fig. 2). Duplication of the core repeat sequence element has not been observed in the other three taxa. The presence or absence of the 5′-subrepeat and core repeat varied geographically and caused most of the length variation seen among the ITS PCR products (Fig. 2). Some lengths include PCR heteroduplexes which have been observed previously (Beebe et al. 2000a). The ITS1 ranges between 822 and 1,631 bp in length among the clones sequenced from this species (Table 1). Longer lengths were observed, but PCR products >2500 bp were not isolated by cloning.

Internal repeats within the rDNA ITS1 from An. farauti and geographic PCR length variation. (a) ITS PCR fragment with an expanded diagram of the ITS1 showing the internal repeats and sequencing primers. Primer sites and orientation are indicated by arrowheads and are labeled: a, F-ITS/18S; b, R-ITS1precore; c, R-HinfI/ITS2; d, R-ITS/28S; e, F5′-ITS1core; f, F-Anf-central; g, F-ITS1postcore; h, R-Anf-central; and i, R3′ITS1core. (b) A 1% agarose gel showing ITS PCR products. Representative samples from 21 An. farauti populations are shown in the following order, from No. 1 to No. 21: Manus, Sepik1, Madang1, Madang2, Lae1, Lae2, Rabaul, Buka, Bougain, Guadal, Vanuatu, Centrl2, Centrl1, Gulf1, Gulf2, WestPrv, Torres, QLD1, QLD2, NT4, NT5. (c) A 1.5% agarose gel showing the PCR products of the 5′-ITS1, using primers F-ITS/18S and R-ITS1precore. Representative samples from the same populations and individuals are shown in the same order as in the gel above. ITS1 copies with a single 5′-subrepeat are 452–454 bp long, and without it they are 161–163 bp long. The flanking lanes of both gels contain a DNA ladder, with lengths shown in kilobases
Copies of the 5′-subrepeat are inserted into the spacer just upstream from a conserved 17- to 19-bp region that comprises the 5′-end of the ITS1. It is flanked by GAn dinucleotide repeats and effectively forms an adjacent recombining region. BLAST searches to published sequence data identified tandem copies of this repeat in the ITS1 of other An. punctulatus species, except in the basal member, An. clowi, and an outgroup species, An. annulipes s.l. (see Beebe and Cooper 2002). However, an unexpected match was also found to the ITS2 of these species, where it occurs as a single sequence element. It appears to be quite conserved, especially given its presence in the ITS2 of An. dirus and An. baimaii from Southeast Asia. Detection of ITS1 copies with or without a single repeat was achieved by PCR (Fig. 2). The abundance of ITS1 copies with a single 5′-subrepeat relative to those without it appeared to differ among populations. Although not strictly quantitative, this illustrates regional variation for the relative abundance of this repeat. It was absent from all clones sequenced from Madang2, Centrl2, and Gulf1, while just one subrepeat-containing clone was isolated from Sepik1, Madang1, and Centrl1. The repeat appears to be fixed in some populations (e.g., Manus), while others are fixed for ITS1 copies without it (e.g., Madang1). Two tandem copies were isolated from two clones (NT5 A2 and A3), which differed by four bases.
Twenty clones contained two tandem copies of a long core repeat, while another had three. This repeat is located 34 bp downstream from the 5′-subrepeat and ends 100 bp before a tandem array of 21-bp repeats. It was isolated from eight localities clustered around northern and southern Papua New Guinea (PNG) (sites 2–8 and 13; Fig. 1). The 5′-subrepeat appears rare in these populations (see Fig. 2), but a copy of this repeat co-occurring with the core repeat was seen in three clones. No core repeat was identical to its adjacent copy and variant nucleotides were not specific to a position within the array.
Almost all variation observed in the 21-bp repeats consisted of substitutional changes in the last seven bases. Copy number varied from four to six repeats, with all deletions and duplications located in the second and third positions of the array. Copy number variation was not restricted to a particular geographic region and tandem copies were often of identical type. The copy number of the 67-bp repeat was also polymorphic in most populations. However, some appeared fixed for one copy (sites 1, 6, 7, 11, 12, 16, and 17; Fig. 1) or two copies (sites 19–26), except for clone NT4 C1 (one copy) and QLD1 C3 (three copies). Five repeat types were present among a total of 251 copies sequenced, and only 5 contained a singleton. Three variable sites occur in the last 14 bp (YAAACATTAGGAMC), to form five repeat types: T/AC, C/AC, T/CC, C/CC, and T/A–. These were widely dispersed, except for T/A–, which was restricted to a region including sites 16–18. Some populations appeared fixed for one type: Queensland (all C/AC), Northern Territory and Vanuatu (all T/AC), and Manus (all T/CC). Three-quarters of the tandemly paired repeats were identical pairs, either C/AC + C/AC or T/AC + T/AC. The nonidentical tandem pair, C/AC + T/CC, was restricted to sites 4 and 5, while the remaining tandem pairs (T/AC + C/AC and C/AC + T/AC) were widely dispersed.
ITS1 Alignments and Sequence Variation
Three alignments were produced from the sequence data. An alignment of the whole ITS1 had 105 unique sequences and 1228 sites, excluding the 72 An. farauti clones without a 5′-subrepeat (supplementary data S1). The three clones with a 5′-subrepeat and two core repeats were also excluded given the potential effects of recombination for phylogenetic analysis and the problem of positional homology. The ITS1 alignment contained an additional 50 sites, with gaps coded as present = 1 or absent = 0. An alignment of the 132 clones with a 5′-subrepeat had 39 unique sequences and 369 sites (Supplementary Data S2). Identical 5′-subrepeats accounted for 84 of these, and 1- to 2-bp indels formed another 8 duplicates. These sequences consisted of large groups from either single or adjacent populations. One exception included WestPrv A2, with 14 other identical sequences from Bougainville and Buka Island, while another group was a mix from the Northern Territory and Queensland. An alignment of the core region from all clones had 163 unique sequences and 863 sites (Supplementary Data S3). Fewer identical (30) and duplicate (10) sequences were present, relative to length, yet most of these were also isolated from the same or adjacent population.
Copy number variation made determining the positional homology of repeats problematic, so the following were edited from the ITS1 alignment: the 3′-copy of two 5′-subrepeats present in two clones each from An. irenicus and An. farauti, the 5′-copy of the core repeats, and the 3′-copy of the 67-bp repeat from An. irenicus and An. farauti. Two tandem copies of the 21-bp and terminal length variation at the 5′-end of the spacer, from a 6-bp overlap in An. farauti 6, were also removed. Finally, all length variation was removed from a GTn dinucleotide repeat that was highly variable within individuals and populations.
Visual inspection of the ITS1 alignment showed that the PCR-RFLP genotypes described by Beebe et al. (2000a) are caused by eight conserved and two polymorphic MspI restriction sites. The RFLPs reflect the presence or absence of an internal repeat, restriction sites within repeats, the two polymorphic sites, and the geographic distribution of this variation. The numbers of polymorphic sites overall (sites with gaps excluded) for the 5′-subrepeat and core were 51 and 120, respectively. Seven singletons occur in the 5′-subrepeat and 30 in the core. No singleton gapped sites occur in the former, though three singleton gaps cover 11 sites in the latter: all deletions of short direct repeats in An. farauti. In only three cases were variant nucleotides restricted to a single individual, all located in the core. The average ML distance was 0.0141 for the 116 An. farauti ITS1 sequences containing the 5′-subrepeat and 0.0049, 0.0036, and 0.0037 for the single population samples of An farauti 6, 7, and 8, respectively. Most notably, the average ML distance for all An. farauti 5′-subrepeat sequences was twice that of the core sequences: 0.0229 versus 0.0115.
Comparisons of nucleotide polymorphisms among populations had to be restricted to the core region. Nine sequences from each of 19 populations were compared, including the 1996 samples from Rabaul (Table 2). The isolated Pacific island populations had a lower average ML distance, though this is not a general feature of all island samples. The single interspecific comparison possible here indicates that the core region from both the An. irenicus and the An. farauti populations on the island of Guadalcanal are equally divergent, at 0.0038. Of a total number of 96 segregating sites, only 8 contained more than two different nucleotides. A majority of the length variation in the core region was caused by copy number variation of the 21-bp repeat, GTn dinucleotide repeat, and 67-bp repeat.
Molecular Evolution
The 5′-subrepeat and core partitions of the ITS1 alignment contained 84 and 157 unique site patterns, respectively, while under a single model there were 212. Likelihood mapping indicated that tree-like phylogenies are produced using the whole ITS1, but with 5.9% of quartets unresolved, some internal polytomies would be present. By comparison, a high proportion of star-like trees was generated from the 5′-subrepeat (19.4%) and core alignments (13.6%). Bayesian analysis was therefore conducted using the whole ITS1 alignment only. An analysis of the core alignment was attempted, but these trees were poorly resolved. No evidence for recombination was found after analyzing the ITS1 alignment with the RDP package of recombination detection methods. Homogeneity of base composition among all the aligned sequences was also confirmed by the chi-square test (p < 0.05).
A large difference was observed for the Bayesian estimates of the base frequencies between partitions. The 5′-subrepeat (G > C > T > A) has a 56% decrease in the A content and a 31% increase in the G content compared to the core (A > G > C > T) (Table 3). No overlap was observed between the density distributions of the posterior probabilities for A + G base frequencies when this parameter was unlinked. The effect of this difference also resulted in uncertainty over the inclusion of the base frequency parameter for the whole ITS1 and core among models with an AIC weight >0. Uncertainty about the number of substitutional classes also led to the differences observed between the AIC and the BIC models. There was less uncertainty over the inclusion of I and Γ in these models, and the Bayesian estimates for both p-inv (0.69) and α (0.18) were the same regardless of model type or partitioning. The relative substitution rates under the GTR model do not differ substantially between partitions from estimates taken from the whole ITS1, despite base frequency differences (Table 3). The variance of these rate estimates is also reduced in the combined data.
Phylogenetic Analysis
The overall mean likelihood score from the four single-model Bayesian analyses (−4662.223) was higher than the mixed HKY + Γ + I and GTR + Γ + I model (−4671.903). However, when base frequencies were treated as a separate parameter between partitions this increased to −659.239, and under the GTR + Γ + I and GTR + Γ + I mixed models it increased further (range, −4645.708 to −4646.508). Despite the improvement in likelihood scores with the more parameter-rich models, consistency of the consensus topologies across multiple MCMC runs was only observed under the single model. These trees were more resolved than under any mixed model and clade support values were less variable. Therefore the simplest model was chosen for estimating the phylogeny of the ITS1. Inclusion of the gaps as an additional data partition during analyses resulted in less resolved trees and was therefore discarded.
Six clades are present in the ITS1 phylogeny with a posterior probability >70%; Queensland (QLD), Northern Territory (NT), southwestern PNG (swPNG), Pacific, northern PNG (nPNG), and Manus Island (Fig. 3). The two Australian clades form a single well-supported group with a sister relationship to swPNG, while Manus Island appears to contain the most ancestral ITS1 types. The tree with the highest likelihood among the four single-model analyses (i.e., the MAP tree; Fig. 4) has the same topology as the MJ-consensus tree. The rate of substitution per site shown by the branch lengths also shows how unique the Manus Island samples are relative to the other An. farauti. The divergence between An. farauti and An. farauti 8 is also apparent, particularly to the Centrl1 A3 clone sampled from the same site. The inferred phylogeny of the ITS1 is therefore congruent with the population origin of most, though not all, samples. For example, two clones with an identical 5′-subrepeat (NT4 A2 and QLD1 A2) form a basal polytomy in the Australian clade. A clone from Torres Strait Island (Torres B2) is also consistently grouped within the QLD clade. The most notable exception is the presence of the WestPrv A2 clone among the Pacific clade.

Bayesian 70% majority-rule consensus tree of the ITS1 from four combined analyses under a single GTR + Γ + I model. Bayesian support values are shown for internal branches. Clades within An. farauti are denoted QLD, NT, swPNG, Pacific, nPNG, and Manus. Abbreviated taxon names and collection sites are listed in Table 1

The maximum posterior probability (MAP) tree of the ITS1 from four Bayesian analyses under a single GTR + Γ + I model. Branch lengths are proportional to the rate of substitution per site (see scale bar). Collection sites and taxon labels are as in Fig. 3
Discussion
Our data clearly indicate concerted evolution of the ITS1 at the intraspecific level in An. farauti. This spacer is only partially homogenized within individuals due to variation in the copy number of both short and long tandem repeats, numerous point mutations, and other indels. Individual copies of the ITS1 were isolated from within and among individual mosquitoes, sampled from populations spanning a greater part of the species distribution. Analysis of the DNA sequence data confirmed the regional differentiation of the spacer seen previously using PCR-RFLPs (Beebe et al. 2000a). Variation in the copy number of the internal repeats, in addition to a pattern of restricted geographic distribution for these and point mutations, indicates a high rate of production and turnover of new variation. ML distance comparisons of the core region show that the degree of homogenization among populations is also variable. Bayesian phylogenetic analysis indicated that homogenization of the variant ITS1 repeats has occurred fast enough to reduce this variation within regional populations. In the main, the phylogenetic estimate of the ITS1 sequence data and the geographic origin of the individuals these were isolated from appear to match. However, there were exceptions to this congruence that indicate the spread of variant repeats between populations and the retention of ancestral polymorphism in some individuals.
Internal Repeats
A majority of the differences between otherwise identical spacers was due to variation in the copy number of short repeats, varying from 1 to 3 bp. These minor changes were found in clones sampled from either the same individual or the same population, though sometimes from an adjacent population. This localized distribution indicates a high rate of duplication and loss of these repeats. More significant differences are caused by longer repeats, which generate a variety of spacer length variants within individuals and among geographic isolates (Fig. 2). Large ITS1 internal repeats have been observed in other invertebrates species, such as aphids (Fenton et al. 1998), beetles (von der Schulenburg et al. 2001), and parasitic worms (van Herwerden et al. 1999). Internal repetition and intraindividual variation of this spacer have been described in studies of other Anopheles species (Paskewitz et al. 1993; Fairley et al. 2005).
An interesting feature of the larger repeats in An. farauti is the geographic variation in copy number and the formation of repeat types. Copy number differences of an ITS1 repeat have been observed between two populations of the parasitic worm, Schistosoma mattheei, yet without intraindividual variation (Kane et al. 1996). It is difficult to assess how common this phenomenon may be, and it is also hindered by the requirement for greater population sampling than is commonly employed. We demonstrated that the relative abundance of spacers with or without a 5′-subrepeat varies geographically (Fig. 2). The long core repeat had a more restricted distribution and was only isolated from a cluster of populations around the northern and southern coastline of PNG. Unlike the other internal repeats, it is not shared among other species in the complex and so is likely to have originated recently in this area. Most variation among the 21- and 67-bp repeat arrays consisted of repeat-type rearrangements with some duplication or loss of repeats, though regional variation was only observed in the latter.
Sequence Variation
Intraindividual variation in the copy number of the 5′-subrepeat indicates a degree of recombinant activity that is independent of the adjacent core region. By examining the observed differences in the amount of sequence variation present, we were able to infer a difference in the relative turnover rate for these two regions. Strikingly, there are many more identical 5′-subrepeat sequences, even after accounting for length differences. Most variant repeats form large groups of identical sequences, whereas identical core sequences were rare. For both regions, the geographic location of identical or duplicate sequences was usually in the same or an adjacent population. Despite the relatively similar number of polymorphic sites, the core has three times the number of singletons, even after the exclusion of gapped sites, and has half the average ML distance. Thus the intraspecific data for An. farauti suggest a higher rate of turnover in the genome for the 5′-subrepeat. This would explain both the greater level of differentiation and its high level of homogenization within local populations.
Genetic distance estimates varied considerably among the An. farauti populations. Nucleotide diversity was at its lowest in the most isolated population of Vanuatu and increased fourfold from the neighboring island of Bougainville to the Sepik region of northern PNG. Comparisons to the ITS1 of other mosquito species (e.g., Miller et al. 1996; Fairley et al. 2005) are not very meaningful, especially with the added differences between sampling strategies. However, we were able to compare An. irenicus with the An. farauti also found on Guadalcanal. The larvae of both species are able to tolerate saltwater, though An. farauti prefers the coastal habitat (Foley and Bryan 2000; Beebe and Cooper 2002), and both are considered sister species (Beebe and Cooper 2002). Interestingly, the average ML distance of the core region was identical and so the low level of variation, compared to other An. farauti populations, may be reflective of some shared demographic and historical factors. These islands are at the edge of the species’ distribution, where they are likely to have experienced founder events and limits to their population size.
Other reasons, beside differences in population dynamics, have been suggested to explain observed differences in the level of homogenization among populations. Population differences in the ITS2 of the South American An. nuneztovari were attributed to the presence of as yet unidentified cryptic species (Fritz et al. 1994). However, observed differences in nucleotide diversity among populations are likely to be poor indicators of species-level relationships. Extensive work has been carried out on identification of cryptic species in the An. punctulatus group, which have been characterized using rDNA (ITS2 and 18S) and mtDNA data (Beebe and Saul 1995; Foley et al. 1998; Beebe et al. 2000b). Ecological, behavioral, and habitat characteristics also indicate that An. farauti populations are conspecific (Beebe et al. 2000a; Beebe and Cooper 2002).
Molecular Evolution
Spacer heterogeneity can be generated through the presence of pseudogenic copies that have escaped homogenization (Li and Wilkerson 2007). This is unlikely here because the 5.8S rRNA gene and a partial 18S sequence were homogenized despite variation in the spacer. Results of the chi-square homogeneity test also showed that the ITS1 sequences have a similar base composition. Variation in the nucleotide content was primarily due to the presence or absence of the larger repeats. Obviously recombination could have caused some variation, though this was not detected in the ITS1 alignment. Likelihood mapping was used to visualize the phylogenetic resolution within and between the two data partitions, given a possible lack of sensitivity in the recombination detection methods. This indicated some incongruence between the partitions, but combining them led to more resolved quartets. Other tests of incongruence, such as the partition homogeneity test (Swofford 2002), could not be used given the large number of sequences and short length of the 5′-subrepeat. Sequence length would also have contributed substantially to the greater resolution of the whole ITS1 alignment, as it is known to be an important factor (Strimmer and von Haeseler 1997).
Base frequency estimates were significantly different between the 5′-subrepeat and the core. This is likely to be reflective of their separate origins given that the BLAST results indicated that the 5′-subrepeat was originally part of a G + C-rich region in the ITS2. Bayesian estimates of the rate variation and proportion of invariants were the same between partitions, which indicates the involvement of similar mutational processes across the spacer. Simulation studies have shown that rate variation has a significant influence on analyses of within-species data, as in the case of D-loop mitochondrial DNA variation in humans (Aris-Brosou and Excoffier 1996). In addition, its estimation is known to be highly sensitive to the number of taxa used (Sullivan et al. 1999). Despite the high level of rate variation (α = 0.18), we were confident in this estimate given its stability among models and our increased taxon sampling. The use of complex evolutionary models can be associated with increased uncertainty for the topology of phylogenetic trees than simpler models (Nylander et al. 2004). As such, checking for topological consistency among analyses is an important step in the evaluation of each model (Castoe et al. 2004). In this case, despite some large differences in base frequencies and the increase in likelihood scores after partitioning of the data, only the simpler model resulted in the same consensus topologies among multiple MCMC runs.
Phylogenetic Analysis
The divergence of repetitive sequences has been hypothesized to be a general feature of genetically isolated populations and associated with the process of allopatric divergence (Elder and Turner 1995). Divergence of the ITS1 in An. farauti had been previously demonstrated and several climatic and geographic barriers exist throughout its distribution that are likely to restrict gene flow between populations (Beebe et al. 2000a; Beebe and Cooper 2002). The inferred ITS1 phylogeny indicates that the homogenization of the spacer has occurred fast enough to reduce variation within regional populations. Six phylogeographic clades are present which received >70% Bayesian support (Fig. 3). Although this level of phylogenetic resolution has not been seen in other ITS1 intraspecific studies to date, it may be due to the increased length and greater number of informative characters. For example, only 16 variable sites were observed in the 582-bp-long ITS1 of brown trout (Presa et al. 2002).
Several consistent relationships are present in the consensus tree, including the ancestral status of Manus Island and an Australian and swPNG clade (Fig. 3). The >90% support for an Australian and swPNG clade is likely to reflect relatively recent allopatric divergence after the flooding of the Gulf of Carpentaria and Torres Strait, only 6000–8000 years ago (Beebe and Cooper 2002). However, the uncertainty represented by the polytomy of the Pacific clade may be due to a lower average ML distance in these samples. We considered a priori that the An. farauti 8 sample might represent an additional taxon within the complex because of differences among the internal repeats and a 5.8S substitution. All analyses here supported a sister-relationship between this taxon and An. farauti. More specimens have since been identified and can be differentiated by PCR-RFLP analysis of the ITS1. These appear more common farther inland from the coast and form an interesting parallel to the sister taxa An. irenicus and An. farauti on Guadalcanal Island. Both species are saltwater tolerant, though An. irenicus occurs on the coast and inland (Beebe et al. 2000b). Ecological niche modeling of An. farauti suggests that the higher atmospheric moisture near seawater and elevation are the most significant factors influencing its distribution (Sweeney et al. 2006), which does not appear to be the case for An. irenicus. This biological difference and ITS1 divergence provide more supporting evidence for An. farauti 8.
The effect of sequence polymorphisms in ancestral populations on topological error is considered to be substantial over short periods of evolutionary time (Pamilo and Nei 1988). This is an important factor which determines whether a gene tree will be congruent with a population tree. Ancestral polymorphisms have been reported in other ITS1 studies (e.g., Rich et al. 1997; Presa et al. 2002), and the ITS1 sampled from a Western Province individual (WestPrv A2) appears to be another example. Its 5′-subrepeat was identical to many in the Pacific clade and its core differed by just a single transition. These adjacent spacer regions have therefore remained together since the swPNG and Pacific clades split, perhaps because it lies at the end of the rDNA loci of this individual. Like pseudogenic copies which translocate to other loci, repeats can also ‘escape’ from homogenization if located at the end of a tandem array, since recombination is less likely to occur there (McAllister and Werren 1999). The presence of the Torres B2 clone within the Australian clade indicates either some recent contact, given its proximity, another example of an ancestral variant, or possibly a recombinant. The presence of a shared identical 5′-subrepeat in the Northern Territory and Queensland may be due to some historical gene flow and subsequent recombination with different core regions. This did cause some incongruence, as indicated by the two clones (QLD1 A2 and NT4 A2) which form a basal polytomy in the Australian clade (Fig. 3).
The ITS1 of the An. farauti is unique given the level of intraindividual variation and internal repetition. It also contains the only known case, to our knowledge, of an insertion that appears to have derived from a duplication of an ITS2 sequence. Internal repetition of the ITS1 and insertion sequences in the 28S rRNA gene is common in arthropods. Indeed rDNA may be a suitable ‘habitat’ for the persistence of these repeat elements (Penton and Crease 2004). From our sample of the extant variation it appears that the 5′-subrepeat is more homogenized than the remainder of the spacer. This could be explained by the higher rate of turnover within the rDNA locus for this repeat relative to the core region. Indeed, the intraspecific concerted evolution of the ITS1 as a whole is likely to be a function of a high rate of turnover within and between the rDNA loci in these mosquitoes.
The degree of phylogenetic resolution demonstrates that the ITS1 is able to evolve in concert at an intraspecific level despite the presence of intraindividual variation. The fact that this species has a continuous coastal distribution yet discontinuities are present in the spacer makes this an even more interesting organism to study. In particular, variation in the ITS1 reflects recent patterns of population structure in this species. The high rate of DNA turnover experienced by satellite DNA (e.g., Pons and Gillespie 2004) and a recent example from a mating-specific gene family (Cvitanich et al. 2006) has proved to be a useful characteristic for improving phylogenetic resolution. The combined effect of a high rate of turnover and rapid divergence means that repetitive sequences are more likely to reflect recent evolutionary patterns of population structure and should prove useful tools despite the increased sampling required.
References
Aris-Brosou S, Excoffier L (1996) The impact of population expansion and mutation rate heterogeneity on DNA sequence polymorphism. Mol Biol Evol 13:494–504
Averbeck KT, Eickbush TH (2005) Monitoring the mode and tempo of concerted evolution in the Drosophila melanogaster rDNA locus. Genetics 171:1837–1846
Beebe NW, Cooper RD (2002) Distribution and evolution of the Anopheles punctulatus group (Diptera: Culicidae) in Australia and Papua New Guinea. Int J Parasitol 32:563–574
Beebe NW, Saul A (1995) Discrimination of all members of the Anopheles punctulatus complex by polymerase chain reaction-restriction fragment length polymorphism analysis. Am J Trop Med Hyg 53:478–481
Beebe NW, Cooper RD, Foley DH, Ellis JT (2000a) Populations of the south-west Pacific malaria vector Anopheles farauti s.s. revealed by ribosomal DNA transcribed spacer polymorphisms. Heredity 84:244–253
Beebe NW, Cooper RD, Morrison DA, Ellis JT (2000b) A phylogenetic study of the Anopheles punctulatus group of malaria vectors comparing rDNA sequence alignments derived from the mitochondrial and nuclear small ribosomal subunits. Mol Phylogenet Evol 17:430–436
Brosius J (2003) The contribution of RNAs and retroposition to evolutionary novelties. Genetica 118:99–116
Castoe TA, Doan TM, Parkinson CL (2004) Data partitions and complex models in Bayesian analysis: the phylogeny of gymnophthalmid lizards. Syst Biol 53:448–469
Cvitanich C, Salcido M, Judelson HS (2006) Concerted evolution of a tandemly arrayed family of mating-specific genes in Phytophthora analyzed through inter- and intraspecific comparisons. Mol Gen Genomics 275:169–184
Dover GA (2000) How genomic and developmental dynamics affect evolutionary processes. BioEssays 22:1153–1159
Dover GA (2002) Molecular drive. Trends Genet 18:587–589
Elder JFJ, Turner BJ (1994) Concerted evolution at the population level: pupfish HindIII satellite DNA sequences. Proc Natl Acad Sci USA 91:994–998
Elder JFJ, Turner BJ (1995) Concerted evolution of repetitive DNA sequences in eukaryotes. Q Rev Biol 70:297–320
Fairley TL, Kilpatrick CW, Conn JE (2005) Intragenomic heterogeneity of internal transcribed spacer rDNA in neotropical malaria vector Anopheles aquasalis (Diptera: Culicidae). J Med Entomol 42:795–800
Famà P, Olsen JL, Stam WT, Procaccini G (2000) High levels of intra- and inter-individual polymorphism in the rDNA ITS1 of Caulerpa racemosa (Chlorophyta). Eur J Phycol 35:349–356
Fenton B, Malloch G, Germa F (1998) A study of variation in rDNA ITS regions shows that two haplotypes coexist within a single aphid genome. Genome 41:337–345
Foley DH, Bryan JH (2000) Shared salinity tolerance invalidates a test for the malarial vector Anopheles farauti s.s. on Guadalcanal. Med Vet Entomol 14:102–104
Foley DH, Bryan JH, Yeates D, Saul A (1998) Evolution and systematics of Anopheles: insights from a molecular phylogeny of Australian mosquitoes. Mol Phylogenet Evol 9:262–275
Fritz GN, Conn J, Cockburn A, Seawright J (1994) Sequence analysis of the ribosomal DNA internal transcribed spacer 2 from populations of Anopheles nuneztovari (Diptera: Culicidae). Mol Biol Evol 11:406–416
Gandolfi A, Bonilauri P, Rossi V, Menozzi P (2001) Intraindividual and intraspecies variability of ITS1 sequences in the ancient asexual Darwinula stevensoni (Crustacea: Ostracoda). Heredity 87:449–455
Gonzalez IL, Sylvester JE (2001) Human rDNA: evolutionary patterns within the genes and tandem arrays derived from multiple chromosomes. Genomics 73:255–263
Graur D, Li W-H (2000) Fundamentals of molecular evolution. Sinauer Associates, Sunderland, MA
Harris DJ, Crandall KA (2000) Intragenomic variation within ITS1 and ITS2 of freshwater crayfishes (Decapoda: Cambaridae): implications for phylogenetic and microsatellite studies. Mol Biol Evol 17:284–291
Kane RA, Ridgers IL, Johnston DA, Rollinson D (1996) Repetitive sequences within the first internal transcribed spacer of ribosomal DNA in schistosomes contain a chi-like site. Mol Biochem Parasitol 75:265–269
Li C, Wilkerson RC (2007) Intragenomic rDNA ITS2 variation in the neotropical Anopheles (Nyssorhynchus) albitarsis complex (Diptera: Culicidae). J Hered 98:51–59
Liao D (1999) Concerted evolution: molecular mechanism and biological implications. Am J Hum Genet 64:24–30
Luchetti A, Cesari M, Carrara G, Cavicchi S, Passamonti M, Scali V, Mantovani B (2003) Unisexuality and molecular drive: Bag320 sequence diversity in Bacillus taxa (Insecta: Phasmatodea). J Mol Evol 56:587–596
Martin D, Rybicki E (2000) RDP: detection of recombination amongst aligned sequences. Bioinformatics 16:562–563
McAllister BF, Werren JH (1999) Evolution of tandemly repeated sequences: what happens at the end of an array. J Mol Evol 48:469–481
Miller BR, Crabtree MB, Savage HM (1996) Phylogeny of fourteen Culex mosquito species, including the Culex pipiens complex, inferred from the internal transcribed spacers of ribosomal DNA. Insect Mol Biol 5:93–107
Minin V, Abdo Z, Joyce P, Sullivan J (2003) Performance-based selection of likelihood models for phylogenetic estimation. Syst Biol 52:674–683
Nylander JAA, Ronquist F, Huelsenbeck JP, Nieves-Aldrey JL (2004) Bayesian phylogenetic analysis of combined data. Syst Biol 53:47–67
Onyabe DY, Conn JE (1999) Intragenomic heterogeneity of a ribosomal DNA spacer (ITS2) varies regionally in the neotropical malarial vector Anopheles nuneztovari (Diptera: Culicidae). Insect Mol Biol 8:435–442
Pamilo P, Nei M (1988) Relationships between gene trees and species trees. Mol Biol Evol 5:568–583
Paskewitz SM, Wesson DM, Collins FH (1993) The internal transcribed spacers of ribosomal DNA in five members of the Anopheles gambiae species complex. Insect Mol Biol 2:247–257
Penton EH, Crease TJ (2004) Evolution of the transposable element Pokey in the ribosomal DNA of species in the subgenus Daphnia (Crustacea: Cladocera). Mol Biol Evol 21:1727–1739
Pons J, Gillespie RG (2004) Evolution of satellite DNAs in a radiation of endemic Hawaiian spiders: does concerted evolution of highly repeated sequences reflect evolutionary history? J Mol Evol 59:632–641
Posada D, Crandall KA (1998) Modeltest: testing the model of DNA substitution. Bioinformatics 14:817–818
Posada D, Buckley TR (2004) Model selection and model averaging in phylogenetics: advantages of Akaike information criterion and Bayesian approaches over likelihood ratio tests. Syst Biol 53:793–808
Presa P, Pardo BG, Martínez P, Bernatchez L (2002) Phylogeographic congruence between mtDNA and rDNA ITS markers in brown trout. Mol Biol Evol 19:2161–2175
Rambaut A, Drummond AJ (2004) Tracer v1.3. Available at: http://evolve.zoo.ox.ac.uk/
Rannala B, Yang Z (1996) Probability distribution of molecular evolutionary trees: a new method of phylogenetic inference. J Mol Evol 43:304–311
Reineke A, Karlovsky P, Zebitz CPW (1998) Preparation and purification of DNA from insects for AFLP analysis. Insect Mol Biol 7:95–99
Rich SM, Rosenthal BM, Telford SR, Spielman A, Hartl DL, Ayala FJ (1997) Heterogeneity of the internal transcribed spacer (ITS-2) region within individual deer ticks. Insect Mol Biol 6:123–129
Ronquist F, Huelsenbeck JP (2003) MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19:1572–1574
Sambrook J, Russell DW (2001) Molecular cloning: a laboratory manual, 3rd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor NY
Schindelhauer D, Schwarz T (2002) Evidence for a fast, intrachromosomal conversion mechanism from mapping of nucleotide variants within a homogenous α-satellite DNA array. Genome Res 12:1815–1826
Schizas NV, Street GT, Coull BC, Chandler GT, Quattro JM (1999) Molecular population structure of the marine benthic copepod Microarthridion littorale along the southeastern Gulf coasts of the USA. Mar Biol 135:399–405
Schmidt HA, Strimmer K, Vingron M, von Haeseler A (2002) TREE-PUZZLE: maximum likelihood phylogenetic analysis using quartets and parallel computing. Bioinformatics 18:502–504
Schön I, Butlin RK, Griffiths HI, Martens K (1998) Slow molecular evolution in an ancient asexual ostracod. Proc R Soc Lond B 265:235–242
Strimmer K, von Haeseler A (1997) Likelihood-mapping: a simple method to visualize phylogenetic content of a sequence alignment. Proc Natl Acad Sci USA 94:6815–6819
Sullivan J, Swofford DL, Naylor GJP (1999) The effect of taxon sampling on estimating rate heterogeneity parameters of maximum likelihood models. Mol Biol Evol 16:1347–1356
Sweeney AW, Beebe NW, Cooper RD, Bauer JT, Peterson AT (2006) Environmental factors associated with distribution and range limits of malaria vector Anopheles farauti in Australia. J Med Entomol 43:1068–1075
Swofford DL (2002) PAUP*. Phylogenetic analysis using parsimony (*and other methods). Version 4. Sinauer Associates, Sunderland, MA
van Herwerden L, Blair D, Agatsuma T (1999) Intra- and interindividual variation in the ITS1 of Paragonimus westermani (Trematoda: Digenea) and related species: implications for phylogenetic studies. Mol Phylogenet Evol 12:67–73
von der Schulenburg JHG, Hancock JM, Pagnamenta A, Sloggett JJ, Majerus MEN, Hurst DD (2001) Extreme length and length variation in the first ribosomal internal transcribed spacer of ladybird beetles (Coleoptera: Coccinellidae). Mol Biol Evol 18:648–660
Wendel JF, Schnabel A, Seelanan T (1995) Bidirectional interlocus concerted evolution following allopolyploid speciation in cotton (Gossypium). Proc Natl Acad Sci USA 92:280–284
Williams SM (1990) The opportunity for natural selection on multigene families. Genetics 124:439–441
Acknowledgments
The authors would like to thank David Ayre for his useful suggestions. This research was supported by a graduate student grant from the University of Wollongong to J.E.B., and the National Health and Medical Research Council of Australia (NHMRC) 117102 to N.W.B., and was published with the approval of the Director General of Defence Health Services.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
These files are unfortunately not in the Publisher's archive anymore:
Supplementary Data S1. Alignment of the ITS1 (NEX 136 kb)
Supplementary Data S2. Alignment of the 5′-subrepeat (NEX 51 kb)
Supplementary Data S3. Alignment of the core region (NEX 178 kb)
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Bower, J.E., Dowton, M., Cooper, R.D. et al. Intraspecific Concerted Evolution of the rDNA ITS1 in Anopheles farauti Sensu Stricto (Diptera: Culicidae) Reveals Recent Patterns of Population Structure. J Mol Evol 67, 397–411 (2008). https://doi.org/10.1007/s00239-008-9161-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-008-9161-x