
Abstract
Circulating cell-free DNA (ccfDNA) has great potential for non-invasive diagnostics, and prediction and monitoring of treatment response, but its amount is usually limited. Therefore, the choice of methods to extract and characterize ccfDNA is crucial. In the current study, we performed the most comprehensive comparison of methods for ccfDNA extraction (11 methods), quantification (3 methods), and estimation of the integrity index (2 methods) from small quantities of different kinds of plasma. The QIAamp® Circulating Nucleic Acid Kit and the Norgen Plasma/Serum Circulating DNA Purification Mini Kit showed the best accuracy and reproducibility, but the Norgen kit allowed to extract a higher amount of ccfDNA. This workflow provides a reliable protocol for the multiple applications of ccfDNA in biomedicine.

Workflow for the evaluation of methods to isolate, quantify and characterize circulating cell-free DNA from small volumes of plasma
Similar content being viewed by others


Introduction
Circulating cell-free DNA (ccfDNA) from plasma or serum bears great promise as a sensitive biomarker for diagnosis, prognosis, prediction, and monitoring of the response to therapeutic regimen for cancer patients [1, 2] and potentially other complex diseases [3]. The availability of a sufficient amount of amplifiable DNA is primordial for its use as biomarker [4]. The quantity of ccfDNA in plasma or serum is, however, very low and depends among other parameters on the type and stage of cancer as well as the age and sex of patient [5–7]. The concentration of isolated ccfDNA also depends on the efficiency of the employed extraction method [4, 8]. In addition, methods to accurately determine the quantity and quality of the ccfDNA are crucial to reliably compare and normalize the ccfDNA for its use as biomarker [4, 9].
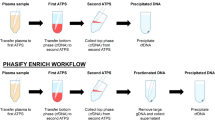
In the present work, we developed a complete workflow for the extraction, quantification and estimation of the integrity of ccfDNA from small volumes of plasma. Eleven extraction methods were compared using commercial plasma samples from healthy male and female individuals as well as from lung, breast, pancreatic, and colorectal cancer patients. The isolated ccfDNA was quantified using two newly developed quantitative real-time PCR (qPCR) assays targeting repetitive genomic elements (Kpn, DHFRP2) as well as a fluorometric assay. Finally, the integrity index was calculated using either qPCR assays to amplify the APP gene or gel electrophoresis to amplify products of the TP53 gene [10–12].
Methods
Samples
Eleven methods to extract ccfDNA were compared using ten commercial human plasma samples (Seralab, Haywards Heath, UK): stage IV colon cancer (male, 61 years, and female, 81 years), stage IV pancreatic cancer (male, 79 years, and female, 48 years), stage IV lung cancer (male, 69 years, and female, 75 years), stage IV breast cancer (two females, 64 years), and healthy human plasma (male, 24 years, and female, 18 years). All plasma samples contained K2-EDTA. Plasma samples were processed using an identical procedure and stored immediately at −80 °C until their extraction.
Extraction
The QIAamp® Circulating Nucleic Acid Kit (Qiagen, Courtaboeuf, France), the NucleoSpin® Plasma XS (Macherey-Nagel, Hoerdt, France), the Chemagic NA Extraction Kit (Perkin-Elmer, Courtaboeuf, France), and the Norgen Plasma/Serum Circulating DNA Purification Mini Kit and the Norgen Plasma/Serum Cell-Free Circulating DNA Purification Mini Kit (Norgen, Thorold, Canada) were used. Experiments were performed using the minimally required quantity of plasma according to the manufacturer’s protocol, with the exception of the Chemagic NA Extraction Kit for which the input of plasma was required to be decreased by tenfold. CcfDNA was extracted from 1000 or 500 μL of plasma for the QIAamp® kit, 500 μL for Chemagic, 240 μL of plasma for NucleoSpin®, and 200 μL of plasma for the two Norgen kits. In addition, we evaluated a number of published protocols: several phenol-chloroform (PC) protocols according to Schmidt et al. [13], Yuan et al. [14], and Hufnagl et al. [15] and a Triton/Heat/Phenol (THP) protocol according to Xue et al. [16]. Experiments were performed starting from 500 μL of plasma following the published protocols, except that for the PC methods, 500 μL of plasma was used instead of 1 mL. Quantities are reported as nanograms per milliliter of plasma to account for the different extraction volumes.
Quantification
CcfDNA was quantified using the Qubit® dsDNA HS Assay (Life Technologies, Illkirch, France) and two newly developed quantitative PCR assays targeting a repetitive sequence present in multiple copies throughout the genome (Kpn) and a microsatellite sequence (DHFRP2). qPCRs were performed on a LightCycler 480 thermocycler (Roche Applied Science, Penzberg, Germany). Primers were purchased from BioTeZ (Berlin, Germany) and modified primers from TIB MOLBIOL (Berlin, Germany).
For Kpn, a 55-bp product was amplified with 5′-TGCCGCAATAAACATAC-3′ as forward primer and 5′-GGACTATAAATCATGCTGC-3′ as reverse primer. For the 165-bp PCR product of DHFRP2, the forward primer was 5′-ATTGTAAGACTTTTGGAGCCATTT-3′ and the reverse primer was 5′-TTCAGGGAGAATGAGATGGGC-3′. For the optimization of the assays and to mimic ccfDNA, 3 μg of human genomic DNA (Promega, Charbonnières-les-Bains, France) in a volume of 60 μL was fragmented using a Covaris E210 (Brighton, UK) with 10 % of duty cycle intensity 3, burst 200 for three times for 3 s. Concentration and fragment size distribution of this fragmented DNA were analyzed on a 2100 Bioanalyzer using a DNA 1000 chip (Agilent Technologies, Les Ulis, France). Standard curves were made using serial dilution of this fragmented DNA between 0.01 and 3 ng/μL (from 0.01 to 0.1 ng/μL in steps of 0.01 ng/μL and from 0.1 to 3 ng/μL in steps of 0.1 ng/μL).
The PCR mix contained 5 μL of 2× Roche SYBR qPCR Master Mix (Roche Applied Science, Penzberg, Germany), 0.2 μL of 10 μM of each primer, 3.8 μL of water, and 1 μL of DNA in a final volume of 10 μL. PCR was performed at 95 °C for 10 min, followed by 50 cycles of 95 °C for 10 s, 60 °C for 45 s, and 72 °C for 15 s. Each sample was analyzed in triplicate, and each standard curve was generated in the same 384-well plate. The standard curve was automatically generated using the LightCycler® software version 1.5.162 (Roche Applied Science, Penzberg, Germany). The software calculated for each sample the concentration and the threshold cycle (CP). For each triplicate, the average concentration, the quantity of isolated ccfDNA per milliliter of plasma, the average \( {2}^{-{\mathrm{C}}_{\mathrm{P}}} \), and its standard deviation and the coefficient of variation (CV) were determined. A CV <15 % was required for the validation of the triplicate.
Integrity index
The size distribution of ccfDNA was evaluated using two previously reported methods: two qPCR assays used to amplify the APP gene (67 and 180 bp) [10, 11] and two semi-quantitative PCR amplifications (400 and 800 bp) of the TP53 gene, which were subsequently analyzed by gel electrophoresis [12]. An integrity index of ccfDNA was calculated using CP180/CP67 (CP = cycle threshold of the qPCR assay) for APP and I 800/I 400 (I = intensity of the gel band) for TP53. The DNA fragmented as described above was used as a positive control. Reaction conditions were modified from published protocols. All reactions were performed in triplicate. Conditions for the qPCR reaction amplifying APP were 1× HotStar Taq buffer (Qiagen, Courtaboeuf, France) supplemented with 1.6 mM MgCl2, 0.2 mM of each dNTP, 0.08 U of HotStar Taq polymerase, 0.2 mM of forward and reverse primers, 0.1 nM of probe (with 5′-FAM and BBQ-3′), and 0.250 ng of ccfDNA in a 25 μL volume [10, 11]. PCR cycling conditions included an initial denaturation step for 10 min at 95 °C, followed by 55 cycles of 15 s at 95 °C, 60 s at 60 °C, and 60 s at 72 °C, followed by a melting curve at 20 acquisition per degree from 65 to 95 °C and a final cooling at 40 °C. As for the above-described qPCRs, CP and CV values were calculated using the LightCycler software. The standard curves obtained with the fragmented DNA showed a mean squared error (MSE) 0.0247, efficiency of 1.986, slope of −3.356, and Y-intercept of 29.82 for the 67-bp fragment and a MSE of 0.0161, efficiency of 1.901, slope of −3.584, and Y-intercept of 31.5 for the 180-bp fragment. The detection limit for the 67- and 180-bp fragments was 0.025 and 0.05 ng/μL, respectively. Starting with 0.25 ng ccfDNA, the mean integrity index of fragmented DNA was 1.09 (SD 0.008).
Conditions for the amplification of the TP53 gene were as follows: 1× HotStar Taq buffer supplemented with 1.6 mM MgCl2, 0.4 mM of each dNTP, 0.08 U of HotStar Taq polymerase, 0.2 mM of forward and reverse primers, and 0.250 ng of ccfDNA in 25 μL [12]. PCR cycling conditions included an initial denaturation step for 15 min at 95 °C, followed by 50 cycles of 30 s at 95 °C, 30 s at 60 °C, and 15 s at 72 °C and a final step for 5 min at 72 °C. Ten microliters of PCR products and 10 μL of loading buffer were deposited on a 2 % agarose gel containing SYBR® Safe DNA Gel Stain (Life Technologies, Illkirch, France) and separated by electrophoresis. Band intensities were measured with the GenoTools software (Synoptics, Cambridge, UK).
Statistical analyses
Statistical significant differences in the amount of DNA obtained using the different extraction methods were assessed by two-by-two comparisons from all the plasma samples extracted with a given kit using a paired t test. Accuracy and reproducibility of the quantification results were determined by analysis of the root-mean-square error (RMSE), which aggregates the error of the individual measurements into a single measure of predictive power. The smaller the value, the higher is the accuracy of the used method.
Results and discussion
ccfDNA extraction
Eleven extraction methods were compared for the isolation of ccfDNA from small volumes of different plasma samples (Fig. 1, Electronic Supplementary Material (ESM) Table S1). Although, the quantity of the obtained ccfDNA varied in function of the disease, some methods showed increased performance, notably the NucleoSpin®, QIAamp®, and both Norgen kits, and to a lesser extent, the “home-made” protocols according to Hufnagl et al. [15] and Yuan et al. [14] (Fig. 1, ESM Tables S1 and S2). In addition, the two published protocols required significantly more time (2–3 days) compared to the commercial kits (2–3 h) but may represent valuable alternatives if cost-efficiency might be of utmost importance.

Comparison of 11 extraction methods of ccfDNA. The amount of ccfDNA obtained from stage IV colon (blue), stage IV lung (red), stage IV pancreatic (green), and plasma (purple) from healthy male was quantified using the Kpn qPCR assay. The standard deviation of triplicates from the same isolated ccfDNA sample is indicated. The Chemagic extraction protocol was modified from the manufacturer’s instructions as ten times less than the initial recommended plasma volume used, which might explain the poor performance of the kit
The Chemagic extraction protocol was modified from the manufacturer’s instructions as ten times less than the initial recommended plasma volume used, which might have led to suboptimal conditions for the extraction protocol and explain the poor performance of the kit under the modified conditions (ESM Table S1).
The QIAamp® kit has frequently been used and shown to perform well compared to the three other commercial extraction kits [4, 9]. This kit has previously been identified as the most efficient with an improved representation of small fragmented DNA [4]. It was also shown to yield the highest recovery of ccfDNA and allow an efficient recovery of a spike-in when compared to other commercial kits [9]. However, tested sample volumes were normally above 1 mL of plasma and the Norgen Kits were not included in these evaluations. The Norgen Plasma/Serum Circulating DNA Purification Mini Kit and the QIAamp® kit gave similar and reproducible results for the three methods of quantification, with a slightly better performance for the QIAamp® kit as assessed by the analysis of the RMSE (Figs. 1 and 2, ESM Tables S1 and S2). However, the Norgen kit yielded approximately twice as much ccfDNA (p = 0.006 using the yield measured by the Kpn assay, paired t test).

Comparison of the two aliquots of ccfDNA from stage IV colon, stage IV breast, stage IV lung, stage IV pancreatic cancer, and healthy female plasma isolated using (a) the Norgen Plasma/Serum Circulating DNA purification Mini Kit or (b) the QIAamp® Circulating Nucleic Acid Kit. Quantification of the amount of ccfDNA was determined by the Kpn assay (blue), the DHFRP2 qPCR assay (red), and the Qubit assay. The standard deviation of the quantification from triplicates of the same isolated ccfDNA sample is indicated
The reproducibility of the extraction methods was further evaluated using two aliquots of the same female plasma (Fig. 2, ESM Table S2) and the six most promising protocols from the initial analysis: the NucleoSpin®, QIAamp®, and both Norgen kits as well as the protocols according to Hufnagl et al. [15] and Yuan et al. [14]. Analysis of the root-mean-square error showed that the QIAamp® kit and the Norgen Plasma/Serum Circulating DNA Purification Mini Kit performed best in extracting ccfDNA from different kinds of plasma: healthy, different cancers, both sexes, and different ages of patients compared to the other kits (Fig. 2, ESM Table S2). In addition, the Norgen kit required an input of only 200 μL of plasma for successful isolation.
Quantification
We further evaluated methods for the quantification of ccfDNA using qPCR assays targeting Kpn and DHFRP2, respectively, and a fluorometric assay (Fig. 2, ESM Tables S1 and S2).
The standard curves for qPCR quantification of the repetitive elements were established for each sample plate. Typical values from the logarithmic regression of the standard curves were a MSE of 0.00811, efficiency of 1.831, slope of −3.806, and Y-intercept of 14.75 for Kpn and a MSE of 0.0161, efficiency of 1.763, slope of −4.062, and Y-intercept of 28.01 for DHFRP2. The Kpn qPCR assay showed an improved efficiency at low concentrations of ccfDNA and a lower limit of detection (0.01 ng/μL) compared to the DHFRP2 qPCR assay (0.08 ng/μL). Furthermore, the standard deviation calculated from triplicates of ccfDNA was higher for the DHFRP2 assay (ESM Tables S1 and S2).
For several of the protocols including our implementations of the THP method according to Xue et al. and the Chemagic kit (ESM Table S1), qPCR reactions were found to be inhibited and only the Qubit assay yielded interpretable results showing the presence of isolated DNA, which could preclude the use of these protocols for PCR-based downstream assays. For protocols yielding reproducibly high amounts of DNA, the quantification using the Qubit assay gave similar results compared to the two qPCR assays for ccfDNA extracted (Fig. 2, ESM Tables S1 and S2). Consequently, quantification using this fluorometric assay could be sufficient prior to normalization for these extraction methods if only a limited amount of DNA is available.
Integrity index
An integrity index was calculated using the threshold cycle (CP) ratio CP180/CP67 for the APP gene and the intensity ratio I 800/I 400 for the TP53 gene from ccfDNA either extracted with the Norgen Plasma/Serum Circulating DNA Purification Mini Kit or the QIAamp® kit. CcfDNA extracted with these two kits was used as they provided a sufficient quantity of ccfDNA to perform the different experiments in triplicates.
The integrity index of ccfDNA calculated from the APP gene was 0.95 and 0.97 for colon cancer using respectively the Norgen or QIAamp® kits, 1.05 and 1.03 for breast cancer, 1.07 and 1.03 for pancreatic cancer, and 1.08 and 1.07 for healthy female plasma. The standard deviation, calculated from two aliquots of these plasma samples, was between 0.002 and 0.02 for the Norgen kit and between 0.01 and 0.1 for the QIAamp® kit. The integrity index of ccfDNA extracted from the aliquot of plasma using these two extraction methods was thus very similar. There was no bias in size selection for the two extraction methods, but the Norgen kit showed a slightly more reproducible pattern.
The integrity index of ccfDNA from the female plasma was also calculated for the TP53 gene, but the standard deviation measured using the two aliquots of the same sample was substantially higher. Although qPCR assays are more reproducible to determine the integrity index compared to separation by a classical electrophoresis gel, the integrity index of ccfDNA from the TP53 gene allows assessing a broader range of fragment sizes as it is calculated from longer fragments compared to the APP assay. Further optimization of the assay including improved cycling conditions and a lower number of PCR cycles could improve the quantification if required.
The extracted, quality-controlled, and normalized ccfDNA samples could be subsequently analyzed by next-generation sequencing or qPCR [2, 17, 18] or using an enrichment method such as E-ice-COLD-PCR [19] for the detection of somatic mutation in plasma samples.
Conclusion
In summary, the Norgen Plasma/Serum Circulating DNA Purification Mini Kit enabled to isolate a sufficient quantity of ccfDNA from different sources of plasma (cancer patients and healthy individuals of both sexes and different ages) using small quantities of plasma. The quantification of ccfDNA could be determined by the Kpn and DHFRP2 qPCR assays. However, a simple fluorometric assay yielded similar results to estimate the concentration and is less costly and time intensive. The integrity index was determined for the APP and TP53 genes using qPCR assays or analysis of PCR products by gel electrophoresis, respectively. The qPCR showed increased reproducibility for the index integrity, but the PCR separated by gel electrophoresis could be used to qualify longer fragment. In summary, we present in the current manuscript a complete workflow to isolate, quantify, and characterize ccfDNA from small volumes of plasma or serum to detect somatic mutations. It could also potentially be used to analyze DNA methylation patterns in ccfDNA.
References
Esposito A, Bardelli A, Criscitiello C, Colombo N, Gelao L, Fumagalli L, Minchella I, Locatelli M, Goldhirsch A, Curigliano G (2014) Monitoring tumor-derived cell-free DNA in patients with solid tumors: clinical perspectives and research opportunities. Cancer Treat Rev 40:648–655
Murtaza M, Dawson SJ, Tsui DW, Gale D, Forshew T, Piskorz AM, Parkinson C, Chin SF, Kingsbury Z, Wong AS, Marass F, Humphray S, Hadfield J, Bentley D, Chin TM, Brenton JD, Caldas C, Rosenfeld N (2013) Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature 497:108–112
Chan RW, Jiang P, Peng X, Tam LS, Liao GJ, Li EK, Wong PC, Sun H, Chan KC, Chiu RW, Lo YM (2014) Plasma DNA aberrations in systemic lupus erythematosus revealed by genomic and methylomic sequencing. Proc Natl Acad Sci U S A 111:E5302–E5311
Devonshire AS, Whale AS, Gutteridge A, Jones G, Cowen S, Foy CA, Huggett JF (2014) Towards standardisation of cell-free DNA measurement in plasma: controls for extraction efficiency, fragment size bias and quantification. Anal Bioanal Chem 406:6499–6512
Jung K, Fleischhacker M, Rabien A (2010) Cell-free DNA in the blood as a solid tumor biomarker—a critical appraisal of the literature. Clin Chim Acta 411:1611–1624
Fleischhacker M, Schmidt B (2007) Circulating nucleic acids (CNAs) and cancer—a survey. Biochim Biophys Acta 1775:181–232
Elshimali YI, Khaddour H, Sarkissyan M, Wu Y, Vadgama JV (2013) The clinical utilization of circulating cell free DNA (CCFDNA) in blood of cancer patients. Int J Mol Sci 14:18925–18958
Fong SL, Zhang JT, Lim CK, Eu KW, Liu Y (2009) Comparison of 7 methods for extracting cell-free DNA from serum samples of colorectal cancer patients. Clin Chem 55:587–589
Page K, Guttery DS, Zahra N, Primrose L, Elshaw SR, Pringle JH, Blighe K, Marchese SD, Hills A, Woodley L, Stebbing J, Coombes RC, Shaw JA (2013) Influence of plasma processing on recovery and analysis of circulating nucleic acids. PLoS One 8:e77963
Pinzani P, Salvianti F, Zaccara S, Massi D, De Giorgi V, Pazzagli M, Orlando C (2011) Circulating cell-free DNA in plasma of melanoma patients: qualitative and quantitative considerations. Clin Chim Acta 412:2141–2145
Salvianti F, Pinzani P, Verderio P, Ciniselli CM, Massi D, De Giorgi V, Grazzini M, Pazzagli M, Orlando C (2012) Multiparametric analysis of cell-free DNA in melanoma patients. PLoS One 7:e49843
Zaher ER, Anwar MM, Kohail HMA, El-Zoghby SM, Abo-El-Eneen MS (2012) Value of circulating DNA concentration and integrity as a screening test for detection of cancer in an Egyptian cohort. Alex J Med 48:187–196
Schmidt B, Weickmann S, Witt C, Fleischhacker M (2005) Improved method for isolating cell-free DNA. Clin Chem 51:1561–1563
Yuan H, Zhu ZZ, Lu Y, Liu F, Zhang W, Huang G, Zhu G, Jiang B (2012) A modified extraction method of circulating free DNA for epidermal growth factor receptor mutation analysis. Yonsei Med J 53:132–137
Hufnagl C, Stöcher M, Moik M, Geisberger R, Greil R (2013) A modified phenol-chloroform extraction method for isolating circulating cell free DNA of tumor patients. J Nucleic Acids Investig 4:1
Xue X, Teare MD, Holen I, Zhu YM, Woll PJ (2009) Optimizing the yield and utility of circulating cell-free DNA from plasma and serum. Clin Chim Acta 404:100–104
Thierry AR, Mouliere F, El Messaoudi S, Mollevi C, Lopez-Crapez E, Rolet F, Gillet B, Gongora C, Dechelotte P, Robert B, Del Rio M, Lamy PJ, Bibeau F, Nouaille M, Loriot V, Jarrousse AS, Molina F, Mathonnet M, Pezet D, Ychou M (2014) Clinical validation of the detection of KRAS and BRAF mutations from circulating tumor DNA. Nat Med 20:430–435
Lebofsky R, Decraene C, Bernard V, Kamal M, Blin A, Leroy Q, Rio Frio T, Pierron G, Callens C, Bieche I, Saliou A, Madic J, Rouleau E, Bidard FC, Lantz O, Stern MH, Le Tourneau C, Pierga JY (2015) Circulating tumor DNA as a non-invasive substitute to metastasis biopsy for tumor genotyping and personalized medicine in a prospective trial across all tumor types. Mol Oncol 9:783–90
How-Kit A, Lebbe C, Bousard A, Daunay A, Mazaleyrat N, Daviaud C, Mourah S, Tost J (2014) Ultrasensitive detection and identification of BRAF V600 mutations in fresh frozen, FFPE, and plasma samples of melanoma patients by E-ice-COLD-PCR. Anal Bioanal Chem 406:5513–5520
Acknowledgments
The authors would like to thank Chao Feng (CNG) for his help with the statistical analysis and Steven McGinn (CNG) for the critical reading of the manuscript. This work was partly supported by the EU FP7 under grant agreement number 241669 (the CAGEKID project, http://www.cng.fr/cagekid).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 237 kb)
Rights and permissions
About this article

Cite this article
Mauger, F., Dulary, C., Daviaud, C. et al. Comprehensive evaluation of methods to isolate, quantify, and characterize circulating cell-free DNA from small volumes of plasma. Anal Bioanal Chem 407, 6873–6878 (2015). https://doi.org/10.1007/s00216-015-8846-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-015-8846-4