
Abstract
A derivatization method that employs diethyl (bromodifluoromethyl) phosphonate (DBDFP) to efficiently tag the endocrine disruptor pentachlorophenol (PCP) and other chlorinated phenols (CPs) along with their reliable detection and analysis by NMR is presented. The method accomplishes the efficient alkylation of the hydroxyl group in CPs with the difluoromethyl (CF2H) moiety in extremely rapid fashion (5 min), at room temperature and in an environmentally benign manner. The approach proved successful in difluoromethylating a panel of 18 chlorinated phenols, yielding derivatives that displayed unique 1H, 19F, and 13C NMR spectra allowing for the clear discrimination between isomerically related CPs. Due to its biphasic nature, the derivatization can be applied to both aqueous and organic mixtures where the analysis of CPs is required. Furthermore, the methodology demonstrates that PCP along with other CPs can be selectively derivatized in the presence of other various aliphatic alcohols, underscoring the superiority of the approach over other general derivatization methods that indiscriminately modify all analytes in a given sample. The present work demonstrates the first application of NMR on the qualitative analysis of these highly toxic and environmentally persistent species.

Rapid and selective derivatization of chlorinated phenols with the difluoromethyl tag for their analysis by NMR spectroscopy
Similar content being viewed by others
Introduction
Chlorinated phenols (CPs), in general, have posed a serious environmental concern due to their high levels of production and toxicity to organisms at extremely low concentrations, earning them a notorious profile and their induction in the list of endocrine disruptor compounds (EDCs) [1]. Due to the wide usage of these compounds in industry as antiseptics, insecticides, wood preservatives, and as important intermediates in the production of pharmaceutical products [2–4], their derivatives as well as their degradation products can be found worldwide in surface and ground waters, bottom sediments, and atmospheric air and solids. In order to mitigate the impact of CPs in the environment and human health, their use has been subject to severe restriction policies and even banning in several countries [4]. Nevertheless, although their use has experienced a steady decline, these chemicals still remain in the environment due to their chemical stability aided by additional physical properties in the water, soil, or sediment that harbors them such as pH and temperature [5].
Methods for the extraction, detection, and monitoring of CPs using numerous analytical techniques exist. The majority of these rely heavily on their intrinsic UV absorption (e.g., LC-MS) [6, 7] while others depend on their semi-volatility brought upon by their proclivity for derivatization (GC-MS) [8]. Although the power of these aforementioned techniques is well established in the analytical chemistry realm, they still represent one-dimensional approaches for the study of these analytes. It is only when we dwell into analyses procured by two-dimensional methods and/or hyphenated GC and LC methods that we truly see their power in studying these species. An approach centered on the well-established and powerful NMR spectroscopy technique offers a multidimensional series of analysis that can all be carried out in one sample and in a non-destructive manner, an absent quality in the previously mentioned methods. However, a NMR approach for the analysis of even CPs could be demandingly challenging, particularly when a mixture of other similar species such as other types of alcohols and a matrix is present. Due to the ubiquitous presence of the proton (1H) in most organic molecules (including those in a given matrix), analysis of such a sample would represent a tedious and complex undertaking. Therefore, a method capable of selectively tagging CPs amid a mixture of other analytes would be an invaluable tool during the data deconvolution process of a given analysis. Furthermore, if this selective tag offers the means to analyze CPs in multiple NMR channels (e.g., 19F, 13C), it would tremendously aid in the unequivocal identification of each CP component in the mixture. After surveying several tagging functionalities for alcohols, particularly acidic phenols like CPs, we selected the difluoromethyl moiety (CF2H) for our studies. The choice of the difluoromethyl tag for labeling CPs was supported by several factors that can be directly attributed to the versatility of the NMR-amenable nuclei available in its structure. For example, using the difluoromethyl tag immediately introduces two readily detectable nuclei (1H and 19F) by NMR, which as a result of their natural abundance enjoy great sensitivity in this form of spectroscopic analysis (Fig. 1). In the NMR field, 19F constitutes the most abundant isotope of the element and, as mentioned briefly previously, it introduces NMR as a highly sensitive method for the detection of fluorine-containing species in a rapid manner (lowest possible number of scans for a given sample). In addition, the large chemical shift range exhibited by the 19F NMR channel allows for the clear resolution of structurally similar derivatized CPs. Lastly, the tag further benefits from possessing another reporting nucleus in the 1H species (CF2 H), whose chemical shift occurs between 6.7 and 6.9 ppm, located further upfield and well outside the resonances of aliphatic and olefinic 1H nuclei. Additionally, analysis of the aromatic 1H signals of the derivatized CP (except for pentachlorophenol, PCP) further aids in their structure determination and discrimination amongst isomers. Studying the derivatization using the 13C NMR channel, although possible as demonstrated in Table 1, would present additional challenges for this type of analysis due to the inherent low sensitivity of this nucleus in NMR spectroscopy. Thus, for the analysis of mixtures, we envision the combined use of 1H and 19F NMR to rapidly and qualitatively assess the presence of acidic phenols (including CPs), followed by further complementary analysis by GC-MS and/or LC-MS. The derivatization protocol described in this work efficiently achieves the difluoromethylation of CPs producing modified species that can be studied by 1H, 19F, and 13C NMR spectroscopy (Fig. 1).

Key features of the difluoromethylation strategy described in this work
Materials and methods
All chemicals were purchased from commercial suppliers and used as received. Acetonitrile, methylene chloride, and the used chlorophenols were purchased from Sigma-Aldrich Chemicals (St. Louis, MO), and diethyl (bromodifluoromethyl) phosphonate was purchased from Matrix Scientific Inc. (Columbia, SC). Deuterated acetonitrile (CD3CN) was purchased from Alfa Aesar (Ward Hill, MA).
Derivatization protocol
The chlorinated phenol (0.08 mmol) was placed in a glass autosampler vial equipped with a small stir bar. The phenol was treated sequentially via pipette with deuterated acetonitrile [9] (CD3CN, 600 μL) and diethyl (bromodifluoromethyl) phosphonate (21.4 μL, 0.12 mmol, 1.5 equiv. to phenol). To the above solution, aqueous saturated potassium hydroxide (300 μL) was added via pipette. The vial was capped and stirred at ambient temperature for 5 min (see Electronic Supplementary Material (ESM), Fig. S1 and Table S1). After the stirring was done, the mixture was allowed to stand to reveal a biphasic mixture and 500 μL of the top layer (CD3CN) was aliquoted into another glass autosampler vial containing anhydrous sodium sulfate (Na2SO4, 50 mg). The dried, organic fraction was filtered into a 5-mm NMR tube through a cotton plug and treated with 100 μL of a 0.17-M solution of hexafluorobenzene in CD3CN (19F NMR internal standard).
NMR analysis
Spectra were obtained using a Bruker Avance III 600-MHz instrument equipped with a Bruker TCI 5-mm cryoprobe (Bruker BioSpin, Billerica, MA) at 30.0 ± 0.1 °C. 1H NMR (600 MHz), 19F NMR (565 MHz), and 13C NMR (150 MHz) were recorded in CD3CN. 1H NMR chemical shifts are calibrated with respect to residual partially deuterated acetonitrile centered at 1.94 ppm. 19F NMR chemical shifts are calibrated with respect to the singlet given by hexafluorobenzene at −164.9 ppm. Lastly, for 13C NMR, the septet centered at 1.79 ppm from CD3CN was used for the spectral calibration.
GC-MS analysis
A 6890 Agilent GC with a 5975 MS detector equipped with a split/splitless injector was used for the analysis. The GC column used for the analysis was an Agilent DB-5MS capillary column (30 m × 0.25 mm id × 0.25 μm i.f.). Ultra high purity helium was used as the carrier gas at 0.8 mL/min. The injector temperature was 250 °C, and the injection volume was 1 μL. The oven temperature program was as follows: 40 °C, held for 3 min, increased at 8 °C/min to 300 °C, held for 3 min. The MS ion source and quadrupole temperatures were 230 and 150 °C, respectively. Electron ionization was used with ionization energy of 70 eV. The MS was operated to scan from m/z 29 to m/z 600 in 0.4 s.
Results and discussion
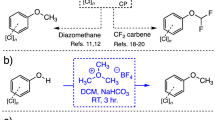
The difluoromethyl (CF2H) moiety has been a key player in the pharmaceutical industry where its role as an isosteric group for the methyl moiety has been, and continues to be, exploited for its superior chemical attributes. Even though its size is comparable to that of the methyl group, its electronic properties are significantly different due to the presence of the fluorine atoms in its makeup. Due to the importance of this group for the aforementioned reason, it is no surprise that many methods have been developed for its introduction in molecular targets [10]. Although some of the methods thus far developed require the use of heating and long reaction times, a method that makes use of diethyl (bromodifluoromethyl) phosphonate (DBDFP) developed by the Zafrani group provides a rapid and environmentally benign option [11]. Due to its high reactivity under basic conditions (pH ∼12), reactions using DBDFP are often carried out at low temperatures (−78 °C) followed by warming up the mixture to room temperature once the reagent has been used to modify a phenolic moiety. The proposed mechanism for the overall transformation is outlined in Scheme 1. Thus, in the highly basic conditions that the reaction is carried out, DBDFP rapidly reacts with the base to produce a bromodifluoromethyl carbanion that spontaneously decomposes, yielding the bromide anion and difluorocarbene. Reaction of the fleeting difluorocarbene with the nucleophilic phenoxide ion produces an anionic intermediate that rapidly protonates to furnish the difluoromethylated phenol [12]. The ease of formation of the phenoxide ion, especially in the case of the chlorinated phenols [13], is greatly favored under these conditions due to their low pK a values (Table 1). Note that most unwanted products will be soluble in the aqueous phase (i.e., chlorophenoxide anion, diethylphosphate) and will not be carried onto the organic phase where the newly derivatized CP now resides. The organic phase can be directly analyzed by NMR and GC-MS after drying over Na2SO4, yielding information that is unique for each derivatized CP (Table 1). The fact that DBDFP can accomplish the efficient derivatization of PCP in a very short amount of time (5 min), compounded to its liquid state at room temperature, makes it an attractive alternative for the derivatization and study of CPs.

Proposed mechanism for the difluoromethylation reaction
After having evaluated the efficiency of the approach in derivatizing CPs, we proceeded to test its selectivity for tagging these species in the presence of other structurally diverse alcohols. To this end, we prepared a mixture in CD3CN consisting of PCP (0.08 mmol) with 12 other structurally diverse alcohols (each spiked at 0.08 mmol) and these included primary (2-methyl-1-butanol, 2-methyl-1-pentanol), secondary (pinacolyl alcohol), tertiary (1-methylcyclopentanol, 3-ethyl-3-pentanol), N-substituted β-amino alcohols (N,N-diisopropylaminoethanol, N,N-dimethylaminoethanol, N-methyldiethanolamine), long-chained alcohols (2-decanol, 1-nonanol), and thioether- and sulfone-containing alcohols (2,2′-thiodiethanol, 2,2′-sulfonyldiethanol) (ESM, Fig. S2 and S3). Once the mixture was treated sequentially with DBDFP and aqueous KOH solution (pH = 12.2), it was stirred for 2 min and the organic layer analyzed by 1H and 19F NMR after drying over Na2SO4. Treatment of the equimolar alcohol mixture with 1, 1.5, 2, and 4 equivalents of DBDFP resulted in the efficient and selective derivatization of only PCP. The use of excess of DBDFP does not cause a surge of interfering signals in the vicinity of the OCF2H-derived triplet at ∼δ = 6.76 ppm, but signals localized in the δ = 0.7–1.1 ppm range arising from the reagent’s hydrolysis and by-products begin to increase in magnitude (ESM, Fig. S2). This observation was further confirmed by the GC-MS analysis of the mixture where 1.5 equivalents of DBDFP was used for the derivatization. The GC chromatogram demonstrated that the only products arising from the procedure belong to PCP and its minor contaminant 2,3,4,6-tetrachlorophenol, while the 12 alcohols in the mixture remained underivatized (ESM, Fig. S3). During our initial assessment of the protocol, we carried out the derivatization for a space of less than 2 min leading to what appeared to be the quantitative derivatization of PCP by NMR analysis. However, it was found that running the procedure for <2 min did not result in the complete derivatization of PCP (ESM, Fig. S3). Therefore, after conducting optimization studies on the protocol, it was found that 1.5 equivalents of DBDFP along with stirring for 5 min is enough to secure the full derivatization of PCP (ESM, Table S1 and Figures S6 to S17). Lastly, the efficiency of the protocol was evaluated by testing it out in a mixture of five CPs. The five CPs used for this study were 4-chlorophenol, 3,5-dichlorophenol, 3,4,5-trichlorophenol, 2,3,4,5-tetrachlorophenol, and PCP. It was found that all five CPs smoothly undergo the derivatization and that each product retains its unique chemical shift (1H and 19F NMR) (ESM, Fig. S4 and S5). Using the chemical shift values for the derivatives in both NMR channels and adding the information that each CP’s aromatic signals provide (i.e., aromatic substitution), one can deduce the exact nature of the original CP in a given mixture. Lastly, an additional, strong attribute of the process is its biphasic nature allowing its adaptation for CP analysis on organic as well as aqueous samples once the derivatization has taken place.
Conclusion
Our experiments have demonstrated the efficacy of diethyl (bromodifluoromethyl) phosphonate at difluoromethylating the endocrine disruptor pentachlorophenol and related CPs for their detection and analysis by multinuclear NMR. The reagent is convenient to use as it is a liquid at room temperature, and once it has reacted, its products are environmentally benign. Furthermore, the derivatization reaction is expedient in nature (5 min) and can be conveniently carried out at room temperature to furnish difluoromethylated derivatives that exhibit unique 1H, 19F, and 13C NMR spectra. Due to its biphasic nature, the derivatization can be applied to both aqueous and organic mixtures. In addition, the protocol demonstrates that CPs can be selectively derivatized in the presence of various kinds of aliphatic alcohols, underscoring the potential superiority of the approach over other general derivatization methods that indiscriminately modify all analytes in a given sample. Due to the ease and mild conditions involved in its execution, this methodology should find wide applicability in the derivatization and qualitative analysis of not only CPs but other environmentally relevant phenolic compounds as well. Most importantly, and as briefly alluded to in the present work, the protocol lends itself as an additional, non-destructive technique for the analysis of acidic phenols in conjunction with other techniques such as GC-MS and LC-MS.
References
Orton F, Lutz I, Kloas W, Routledge EJ (2009) Endocrine disrupting effects of herbicides and pentachlorophenol: in vitro and in vivo evidence. Environ Sci Technol 43:2144–2150
Czaplicka M (2004) Sources and transformations of chlorophenols in the natural environment. Sci Total Environ 322:21–39
Olaniran AO, Igbinosa EO (2011) Chlorophenols and other related derivatives of environmental concern: properties, distribution and microbial degradation processes. Chemosphere 83:1297–1306
Zheng W, Wang X, Yu H, Tao X, Zhou Y, Qu W (2011) Global trends and diversity in pentachlorophenol levels in the environment and in humans: a meta-analysis. Environ Sci Technol 45:4668–4675
Arcand Y, Hawari J, Guiot SR (1995) Solubility of pentachlorophenol in aqueous solutions: the pH effect. Wat Res 29:131–136
Kim D, Han J, Choi Y (2013) On-line solid-phase microextraction of triclosan, bisphenol A, chlorophenols, and selected pharmaceuticals in environmental water samples by high-performance liquid chromatography–ultraviolet detection. Anal Bioanal Chem 405:377–387
Martínez Vidal JL, Belmonte Vega A, Garrido Frenich A, Egea González FJ, Arrebola Liebanas FJ (2004) Determination of fifteen priority phenolic compounds in environmental samples from Andalusia (Spain) by liquid chromatography-mass spectrometry. Anal Bioanal Chem 379:125–130
Rompa M, Kremer E, Zygmunt B (2003) Derivatisation in gas chromatographic determination of acidic herbicides in aqueous environmental samples. Anal Bioanal Chem 377:590–599
The use of acetonitrile as a solvent is important, as once the derivatized sample has been analyzed by NMR, it can be directly employed in GC- and LC-MS analyses with no need for solvent exchange
Mild heating (45–50 °C) for 1–2 hours is needed when the difluorocarbene generating compound fluorosulfonyldifluoroacetic acid is used: Chen QY, Wu SW (1989) A simple convenient method for preparation of difluoromethyl ethers using fluorosulfonyldifluoroacetic acid as a difluorocarbene precursor. J. Fluorine Chem. 44: 433–440; whereas for the agent sodium chlorodifluoroacetate, heating at 100 °C for 16 hours is required: Sifferlen T, Koberstein R, Cottreel E, Boller A, Weller T, Gatfield J, Brisbare-Roch C, Jenck F, Boss C (2013) Structure-activity relationship studies and sleep-promoting activity of novel 1-chloro-5,6,7,8-tetrahydroimidazo[1,5-a]pyrazine derivatives as dual orexin receptor antagonists. Part 2. Bioorg. Med. Chem. Lett. 23: 3857–3863
Zafrani Y, Sod-Moriah G, Segall Y (2009) Diethyl bromodifluoromethylphosphonate: a highly efficient and environmentally benign difluorocarbene precursor. Tetrahedron 65:5278–5283
Flynn RM, Burton DJ, Wiemers DM (2008) Synthetic and mechanistic aspects of the reactions between bromodifluoromethyltriphenylphosphonium bromide and dibromofluoromethyltriphenylphosphonium bromide and trialkylphosphites. J Fluorine Chem 129:583
Due to the close pKa values amongst phenolic species, this method is expected to result in their difluoromethylation. As originally presented in Ref. 11, the propensity of the alkylation increases as the pKa value of a phenol decreases
Acknowledgments
Disclaimer
This document (LLNL-JRNL-666745) was prepared as an account of work sponsored by an agency of the U.S. government. Neither the U.S. government nor Lawrence Livermore National Security, LLC, nor any of their employees makes any warranty, expressed or implied, or assumes any legal liability or responsibility for the accuracy, completeness, or usefulness of any information, apparatus, product, or process disclosed, or represents that its use would not infringe privately owned rights. Reference herein to any specific commercial product, process, or service by trade name, trademark, manufacturer, or otherwise does not necessarily constitute or imply its endorsement, recommendation, or favoring by the U.S. government or Lawrence Livermore National Security, LLC. The views and opinions of authors expressed herein do not necessarily state or reflect those of the U.S. government or Lawrence Livermore National Security, LLC, and shall not be used for advertising or product endorsement purposes.
Auspices statement
This work was performed under the auspices of the U.S. Department of Energy by Lawrence Livermore National Laboratory under Contract DE-AC52-07NA27344.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 3239 kb)
Rights and permissions
About this article

Cite this article
Valdez, C.A., Leif, R.N. Chemical tagging of chlorinated phenols for their facile detection and analysis by NMR spectroscopy. Anal Bioanal Chem 407, 3539–3543 (2015). https://doi.org/10.1007/s00216-015-8625-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-015-8625-2